Thermal Analysis of Crystallization and Phase Transition in Novel Polyethylene Glycol Grafted Butene-1 Copolymers

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
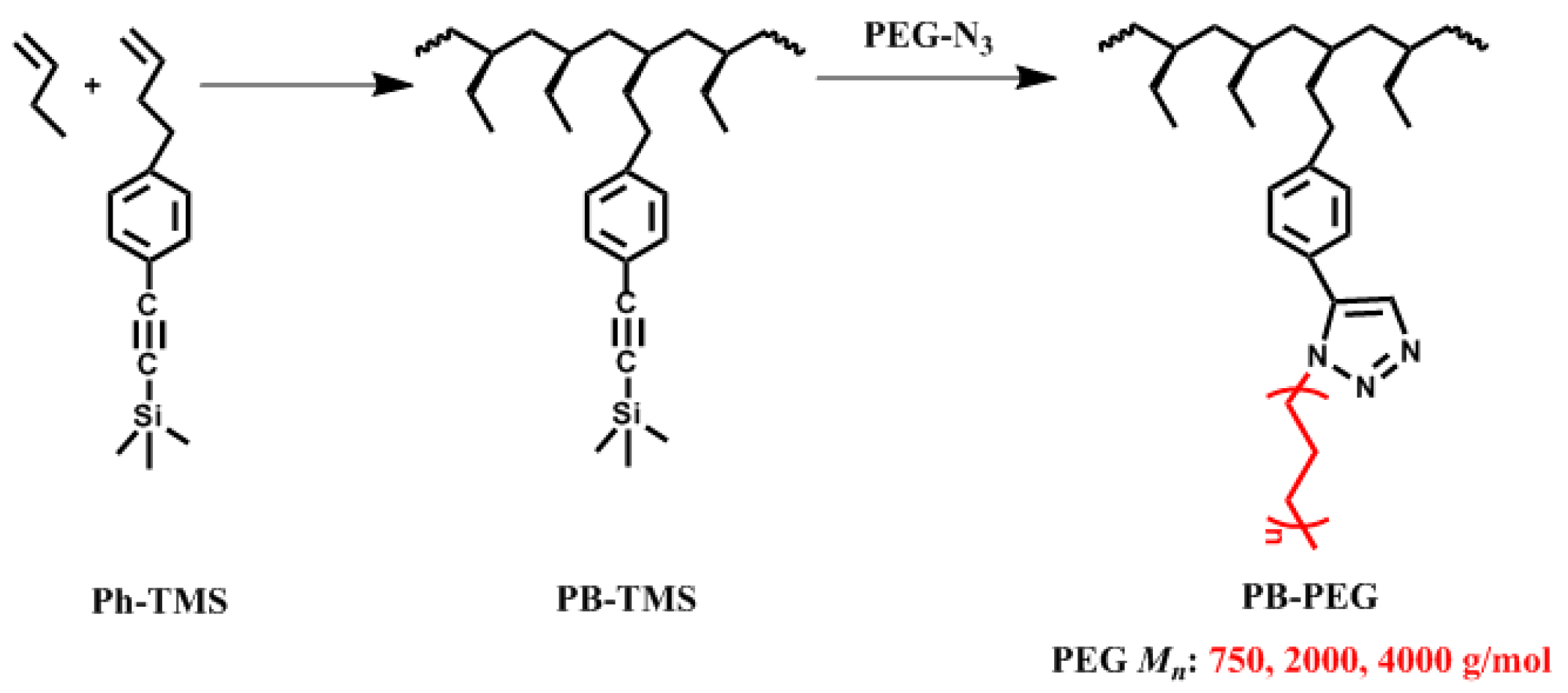
2.2. Copolymerization of Butene-1 with Ph-TMS
2.3. Synthesis of Polybutene-1 Grafted with Polyethylene Glycol
2.4. Methods
2.4.1. Nuclear Magnetic Resonance (1H-NMR) and Gel Permeation Chromatography (GPC) Characterization
2.4.2. Differential Scanning Calorimetry (DSC)
2.4.3. Fourier Transform Infrared Spectroscopy (FTIR)
3. Results and Discussion
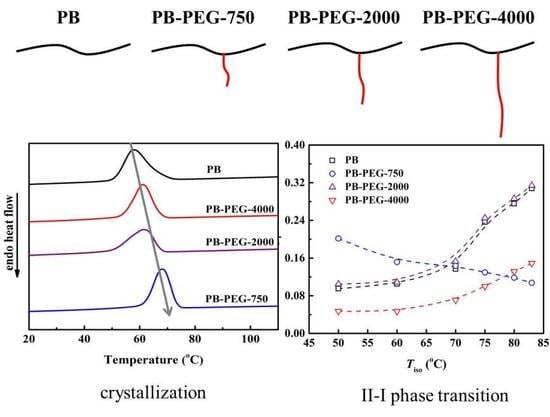
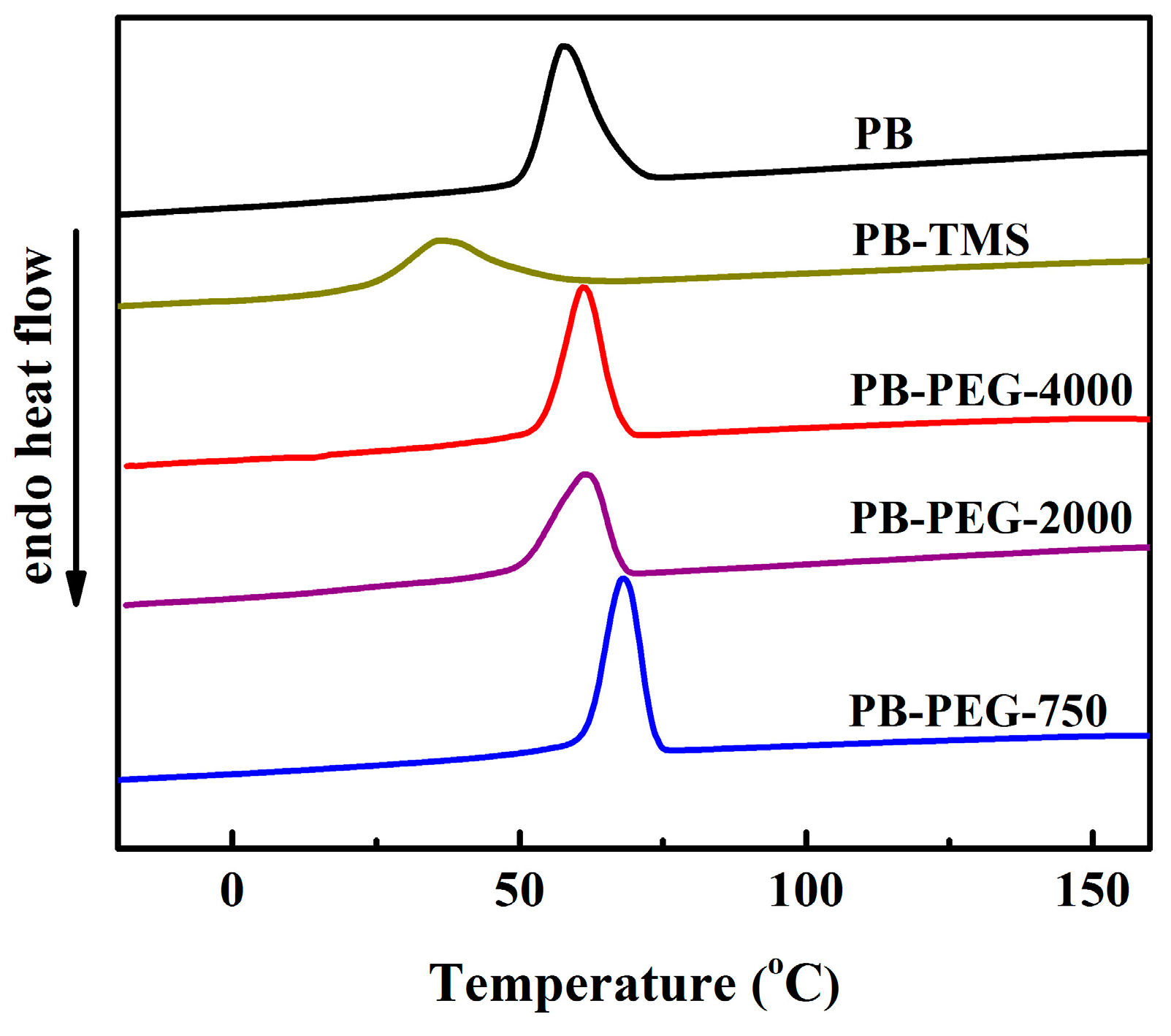
3.1. Non-Isothermal Crystallization
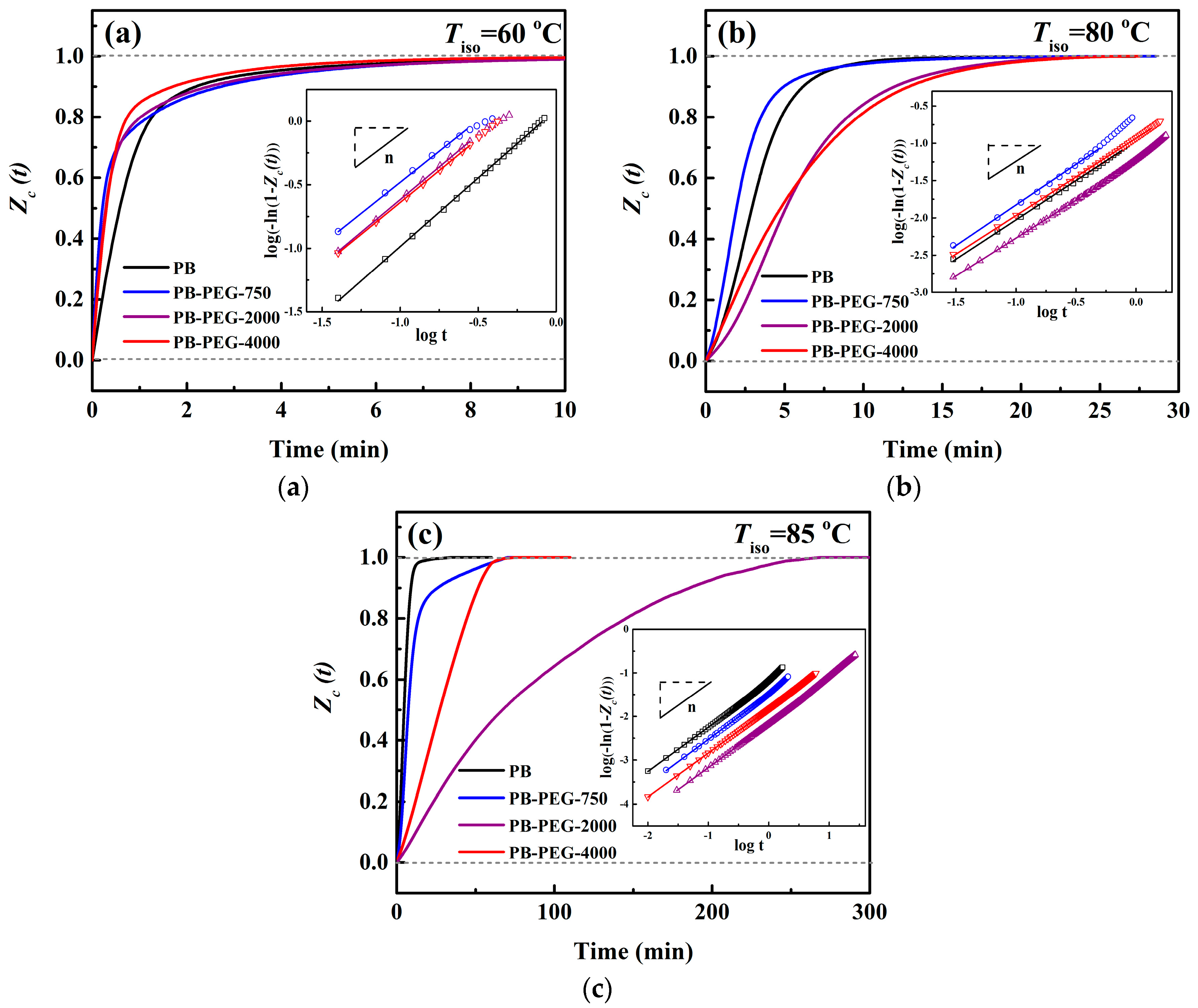
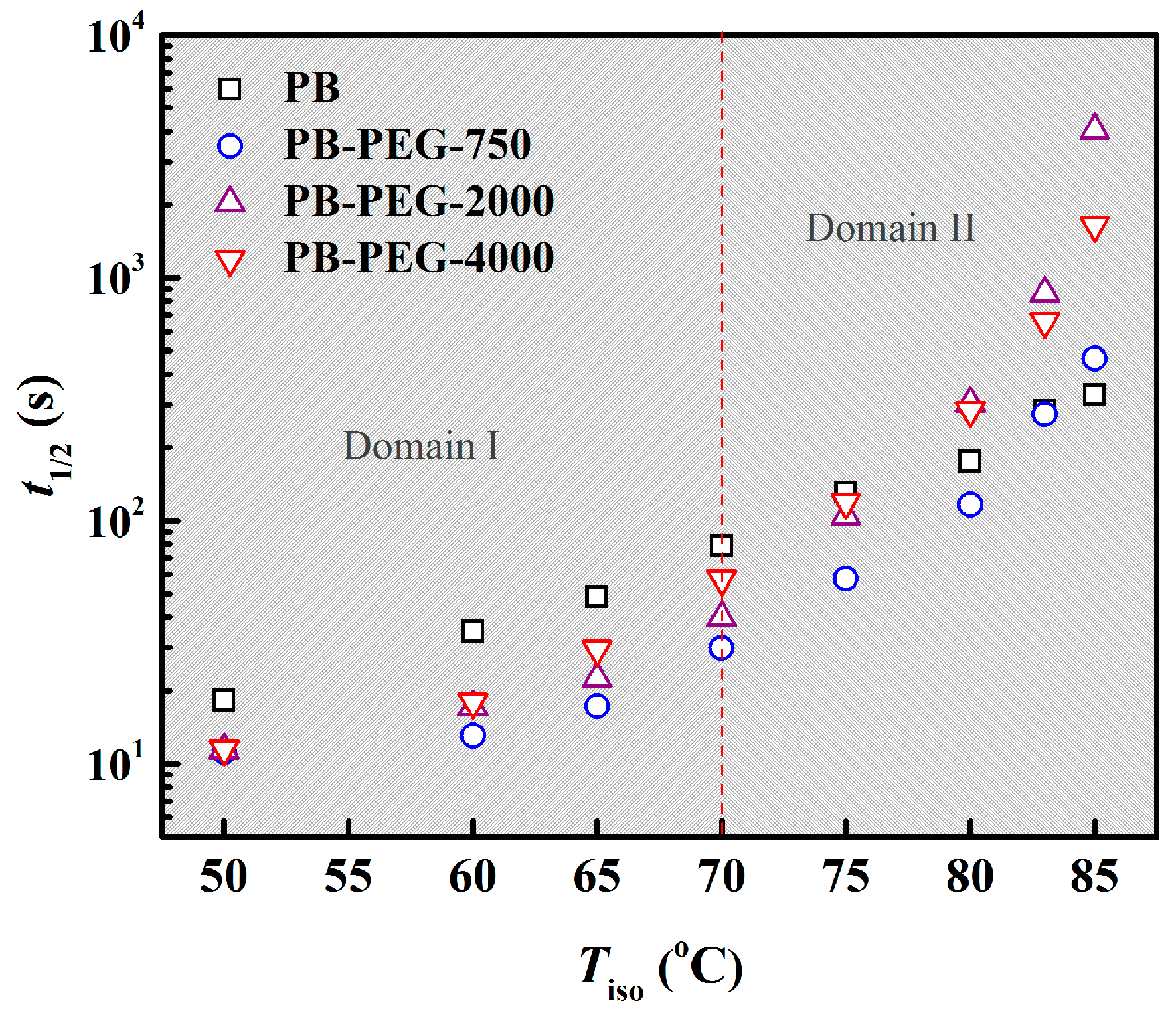
3.2. Isothermal Crystallization Kinetics
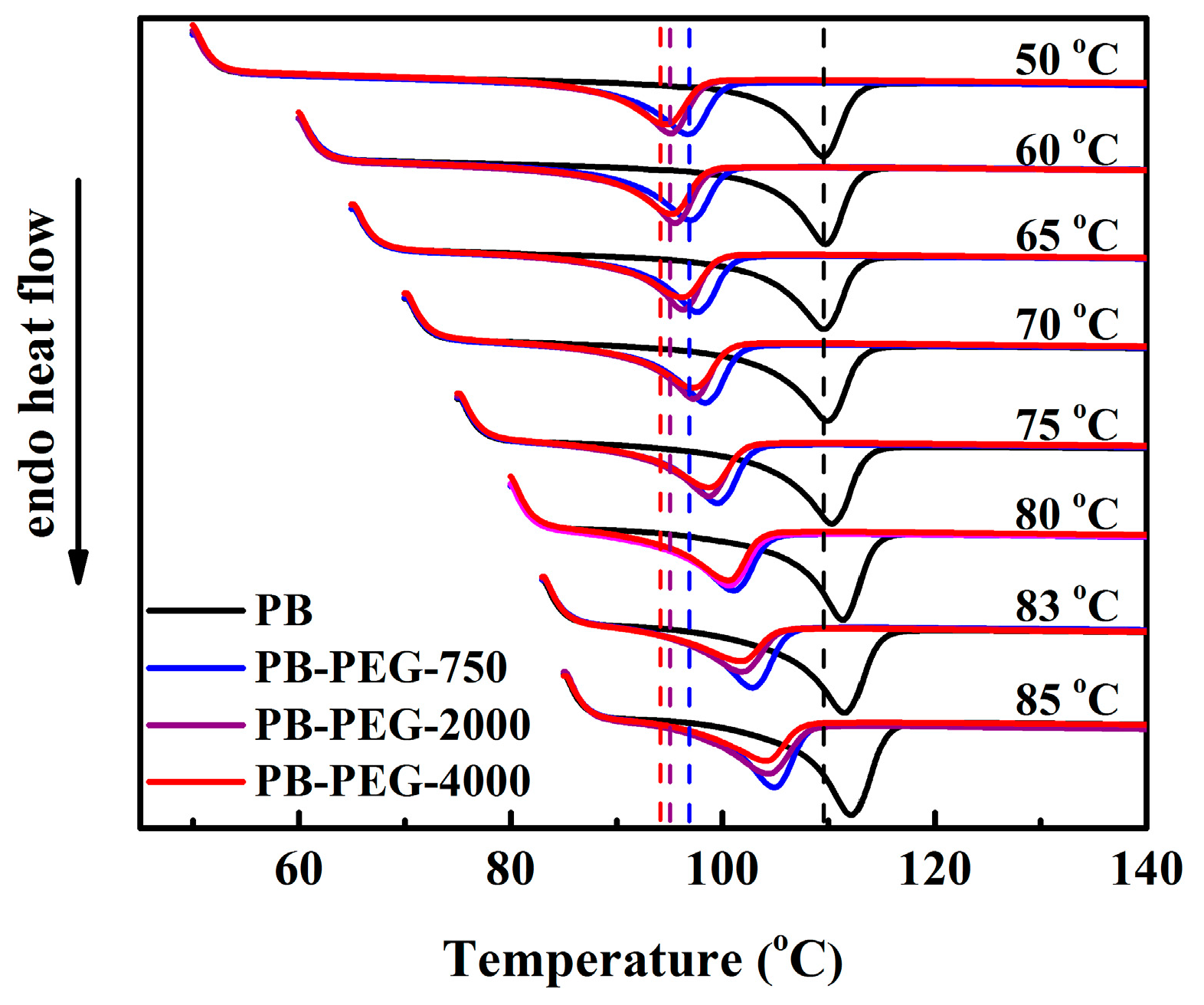
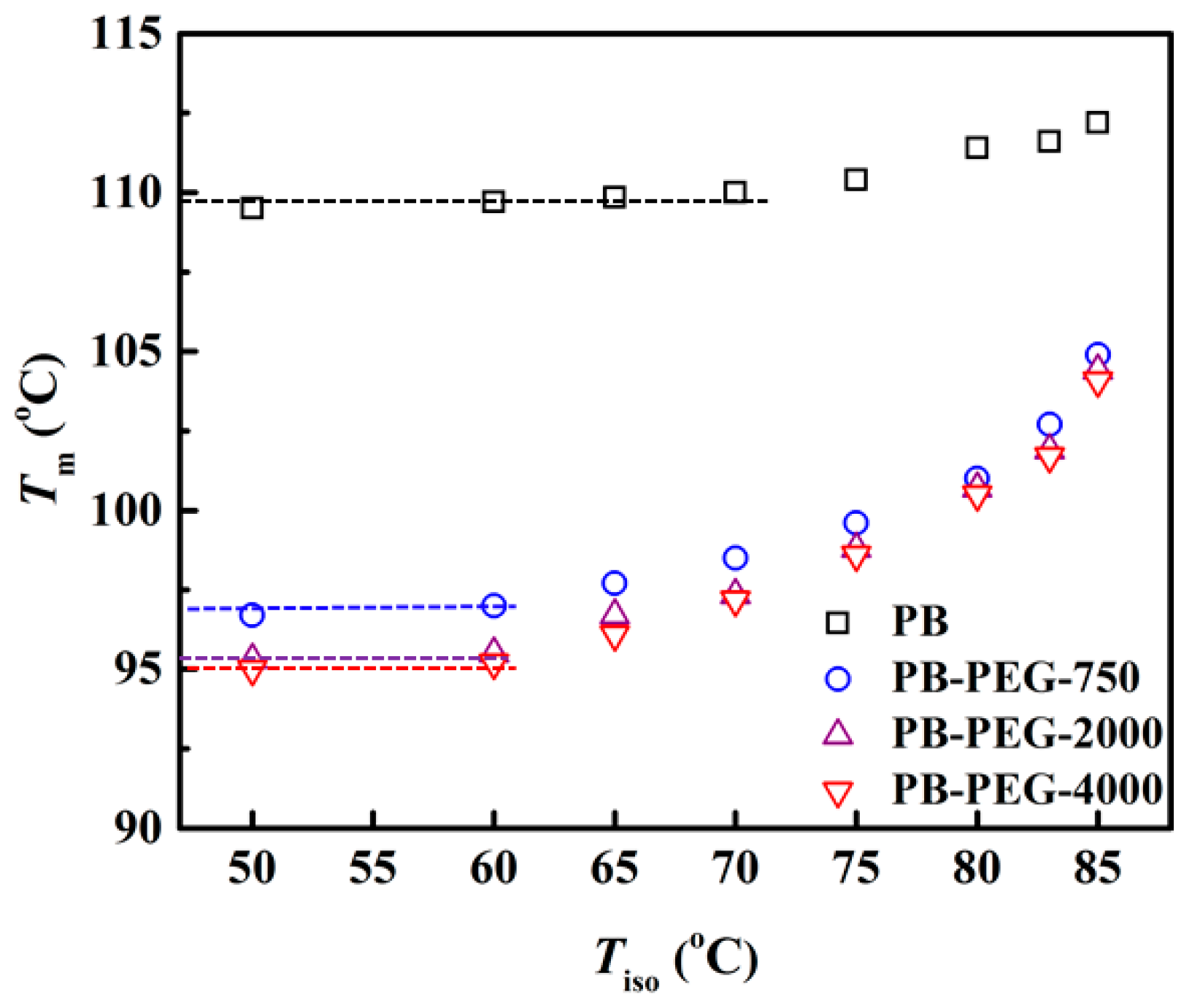
3.3. Melting Behavior
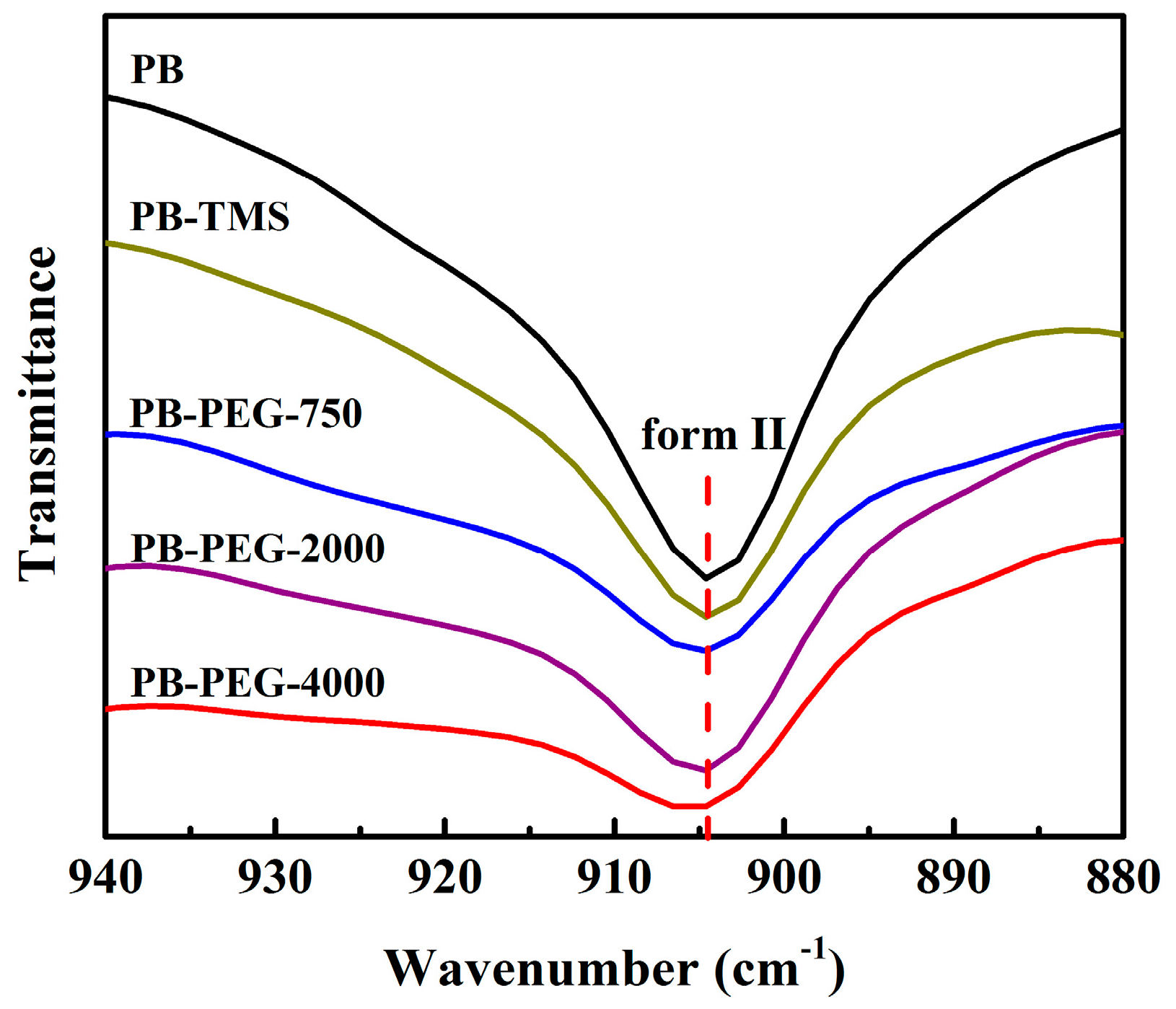
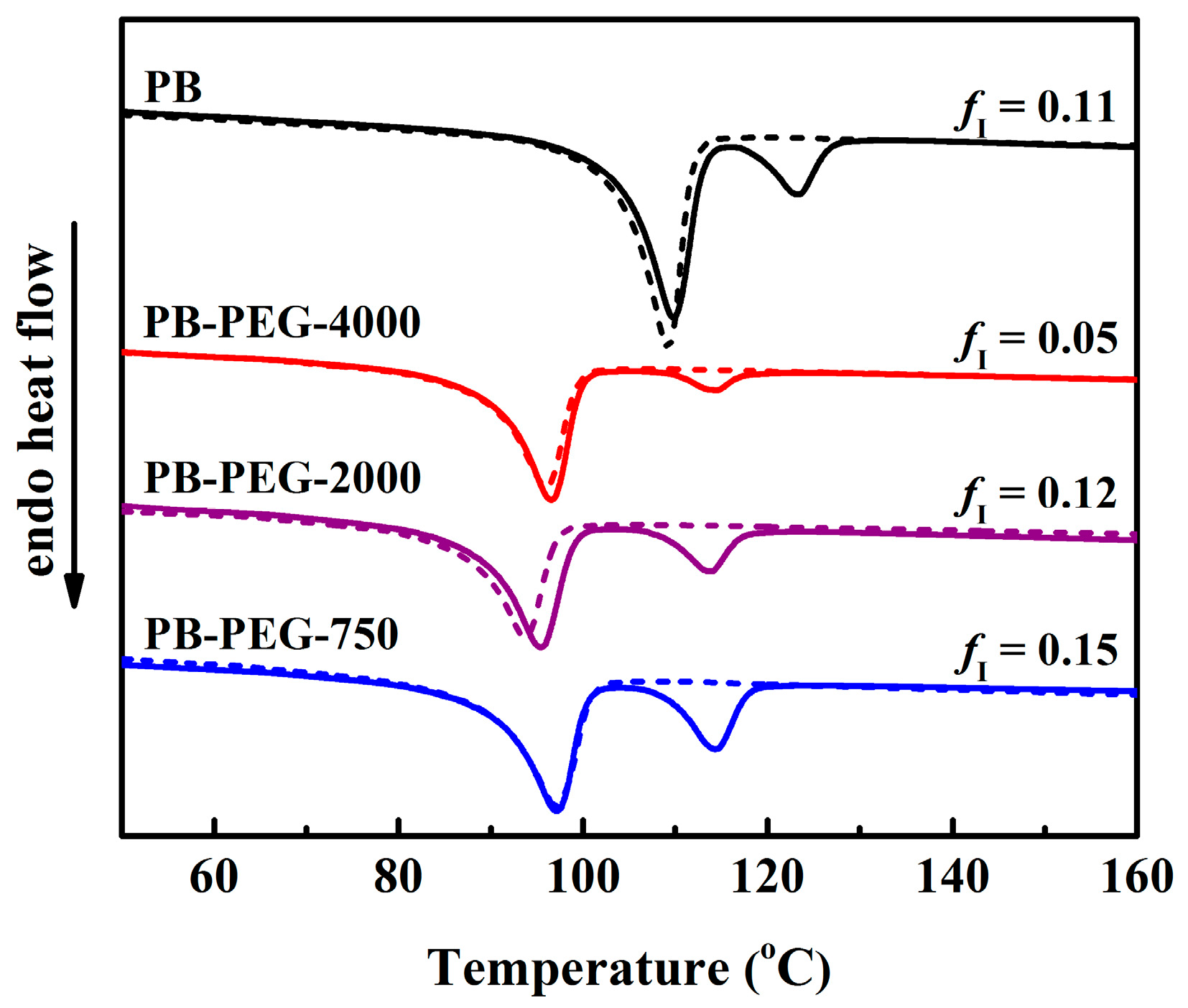
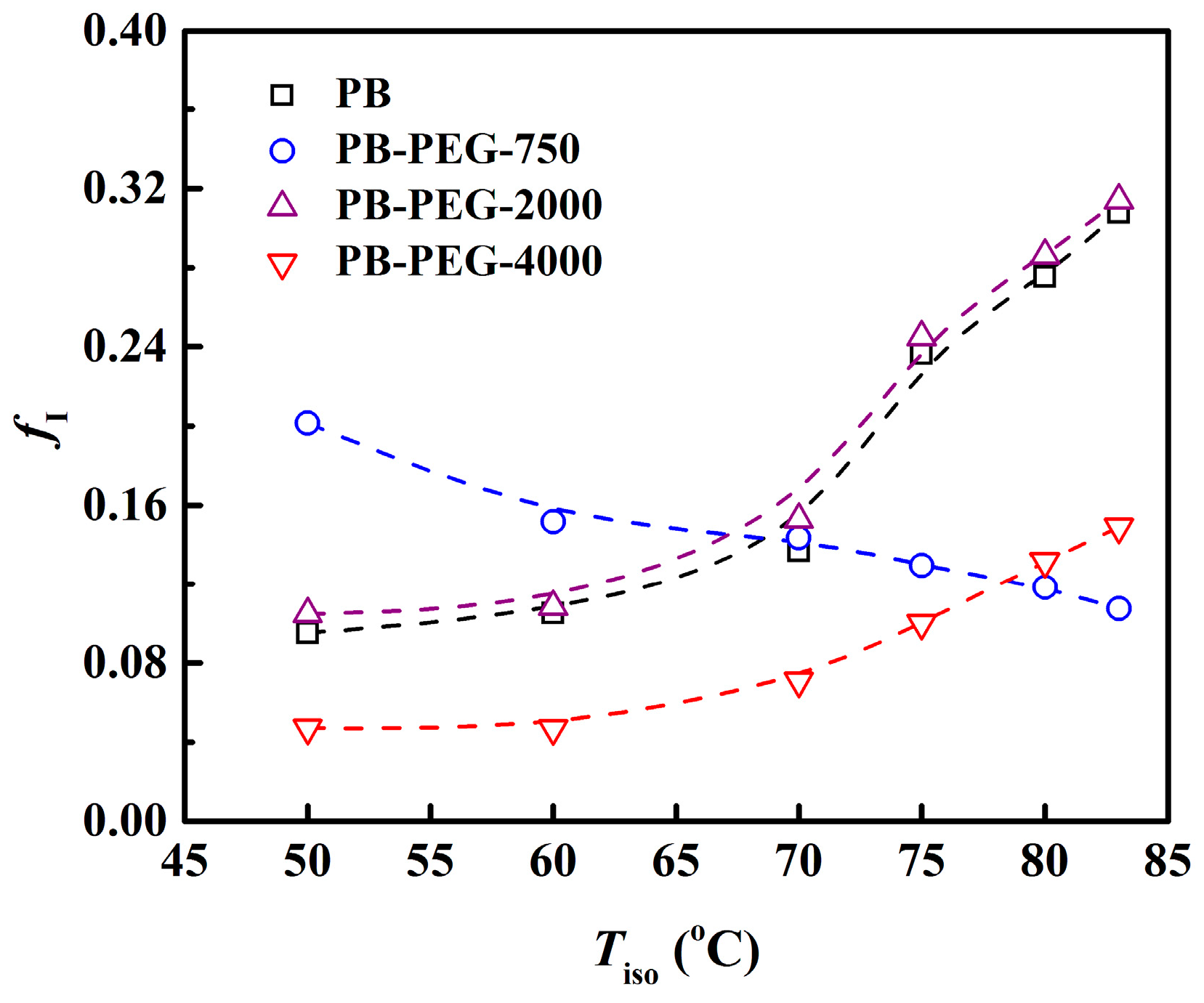
3.4. Polymorphic Form II to Form I Phase Transition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nakafuku, C.; Miyaki, T. Effect of pressure on the melting and crystallization behaviour of isotactic polybutene-1. Polymer 1983, 24, 141–148. [Google Scholar] [CrossRef]
- Luciani, L.; Seppälä, J.; Löfgren, B. Poly-1-butene: Its preparation, properties and challenges. Prog. Polym. Sci. 1988, 13, 37–62. [Google Scholar] [CrossRef]
- Azzurri, F.; Flores, A.; Alfonso, G.C.; Baltá Calleja, F.J. Polymorphism of isotactic poly(1-butene) as revealed by microindentation hardness. 1. kinetics of the transformation. Macromolecules 2002, 35, 9069–9073. [Google Scholar] [CrossRef]
