Influence of Branching on the Configurational and Dynamical Properties of Entangled Polymer Melts

Abstract
:1. Introduction
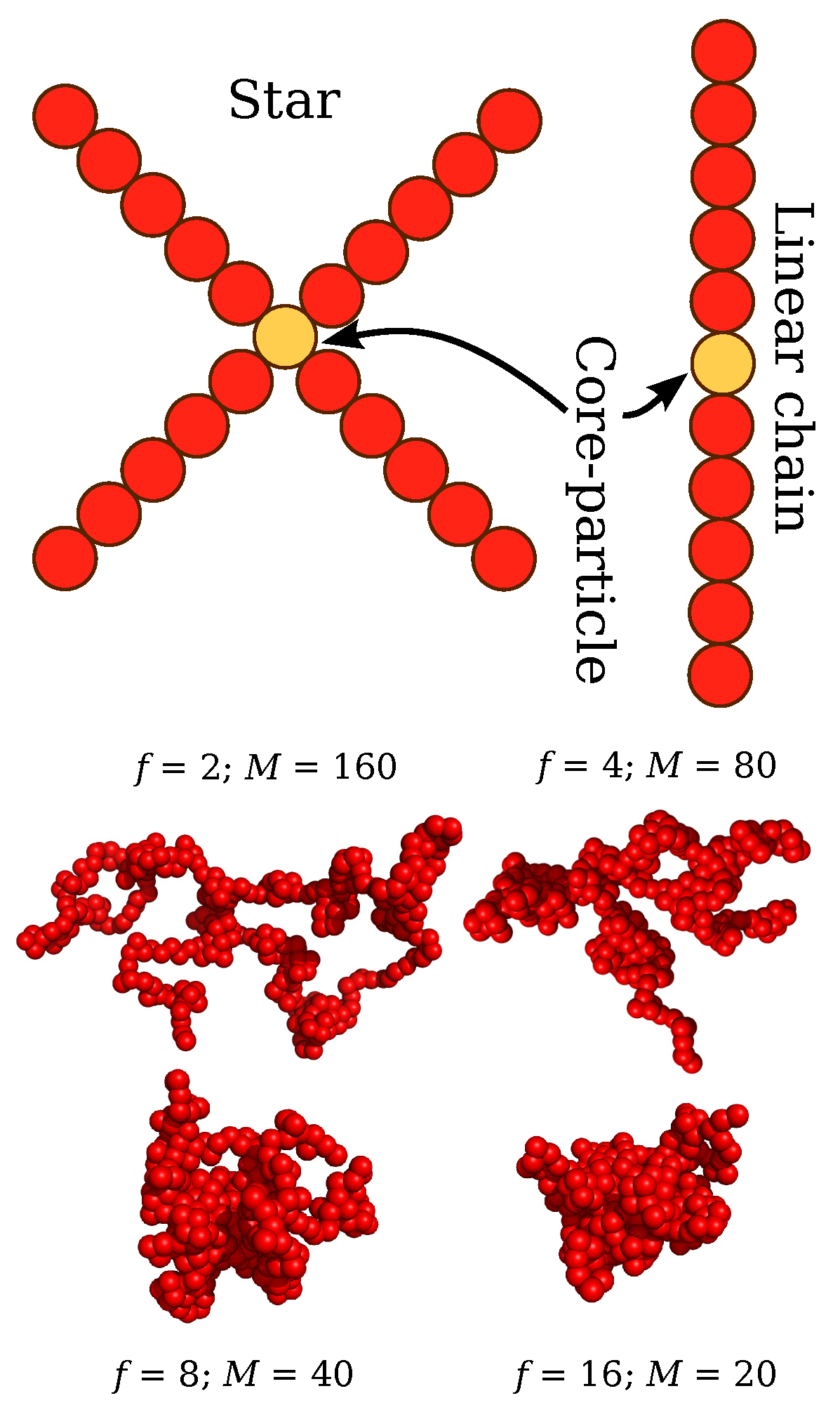
2. Model and Methodology
3. Results and Discussion
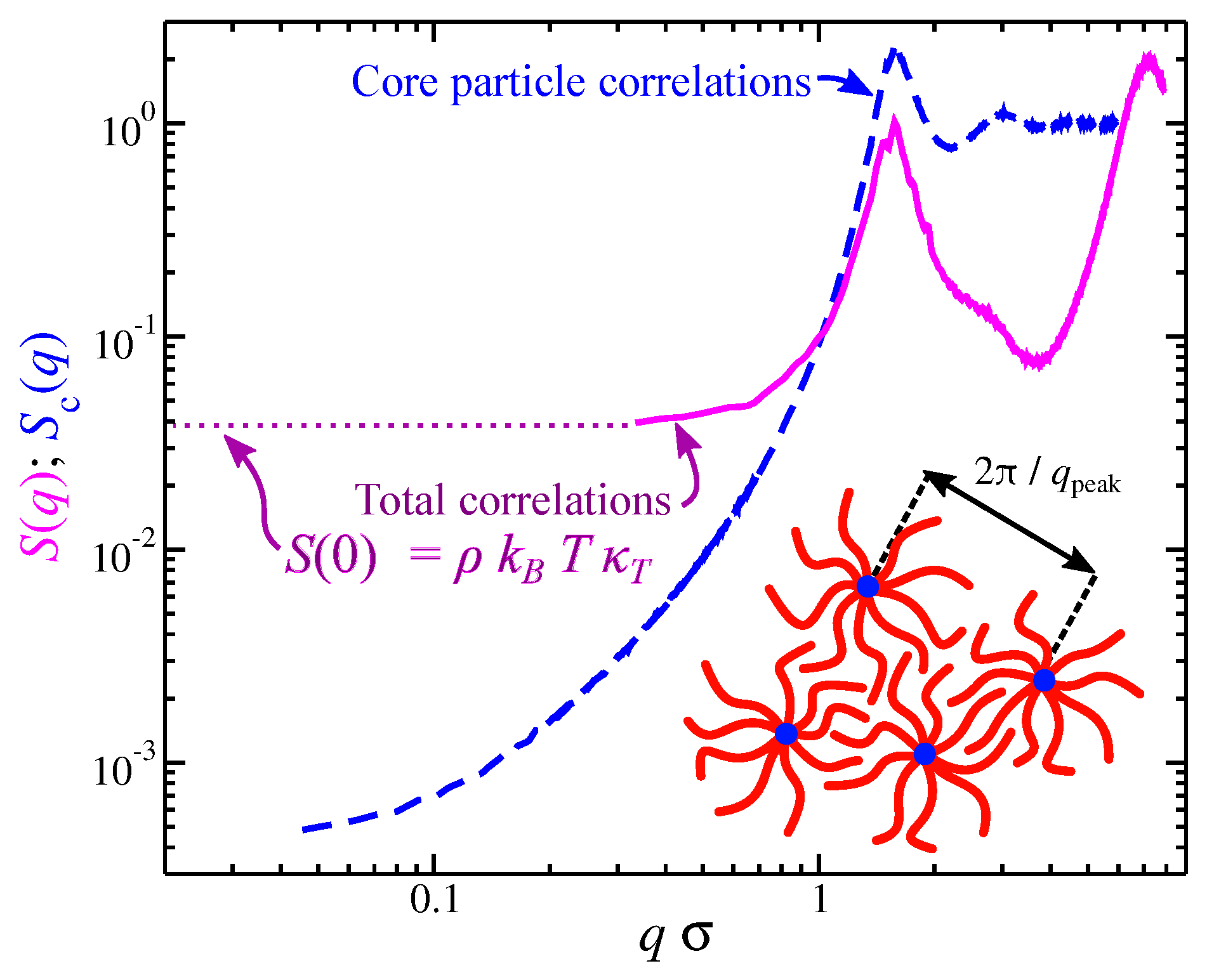
3.1. Packing Length
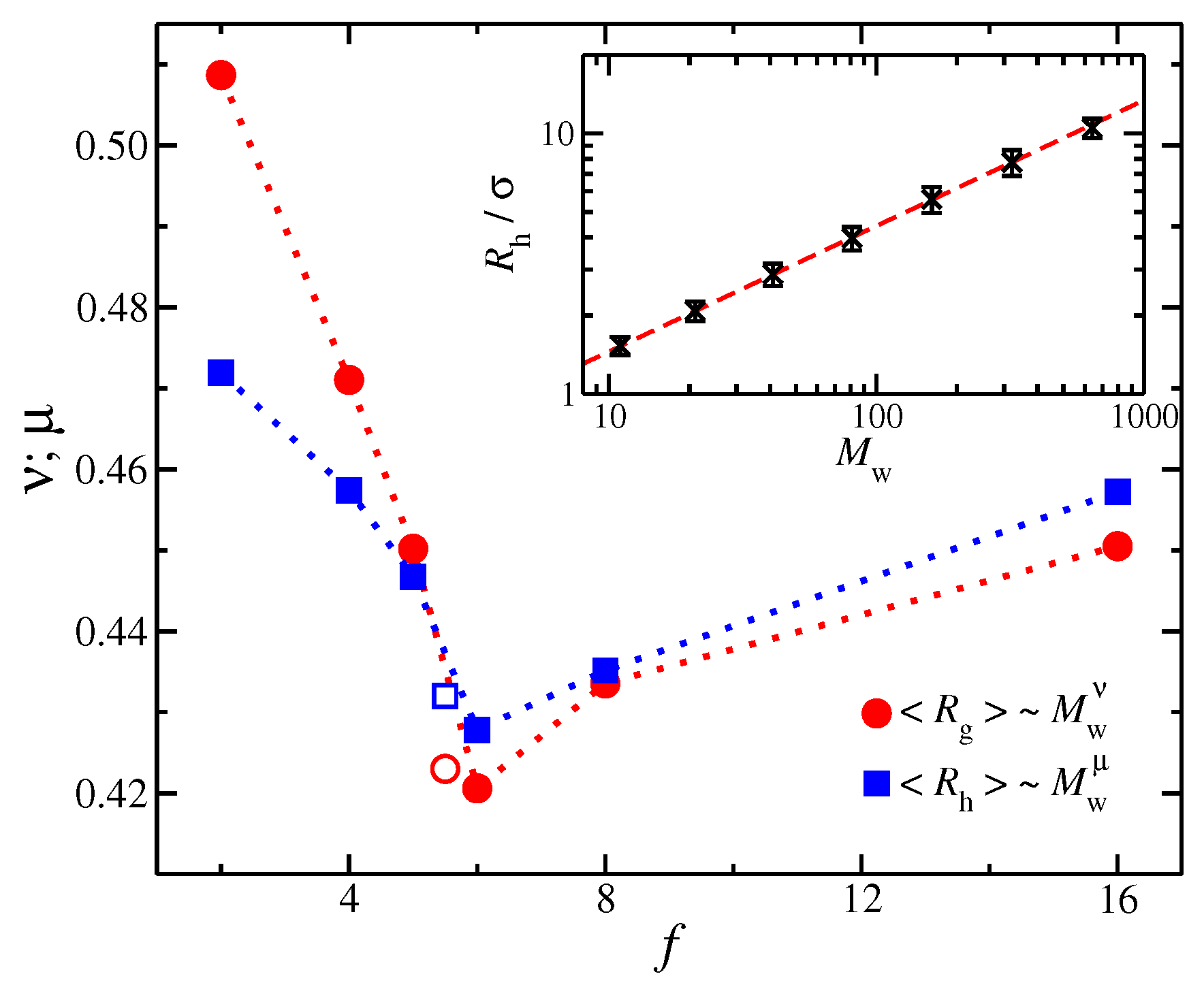
3.2. Influence of Polymer Mass and Scaling of
3.3. Quantification of the Influence of Molecular Topology on Molecular Packing
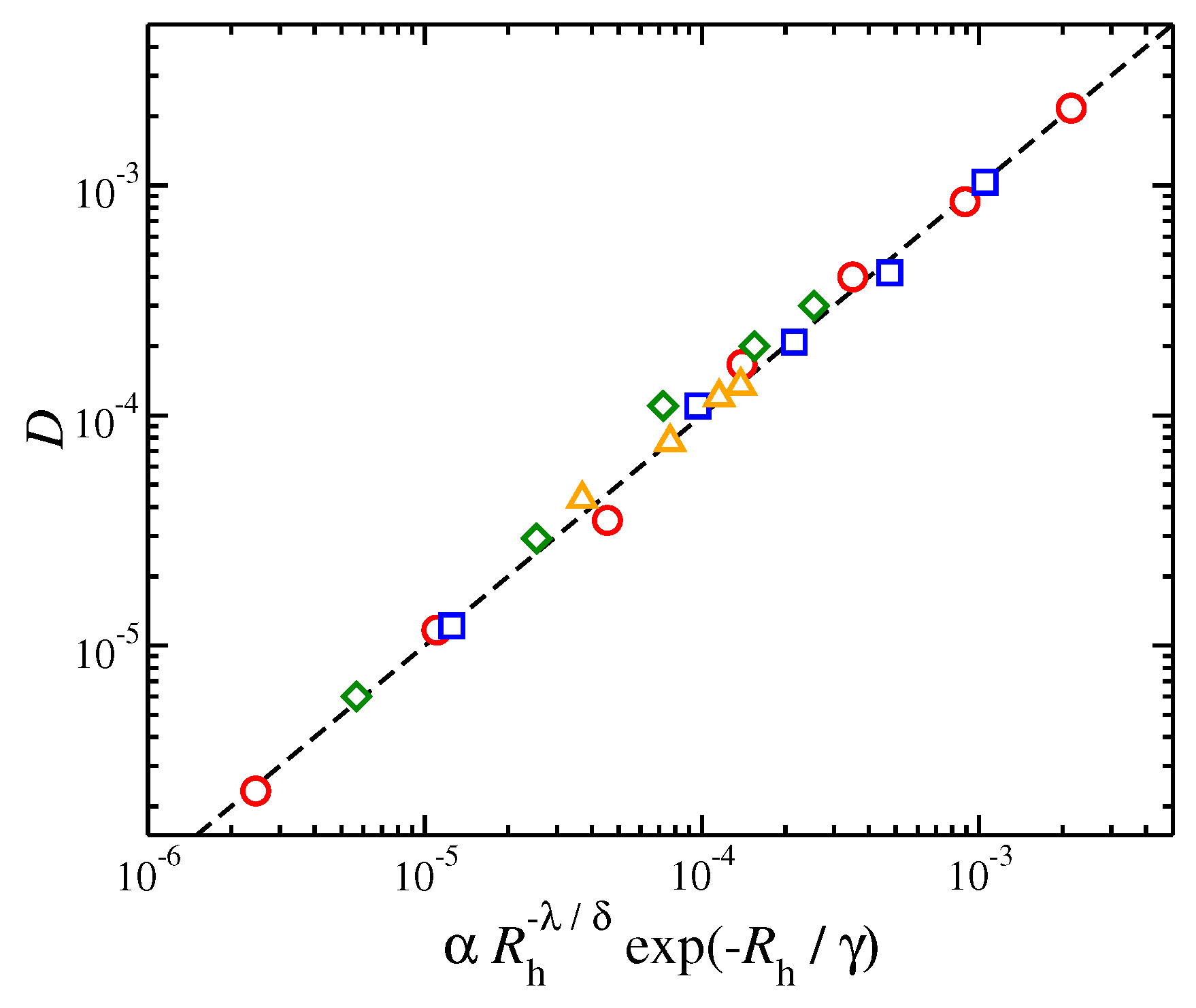
3.4. Application of Stokes-Einstein and Fractional Stokes Einstein Relations to Polymer Melts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Rouse, P.E. A theory of the linear viscoelastic properties of dilute solutions of coiling polymers. J. Chem. Phys. 1953, 21, 1272–1280. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1980. [Google Scholar]
- Flory, P.J. Statistical Mechanics of Chain Molecules; Interscience: New York, NY, USA, 1969. [Google Scholar]
- Freed, K.F.; Edwards, S.F. Polymer viscosity in concentrated solutions. J. Chem. Phys. 1974, 61, 3626–3633. [Google Scholar] [CrossRef]
- Colby, R.H.; Fetters, L.J.; Graessley, W.W. The melt viscosity-molecular weight relationship for linear polymers. Macromolecules 1987, 20, 2226–2237. [Google Scholar] [CrossRef]
- Paul, W.; Smith, G.D.; Yoon, D.Y.; Farago, B.; Rathgeber, S.; Zirkel, A.; Wilner, L.; Richter, D. Chain motion in an unentangled polyethylene melt: A critical test of the rouse model by molecular dynamics simulations and neutron spin echo spectroscopy. Phys. Rev. Lett. 1998, 80, 2346–2349. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N.; Kröger, M.; Ramirez, J.; Öttinger, H.C.; Vlassopoulos, D. Crossover from the rouse to the entangled polymer melt regime: Signals from long, detailed atomistic molecular dynamics simulations, supported by rheological experiments. Macromolecules 2003, 36, 1376–1387. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Heermann, D.W.; Kremer, K. Crossover scaling in semidilute polymer solutions: A Monte Carlo test. J. Phys. II (Paris) 1991, 1, 37–60. [Google Scholar] [CrossRef]
- Paul, W.; Smith, G.D.; Yoon, D.Y. Static and dynamic properties of a n-C100H202 melt from molecular dynamics simulations. Macromolecules 1997, 30, 7772–7780. [Google Scholar] [CrossRef]
- Jeong, C.; Douglas, J.F. Mass dependence of the activation enthalpy and entropy of unentangled linear alkane chains. J. Chem. Phys. 2015, 143, 144905. [Google Scholar] [CrossRef]