- Azzurri, F.; Flores, A.; Alfonso, G.C.; Sics, I.; Hsiao, B.S.; Baltá Calleja, F.J. Polymorphism of isotactic polybutene-1 as revealed by microindentation hardness. Part II: Correlations to microstructure. Polymer 2003, 44, 1641–1645. [Google Scholar] [CrossRef]
- Lou, Y.H.; Liao, Y.L.; Pan, L.; Wang, B.; Li, Y.S.; Ma, Z. Effect of linear and ring-like co-units on the temperature dependence of nucleation and growth in II-I phase transition of butene-1 copolymers. Chin. J. Polym. Sci. 2018, 36, 1269–1276. [Google Scholar] [CrossRef]
- Li, J.Q.; Wang, D.; Cai, X.Q.; Zhou, C.B.; de Claville Christiansen, J.; Sørensen, T.; Yu, D.H.; Xue, M.L.; Jiang, S.C. Conformation selected direct formation of form I in isotactic poly(butene-1). Cryst. Growth Des. 2018, 18, 2525–2537. [Google Scholar] [CrossRef]
- De Rosa, C.; Auriemma, F.; Ruiz de Ballesteros, O.; Esposito, F.; Laguzza, D.; Di Girolamo, R.; Resconi, L. Crystallization properties and polymorphic behavior of isotactic poly(1-butene) from metallocene catalysts: The crystallization of form I from the melt. Macromolecules 2009, 42, 8286–8297. [Google Scholar] [CrossRef]
- Liu, Y.P.; Cui, K.P.; Tian, N.; Zhou, W.Q.; Meng, L.P.; Li, L.B.; Ma, Z.; Wang, X.L. Stretch-induced crystal–crystal transition of polybutene-1: An in situ synchrotron radiation wide-angle X-ray scattering study. Macromolecules 2012, 45, 2764–2772. [Google Scholar] [CrossRef]
- Wang, Y.T.; Liu, P.R.; Lu, Y.; Men, Y.F. Mechanism of polymorph selection during crystallization of random butene-1/ethylene copolymer. Chin. J. Polym. Sci. 2016, 34, 1014–1020. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L.; Righetti, M.C. The three-phase structure of isotactic poly(1-butene). Polymer 2008, 49, 1323–1331. [Google Scholar] [CrossRef]
- Xin, R.; Zhang, J.; Sun, X.l.; Li, H.H.; Ren, Z.J.; Yan, S.K. Polymorphic behavior and phase transition of poly(1-butene) and its copolymers. Polymers 2018, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.N.; Wang, H.; Men, Y.F. Retardance of form II to form I transition in polybutene-1 at late stage: A proposal of a new mechanism. Macromolecules 2018, 51, 2232–2239. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, X.L.; Li, H.H.; Yan, S.K. Orientation-induced crystallization of isotactic polypropylene. Polymer 2013, 54, 4404–4421. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Yang, D.C.; Yan, S.K. Direct formation of form I poly(1-butene) single crystals from melt crystallization in ultrathin films. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2641–2645. [Google Scholar] [CrossRef]
- Tashiro, K.; Hu, J.; Wang, H.; Hanesaka, M.; Saiani, A. Refinement of the crystal structures of forms I and II of isotactic polybutene-1 and a proposal of phase transition mechanism between Them. Macromolecules 2016, 49, 1392–1404. [Google Scholar] [CrossRef]
- Corradini, P.; Napolitano, R.; Petraccone, V.; Pirozzi, B. Conformational and packing energy for the three crystalline forms of isotactic poly-α-butene. Eur. Polym. J. 1984, 20, 931–935. [Google Scholar] [CrossRef]
- Danusso, F.; Gianotti, G. Isotactic polybutene-1: Formation and transformation of modification 2. Die Makromol. Chem. 1965, 88, 149–158. [Google Scholar] [CrossRef]
- Qiao, Y.N.; Wang, Q.; Men, Y.F. Kinetics of nucleation and growth of form II to I polymorphic transition in polybutene-1 as revealed by stepwise annealing. Macromolecules 2016, 49, 5126–5136. [Google Scholar] [CrossRef]
- Zheng, L.R.; Liu, L.; Shao, C.G.; Wang, W.; Wang, B.; Pan, L.; Li, Y.S.; Ma, Z. Phase transition from tetragonal form II to hexagonal form I of butene-1/4-methyl-1-pentene random copolymers: Molecular factor versus stretching stimuli. Macromolecules 2019, 52, 1188–1199. [Google Scholar] [CrossRef]