- de Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- de Gennes, P.G. Kinetics of diffusion-controlled processes in dense polymer systems. II. Effects of entanglements. J. Chem. Phys. 1982, 76, 3322–3326. [Google Scholar] [CrossRef]
- Everaers, R. Topological versus rheological entanglement length in primitive-path analysis protocols, tube models, and slip-link models. Phys. Rev. E 2012, 86, 022801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijkers, F.; Pasquino, R.; Olmsted, P.D.; Vlassopoulos, D. Perspectives on the viscoelasticity and flow behavior of entangled linear and branched polymers. J. Phys. Condens. Matter 2015, 27, 473002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.S.; Kang, K.G.; Katzarova, M.; Hall, R.; Huang, Q.; Lee, S.; Shivokhin, M.; Chang, T.; Venerus, D.C.; Mays, J.; et al. Challenging tube and slip-link models: Predicting the linear rheology of blends of well-characterized star and linear 1, 4-polybutadienes. Macromolecules 2016, 49, 4964–4977. [Google Scholar] [CrossRef]
- de Gennes, P.G. Reptation of stars. J. Chem. Phys. 1975, 36, 1199–1203. [Google Scholar] [CrossRef]
- Milner, S.T.; McLeish, T.C.B. Parameter-free theory for stress relaxation in star polymer melts. Macromolecules 1997, 30, 2159–2166. [Google Scholar] [CrossRef]
- Vlassopoulos, D. Macromolecular topology and rheology: Beyond the tube model. Rheol. Acta 2016, 55, 613–632. [Google Scholar] [CrossRef]
- Jabbarzadeh, A.; Atkinson, J.D.; Tanner, R.I. Effect of molecular shape on rheological properties in molecular dynamics simulation of star, H, comb, and linear polymer melts. Macromolecules 2003, 36, 5020–5031. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Mavrantzas, V.G. Hierarchical modeling of the dynamics of polymers with a nonlinear molecular architecture: Calculation of branch point friction and chain reptation time of H-shaped polyethylene melts from long molecular dynamics simulations. Macromolecules 2005, 38, 8583–8596. [Google Scholar] [CrossRef]
- Lyulin, A.V.; Vorselaars, B.; Mazo, M.A.; Balabaev, N.K.; Michels, M.A.J. Strain softening and hardening of amorphous polymers: Atomistic simulation of bulk mechanics and local dynamics. EPL 2005, 71, 618–624. [Google Scholar] [CrossRef]
- Ionescu, T.C.; Baig, C.; Edwards, B.J.; Keffer, D.J.; Habenschuss, A. Structure formation under steady-state isothermal planar elongational flow of n-eicosane: A comparison between simulation and experiment. Phys. Rev. Lett. 2006, 96, 037802. [Google Scholar] [CrossRef]