- Tang, X.L.; Chen, W.; Li, L.B. The tough journey of polymer crystallization: battling with chain flexibility and connectivity. Macromolecules 2019. [Google Scholar] [CrossRef]
- Androsch, R.; Hohlfeld, R.; Frank, W.; Nase, M.; Cavallo, D. Transition from two-stage to direct melt-crystallization in isotactic random butene-1/propene copolymers. Polymer 2013, 54, 2528–2534. [Google Scholar] [CrossRef]
- Shi, J.Y.; Wu, P.Y.; Li, L.; Liu, T.; Zhao, L. Crystalline transformation of isotactic polybutene-1 in supercritical CO2 studied by in-situ fourier transform infrared spectroscopy. Polymer 2009, 50, 5598–5604. [Google Scholar] [CrossRef]
- Wang, W.; Shao, C.G.; Zheng, L.R.; Wang, B.; Pan, L.; Ma, G.Q.; Li, Y.S.; Wang, Y.M.; Liu, C.T.; Ma, Z. Stretching-induced phase transition of the butene-1/ethylene random copolymer: Orientation and kinetics. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 116–126. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, L.R.; Liu, L.Y.; Li, W.; Li, Y.S.; Ma, Z. Stretching behavior of the butene-1/ethylene random copolymer: A direct correspondence between triggering of II-I phase transition and mechanical yielding. Polym. Cryst. 2019, e10052. [Google Scholar] [CrossRef]
- Hu, J.; Tashiro, K. Relation between higher-order structure and crystalline phase transition of oriented isotactic polybutene-1 investigated by temperature-dependent time-resolved simultaneous WAXD/SAXS measurements. Polymer 2016, 90, 165–177. [Google Scholar] [CrossRef]
- Zhang, X.X.; Li, Y.K.; Sun, Z.Y. Acceleration of crystal transformation from crystal form II to form I in Polybutene-1 induced by nanoparticles. Polymer 2018, 150, 119–129. [Google Scholar] [CrossRef]