- Gee, R.H.; Lacevic, N.; Fried, L.E. Atomistic simulations of spinodal phase separation preceding polymer crystallization. Nature Mater. 2006, 5, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Baig, C.; Mavrantzas, V.G.; Kröger, M. Flow effects on melt structure and entanglement network of linear polymers: Results from a nonequilibrium molecular dynamics simulation study of a polyethylene melt in steady shear. Macromolecules 2010, 43, 6886–6902. [Google Scholar] [CrossRef]
- Yoon, J.; Kim, J.; Baig, C. Nonequilibrium molecular dynamics study of ring polymer melts under shear and elongation flows: A comparison with their linear analogs. J. Rheol. 2016, 60, 673–685. [Google Scholar] [CrossRef]
- Jeong, S.H.; Kim, J.M.; Baig, C. Effect of chain orientation and stretch on the stress overshoot of entangled polymeric materials under start-up shear. Macromolecules 2017, 50, 3424–3429. [Google Scholar] [CrossRef]
- Jeong, S.H.; Kim, J.M.; Baig, C. Rheological behaviors of H-shaped polymers incorporated with short branches under shear and elongational flows via FENE-Rouse model. J. Rheol. 2018, 62, 1115–1124. [Google Scholar] [CrossRef]
- Sefiddashti, M.H.N.; Edwards, B.J.; Khomami, B. Steady shearing flow of a moderately entangled polyethylene liquid. J. Rheol. 2016, 60, 1227–1244. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Kremer, K. Dynamics of polystyrene melts through hierarchical multiscale simulations. Macromolecules 2009, 42, 791–802. [Google Scholar] [CrossRef]
- de Pablo, J.J. Coarse-grained simulations of macromolecules: From DNA to nanocomposites. Annu. Rev. Phys. Chem. 2011, 62, 555–574. [Google Scholar] [CrossRef]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Brini, E.; Algaer, E.A.; Ganguly, P.; Li, C.; Rodríguez-Ropero, F.; van der Vegt, N. Systematic coarse-graining methods for soft matter simulations—A review. Soft Matter 2013, 9, 2108–2119. [Google Scholar] [CrossRef]
- Gartner III, T.E.; Jayaraman, A. Modeling and simulations of polymers: A Roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef]
- Xia, W.; Hansoge, N.K.; Xu, W.S.; Phelan, F.R.; Keten, S.; Douglas, J.F. Energy renormalization for coarse-graining polymers having different segmental structures. Sci. Adv. 2019, 5, eaav4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chremos, A.; Jeong, C.; Douglas, J.F. Influence of polymer architectures on diffusion in unentangled polymer melts. Soft Matter 2017, 13, 5778–5784. [Google Scholar] [CrossRef] [PubMed]
- Juba, D.; Audus, D.J.; Mascagni, M.; Douglas, J.F.; Keyrouz, W. ZENO: Software for calculating hydrodynamic, electrical, and shape properties of polymer and particle suspensions. J. Res. Natl. Inst. Stand. Technol. 2017, 122, 1. [Google Scholar] [CrossRef]
- Hoy, R.S.; Foteinopoulou, K.; Kröger, M. Topological analysis of polymeric melts: Chain-length effects and fast-converging estimators for entanglement length. Phys. Rev. E 2009, 80, 031803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of repulsive forces in determining the equilibrium structure of simple liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Smith, J.S.; Bedrov, D.; Smith, G.D. A molecular dynamics simulation study of nanoparticle interactions in a model polymer-nanoparticle composite. Compos. Sci. Technol. 2003, 63, 1599–1605. [Google Scholar] [CrossRef]
- Plimpton, S.J. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639–4647. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Graessley, W.W. Chain dimensions and entanglement spacings in dense macromolecular systems. J. Polym. Sci. B 1998, 37, 1023–1033. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Milner, S.T. Packing length influence in linear polymer melts on the entanglement, critical, and reptation molecular weights. Macromolecules 1999, 32, 6847–6851. [Google Scholar] [CrossRef]
- Lohse, D.J. The influence of chemical structure on polyolefin melt rheology and miscibility. J. Macromol. Sci. C 2005, 45, 289–308. [Google Scholar] [CrossRef]
- Lodge, T.P.; Muthukumar, M. Physical chemistry of polymers: Entropy, interactions, and dynamics. J. Phys. Chem. 1996, 100, 13275–13292. [Google Scholar] [CrossRef]
- Sung, B.J.; Yethiraj, A. Dynamics of two-dimensional and quasi-two-dimensional polymers. J. Chem. Phys. 2013, 138, 234904. [Google Scholar] [CrossRef] [PubMed]
- Polanowski, P.; Jeszka, J.K.; Sikorski, A. Dynamic properties of linear and cyclic chains in two dimensions. Computer simulation studies. Macromolecules 2014, 47, 4830–4839. [Google Scholar] [CrossRef]
- Halverson, J.D.; Lee, W.B.; Grest, G.S.; Grosberg, A.Y.; Kremer, K. Molecular dynamics simulation study of nonconcatenated ring polymers in a melt. II. Dynamics. J. Chem. Phys. 2011, 134, 204905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reigh, S.Y.; Yoon, D.Y. Concentration dependence of ring polymer conformations from monte carlo simulations. ACS Maro Lett. 2013, 2, 296–300. [Google Scholar] [CrossRef]