- Wanjale, S.D.; Jog, J.P. Poly(1-butene)/clay nanocomposites: Preparation and properties. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 1014–1021. [Google Scholar] [CrossRef]
- Tarallo, O.; Ruiz de Ballesteros, O.; Bellissimo, A.; Scoti, M.; Malafronte, A.; Auriemma, F.; De Rosa, C. Crystallization and mechanical properties of metallocene made 1-butene-pentene and 1-butene-hexene isotactic copolymers. Polymer 2018, 158, 231–242. [Google Scholar] [CrossRef]
- De Rosa, C.; Tarallo, O.; Auriemma, F.; Ruiz de Ballesteros, O.; Di Girolamo, R.; Malafronte, A. Crystallization behavior and mechanical properties of copolymers of isotactic poly(1-butene) with 1-octene from metallocene catalysts. Polymer 2015, 73, 156–169. [Google Scholar] [CrossRef]
- Gianotti, G.; Capizzi, A. Butene-1/propylene copolymers. Influence of the comonomerie units on polymorphism. Die Makromol. Chem. 1969, 124, 152–159. [Google Scholar] [CrossRef]
- Jones, A.T. Cocrystallization in copolymers of α-olefins II—Butene-1 copolymers and polybutene type II/I crystal phase transition. Polymer 1966, 7, 23–59. [Google Scholar] [CrossRef]
- Azzurri, F.; Alfonso, G.C.; Gómez, M.A.; Martì, M.C.; Ellis, G.; Marco, C. Polymorphic transformation in isotactic 1-butene/ethylene copolymers. Macromolecules 2004, 37, 3755–3762. [Google Scholar] [CrossRef]
- Qiao, Y.N.; Men, Y.F. Intercrystalline links determined kinetics of form II to I polymorphic transition in polybutene-1. Macromolecules 2017, 50, 5490–5497. [Google Scholar] [CrossRef]
- Qiao, Y.N.; Yang, F.; Lu, Y.; Liu, P.R.; Li, Y.S.; Men, Y.F. Spontaneous form II to I transition in low molar mass polybutene-1 at crystallization temperature reveals stabilization role of intercrystalline links and entanglements for metastable form II crystals. Macromolecules 2018, 51, 8298–8305. [Google Scholar] [CrossRef]
- Auriemma, F.; De Rosa, C.; Esposito, S.; Mitchell, G.R. Polymorphic superelasticity in semicrystalline polymers. Angew. Chem. Int. Ed. 2007, 46, 4325–4328. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, C.; Ruiz de Ballesteros, O.; Auriemma, F.; Di Girolamo, R.; Scarica, C.; Giusto, G.; Esposito, S.; Guidotti, S.; Camurati, I. Polymorphic behavior and mechanical properties of isotactic 1-butene–ethylene copolymers from metallocene catalysts. Macromolecules 2014, 47, 4317–4329. [Google Scholar] [CrossRef]
- He, L.L.; Wang, B.; Yang, F.; Li, Y.S.; Ma, Z. Featured crystallization polymorphism and memory effect in novel butene-1/1,5-hexadiene copolymers synthesized by post-metallocene hafnium catalyst. Macromolecules 2016, 49, 6578–6589. [Google Scholar] [CrossRef]
- Takamura, M.; Sugimoto, M.; Kawaguchi, S.; Takahashi, T.; Koyama, K. Influence of extrusion temperature on molecular architecture and crystallization behavior of peroxide-induced slightly crosslinked poly(L-lactide) by reactive extrusion. J. Appl. Polym. Sci. 2012, 123, 1468–1478. [Google Scholar] [CrossRef]
- Wang, L.Y.; Jing, X.B.; Cheng, H.B.; Hu, X.L.; Yang, L.X.; Huang, Y.B. Rheology and crystallization of long-chain branched poly(l-lactide) s with controlled branch length. Ind. Eng. Chem. Res. 2012, 51, 10731–10741. [Google Scholar] [CrossRef]
- Fang, H.G.; Zhang, Y.Q.; Bai, J.; Wang, Z.G. Shear-induced nucleation and morphological evolution for bimodal long chain branched polylactide. Macromolecules 2013, 46, 6555–6565. [Google Scholar] [CrossRef]
- Bai, J.; Fang, H.G.; Zhang, Y.Q.; Wang, Z.G. Studies on crystallization kinetics of bimodal long chain branched polylactides. Cryst. Eng. Comm. 2014, 16, 2452. [Google Scholar] [CrossRef]