- Jeong, C.; Douglas, J.F. Relation between polymer conformational structure and dynamics in linear and ring polyethylene blends. Macromol. Theory Simul. 2017, 26, 1700045. [Google Scholar] [CrossRef]
- Douglas, J.F. Weak and strong gels and the emergence of the amorphous solid state. Gels 2018, 4, 19. [Google Scholar] [CrossRef]
- Dolgushev, M.; Hauder, A.L.; Pelagejcev, P.; Wittmer, J.P. Marginally compact fractal trees with semiflexibility. Phys. Rev. E 2017, 96, 012501. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Everaers, R. Computer simulations of melts of randomly branching polymers. J. Chem. Phys. 2016, 145, 164906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everaers, R.; Grosberg, A.Y.; Rubinstein, M.; Rosa, A. Flory theory of randomly branched polymers. Soft Matter 2017, 13, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Everaers, R. Beyond Flory theory: Distribution functions for interacting lattice trees. Phys. Rev. E 2017, 95, 012117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chremos, A.; Douglas, J.F. A comparative study of thermodynamic, conformational, and structural properties of bottlebrush with star and ring polymer melts. J. Chem. Phys. 2018, 149, 044904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chremos, A.; Douglas, J.F. Communication: When does a branched polymer become a particle? J. Chem. Phys. 2015, 143, 111104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, J.F.; Zhou, H.-X.; Hubbard, J.B. Hydrodynamic friction and the capacitance of arbitrarily shaped objects. Phys. Rev. E 1994, 49, 5319–5331. [Google Scholar] [CrossRef]
- Douglas, J.F.; Ishinabe, T. Self-avoiding-walk contacts and random-walk self-intersections in variable dimensionality. Phys. Rev. E 1995, 51, 1791–1817. [Google Scholar] [CrossRef]
- Douglas, J.F. Influence of chain structure and swelling on the elasticity of rubbery materials: Localization model description. Macromol. Symp. 2013, 329, 87–100. [Google Scholar] [CrossRef]
- Douglas, J.F. A dynamic measure of order in structural glasses. Comput. Mater. Sci. 1995, 4, 292–308. [Google Scholar] [CrossRef]
- Louis, A.A.; Bolhuis, P.G.; Hansen, J.P.; Meijer, E.J. Can polymer coils be modeled as “soft colloids”? Phys. Rev. Lett. 2000, 85, 2522–2525. [Google Scholar] [CrossRef]
- Pakula, T. Static and dynamic properties of computer simulated melts of multiarm polymer stars. Comput. Theor. Polym. Sci. 1998, 8, 21–30. [Google Scholar] [CrossRef]