- Wang, J.; Bai, J.; Zhang, Y.; Fang, H.; Wang, Z. Shear-induced enhancements of crystallization kinetics and morphological transformation for long chain branched polylactides with different branching degrees. Sci. Rep. 2016, 6, 26560. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Liu, J.Y.; Li, B.X.; Li, Y.S. Facile functionalization of polyethylene via click chemistry. Macromolecules 2011, 44, 5659–5665. [Google Scholar] [CrossRef]
- Gao, H.F.; Matyjaszewski, K. Synthesis of molecular brushes by “grafting onto” method: Combination of ATRP and click reactions. J. Am. Chem. Soc. 2007, 129, 6633–6639. [Google Scholar] [CrossRef]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Rheingold, A.L.; Liable-Sands, L.M.; Sommer, R.D. Chiral ansa metallocenes with Cp ring-fused to thiophenes and pyrroles: Syntheses, crystal structures, and isotactic polypropylene Catalysts. J. Am. Chem. Soc. 2001, 123, 4763–4773. [Google Scholar] [CrossRef]
- Wang, Z.F.; Dong, X.; Cavallo, D.; Müller, A.J.; Wang, D.J. Promotion of self-nucleation with latent form I nuclei in polybutene-1 and its copolymer. Macromolecules 2018, 51, 6037–6046. [Google Scholar] [CrossRef]
- Yamashita, M. Direct crystal growth of isotactic polybutene-1 trigonal phase in the melt: In-situ observation. J. Cryst. Growth 2008, 310, 1739–1743. [Google Scholar] [CrossRef]
- Li, L.; Liu, T.; Zhao, L.; Yuan, W.K. CO2-induced phase transition of isotactic poly-1-butene with form III upon heating. Macromolecules 2011, 44, 4836–4844. [Google Scholar] [CrossRef]
- Rubin, I.D. Relative stabilities of polymorphs of polybutene-1 obtained from the melt. J. Polym. Sci. Part B Polym. Lett. 1964, 2, 747–749. [Google Scholar] [CrossRef]
- Heck, B.; Siegenführ, S.; Strobl, G.; Thomann, R. A law controlling polymer recrystallization showing up in experiments on s-polypropylene. Polymer 2007, 48, 1352–1359. [Google Scholar] [CrossRef]
- Wang, Y.T.; Lu, Y.; Jiang, Z.Y.; Men, Y.F. Molecular weight dependency of crystallization line, recrystallization line, and melting line of polybutene-1. Macromolecules 2014, 47, 6401–6407. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Incorp. a (mol %) | Mn of PEG (Da) | Mwb (105 Da) | Mw/Mnb |
|---|---|---|---|---|
| PB | 0 | None | 1.98 | 2.38 |
| PB-TMS | 0.49 | None | 2.08 | 2.84 |
| PB-PEG-750 | 750 | 2.59 | 2.50 | |
| PB-PEG-2000 | 2000 | 2.77 | 2.44 | |
| PB-PEG-4000 | 4000 | 3.05 | 2.93 |
| Sample Code | PB | PB-TMS | PB-PEG-4000 | PB-PEG-2000 | PB-PEG-750 |
|---|---|---|---|---|---|
| Tc (°C) | 58.3 | 36.5 | 63.2 | 63.4 | 68.1 |
| Xc (%) | 41.0 | 29.3 | 39.1 | 38.5 | 39.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, C.; Li, Y.; Lou, Y.; Song, D.; Wang, B.; Pan, L.; Ma, Z.; Li, Y. Thermal Analysis of Crystallization and Phase Transition in Novel Polyethylene Glycol Grafted Butene-1 Copolymers. Polymers 2019, 11, 837. https://doi.org/10.3390/polym11050837
An C, Li Y, Lou Y, Song D, Wang B, Pan L, Ma Z, Li Y. Thermal Analysis of Crystallization and Phase Transition in Novel Polyethylene Glycol Grafted Butene-1 Copolymers. Polymers. 2019; 11(5):837. https://doi.org/10.3390/polym11050837
Chicago/Turabian StyleAn, Chuanbin, Yulian Li, Yahui Lou, Dongpo Song, Bin Wang, Li Pan, Zhe Ma, and Yuesheng Li. 2019. "Thermal Analysis of Crystallization and Phase Transition in Novel Polyethylene Glycol Grafted Butene-1 Copolymers" Polymers 11, no. 5: 837. https://doi.org/10.3390/polym11050837
APA StyleAn, C., Li, Y., Lou, Y., Song, D., Wang, B., Pan, L., Ma, Z., & Li, Y. (2019). Thermal Analysis of Crystallization and Phase Transition in Novel Polyethylene Glycol Grafted Butene-1 Copolymers. Polymers, 11(5), 837. https://doi.org/10.3390/polym11050837