- Pakula, T.; Vlassopoulos, D.; Fytas, G.; Roovers, J. Structure and dynamics of melts of multiarm polymer stars. Macromolecules 1998, 31, 8931–8940. [Google Scholar] [CrossRef]
- Chremos, A.; Glynos, E.; Green, P.F. Structure and dynamical intra-molecular heterogeneity of star polymer melts above glass transition temperature. J. Chem. Phys. 2015, 142, 044901. [Google Scholar] [CrossRef]
- Chremos, A.; Panagiotopoulos, A.Z.; Yu, H.-Y.; Koch, D.L. Structure of solvent-free grafted nanoparticles: Molecular dynamics and density-functional theory. J. Chem. Phys. 2011, 135, 114901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chremos, A.; Douglas, J.F. Particle localization and hyperuniformity of polymer-grafted nanoparticle materials. Annanel der Physik 2017, 529, 1600342. [Google Scholar] [CrossRef] [PubMed]
- Torquato, S.; Stillinger, F.H. Local density fluctuations, hyperuniformity, and order metrics. Phys. Rev. E 2003, 68, 041113. [Google Scholar] [CrossRef] [Green Version]
- Torquato, S. Hyperuniform states of matter. Phys. Rep. 2018, 745, 1–95. [Google Scholar] [CrossRef] [Green Version]
- Chremos, A.; Douglas, J.F. Hidden hyperuniformity in soft polymeric materials. Phys. Rev. Lett. 2018, 121, 258002. [Google Scholar] [CrossRef] [PubMed]
- Likos, C.N.; Löwen, H.; Watzlawek, M.; Abbas, B.; Jucknischke, O.; Allgaier, J.; Richter, D. Star polymers viewed as ultrasoft colloidal particles. Phys. Rev. Lett. 1998, 80, 4450–4453. [Google Scholar] [CrossRef]
- Yatsenko, G.; Sambriski, E.J.; Nemirovskaya, M.A.; Guenza, M. Analytical soft-core potentials for macromolecular fluids and mixtures. Phys. Rev. Lett. 2004, 93, 257803. [Google Scholar] [CrossRef]
- Likos, C.N. Soft matter with soft particles. Soft Matter 2006, 2, 478–498. [Google Scholar] [CrossRef]
- Narros, A.; Moreno, A.J.; Likos, C.N. Influence of topology on effective potentials: Coarse-graining ring polymers. Soft Matter 2010, 6, 2435–2441. [Google Scholar] [CrossRef]
- Coslovich, D.; Bernabei, M.; Moreno, A.J. Cluster glasses of ultrasoft particles. J. Chem. Phys. 2012, 137, 184904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flory, P.J.; Krigbaum, W.R. Statistical mechanics of dilute polymer solutions. II. J. Chem. Phys. 1950, 18, 1086–1094. [Google Scholar] [CrossRef]
- Gobush, W.; Sole, K.; Stockmayer, W.H. Statistical mechanics of random-flight chains. V Excluded volume expansion and second virial coefficient for linear chains of varying shape. J. Chem. Phys. 1974, 60, 12–21. [Google Scholar] [CrossRef]
- Bartels, C.R.; Crist Jr, B.; Fetters, L.J.; Graessley, W.W. Self-diffusion in branched polymer melts. Macromolecules 1986, 19, 785–793. [Google Scholar] [CrossRef]
- Hess, W. Tracer diffusion in polymeric mixtures. Macromolecules 1987, 20, 2587–2599. [Google Scholar] [CrossRef]
- Nemoto, N.; Kishine, M.; Inoue, T.; Osaki, K. Tracer diffusion of linear polystyrene in entanglement networks. Macromolecules 1990, 23, 659–664. [Google Scholar] [CrossRef]
- Lungova, M.; Krutyeva, M.; Pyckhout-Hintzen, W.; Wischnewski, A.; Monkenbusch, M.; Allgaier, J.; Ohl, M.; Sharp, M.; Richter, D. Nanoscale motion of soft nanoparticles in unentangled and entangled polymer matrices. Phys. Rev. Lett. 2016, 117, 147803. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; An, L. Simulation studies on architecture dependence of unentangled polymer melts. J. Chem. Phys. 2015, 142, 074903. [Google Scholar] [CrossRef]
- Martin, J.; Wilcoxon, J.P. Critical dynamics of the sol-gel transition. Phys. Rev. Lett. 1988, 61, 373. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.; Adolf, D. Diffusion in branched polymer melts. Macromolecules 1989, 22, 4309–4311. [Google Scholar] [CrossRef]
- Ball, R.C.; McLeish, T.C.B. Dynamic dilution and the viscosity of star-polymer melts. Macromolecules 1989, 22, 1911. [Google Scholar] [CrossRef]
- Fetters, L.J.; Kiss, A.D.; Pearson, D.S.; Quack, G.F.; Vitus, F.J. Rheological behavior of star-shaped polymers. Macromolecules 1993, 26, 647–654. [Google Scholar] [CrossRef]
- Wyart, F.B.; de Gennes, P.G. Viscosity at small scales in polymer melts. Euro. Phys. J. E 2000, 1, 93. [Google Scholar] [CrossRef]
- Ye, X.; Tong, P.; Fetters, L.J. Transport of probe particles in semidilute polymer solutions. Macromolecules 1998, 31, 5785. [Google Scholar] [CrossRef]
- Chen, Y.; Prud’homme, R.K.; Thomas, J.L. Diffusion of mesoscopic probes in aqueous polymer solutions measured by fluorescence recovery after photobleaching. Macromolecules 2002, 35, 8111–8121. [Google Scholar] [CrossRef]
- Sluch, M.I.; Somoza, M.M.; Berg, M.A. Friction on small objects and the breakdown of hydrodynamics in solution: rotation of anthracene in poly (isobutylene) from the small-molecule to polymer limits. J. Phys. Chem. B 2002, 106, 7385–7397. [Google Scholar] [CrossRef]
- Cai, L.-H.; Panyukov, S.; Rubinstein, M. Mobility of nonsticky nanoparticles in polymer liquids. Macromolecules 2011, 44, 7853–7863. [Google Scholar] [CrossRef]
- Douglas, J.F.; Hubbard, J.B. Semiempirical theory of relaxation: Concentrated polymer solution dynamics. Macromolecules 1991, 24, 3163–3177. [Google Scholar] [CrossRef]
- Abadi, M.; Serag, M.F.; Habuchi, S. Entangled polymer dynamics beyond reptation. Nat. Comm. 2018, 9, 5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J. Evidence for reptation in an entangled polymer melt. Nature 1978, 271, 143–145. [Google Scholar] [CrossRef]
- Wang, S.Q. Chain dynamics in entangled polymers: Diffusion versus rheology and their comparison. J. Polym. Sci. 2003, 41, 1589–1604. [Google Scholar] [CrossRef]
- Douglas, J.F.; Leporini, D. Obstruction model of the fractional Stokes–Einstein relation in glass-forming liquids. J. Non-Cryst. Solids 1998, 235, 137–141. [Google Scholar] [CrossRef]
- Lodge, T.P. Reconciliation of the molecular weight dependence of diffusion and viscosity in entangled polymers. Phys. Rev. Lett. 1999, 83, 3218. [Google Scholar] [CrossRef]
- Xu, W.-S.; Douglas, J.F.; Freed, K.F. Influence of cohesive energy on relaxation in a model glass-forming polymer melt. Macromolecules 2016, 49, 8355–8370. [Google Scholar] [CrossRef]
- Sozański, K.; Wiśniewska, A.; Kalwarczyk, T.; Hołlyst, R. Activation energy for mobility of dyes and proteins in polymer solutions: From diffusion of single particles to macroscale flow. Phys. Rev. Lett. 2013, 111, 228301. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| f | M | ||||
|---|---|---|---|---|---|
| 2 | ∞ | 1 | |||
| 2 | ∞ | ||||
| 4 | ∞ | 1 | |||
| 4 | |||||
| 8 | − | 1 | |||
| 16 | − | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chremos, A.; Douglas, J.F. Influence of Branching on the Configurational and Dynamical Properties of Entangled Polymer Melts. Polymers 2019, 11, 1045. https://doi.org/10.3390/polym11061045
Chremos A, Douglas JF. Influence of Branching on the Configurational and Dynamical Properties of Entangled Polymer Melts. Polymers. 2019; 11(6):1045. https://doi.org/10.3390/polym11061045
Chicago/Turabian StyleChremos, Alexandros, and Jack F. Douglas. 2019. "Influence of Branching on the Configurational and Dynamical Properties of Entangled Polymer Melts" Polymers 11, no. 6: 1045. https://doi.org/10.3390/polym11061045
APA StyleChremos, A., & Douglas, J. F. (2019). Influence of Branching on the Configurational and Dynamical Properties of Entangled Polymer Melts. Polymers, 11(6), 1045. https://doi.org/10.3390/polym11061045