Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate


 , ,
, , 
Abstract
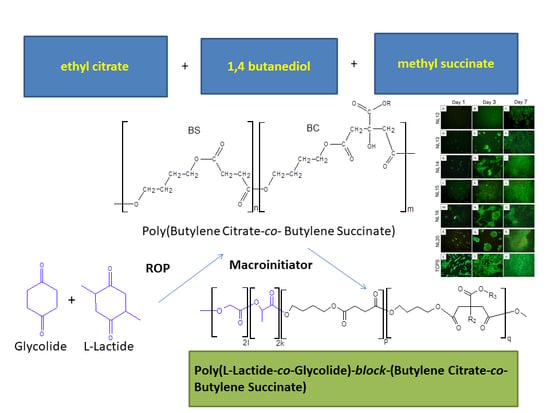
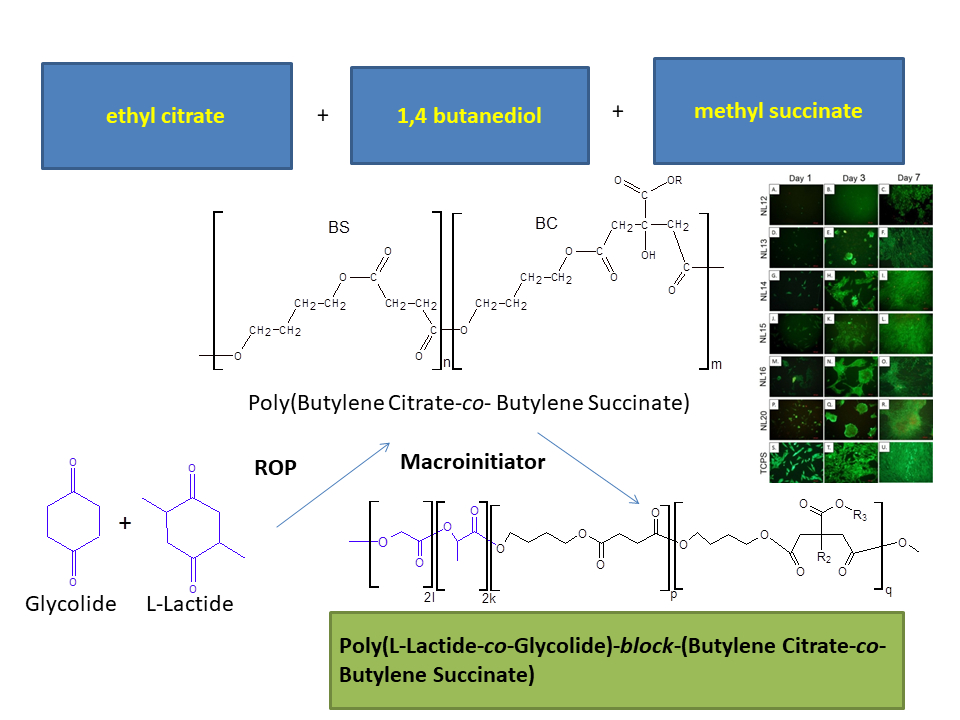
:
1. Introduction
2. Materials and Methods
2.1. Materials
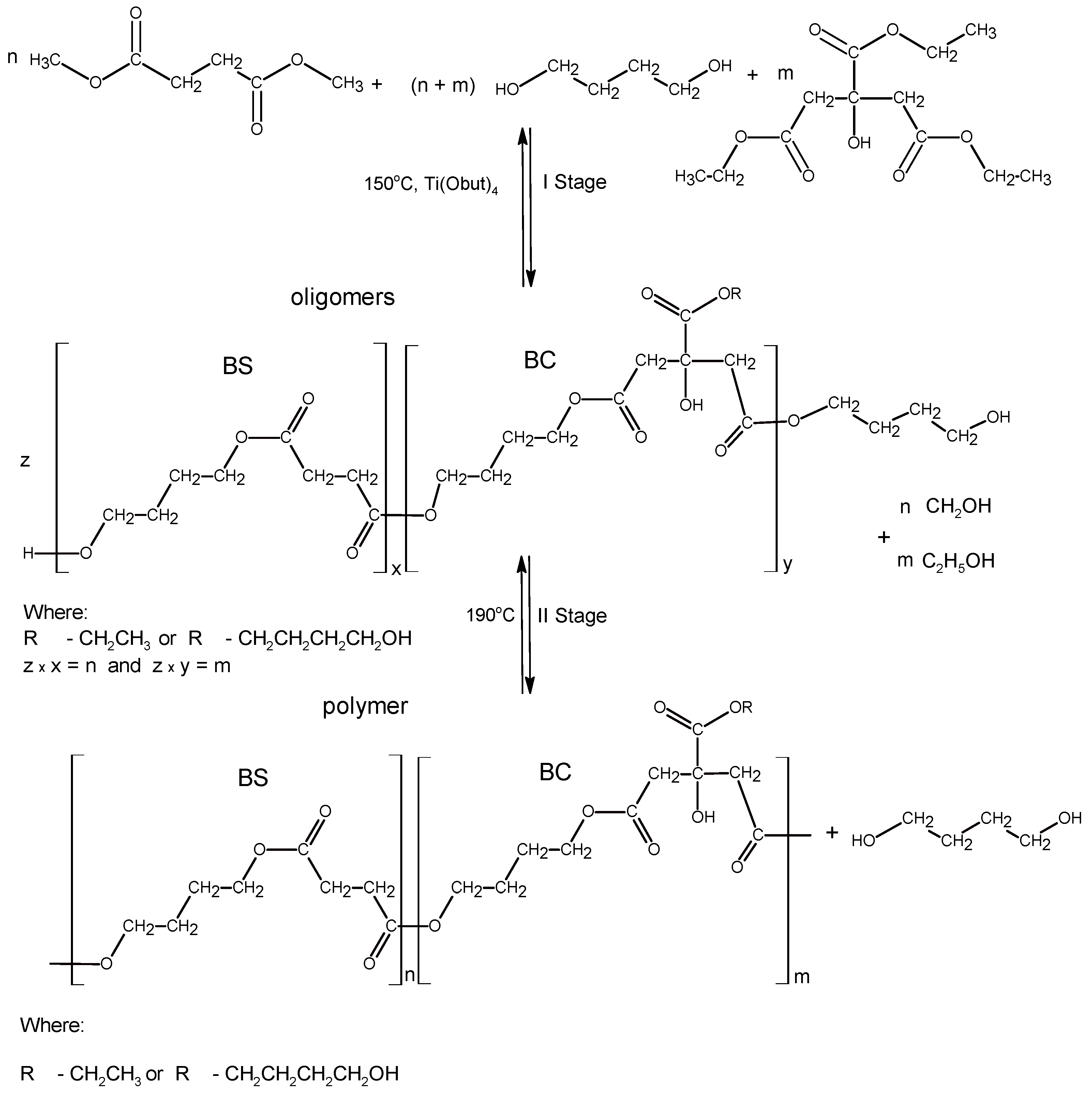
2.2. Macroinitiators Synthesis—Poly(Butylene Succinate–co–Butylene Citrate)
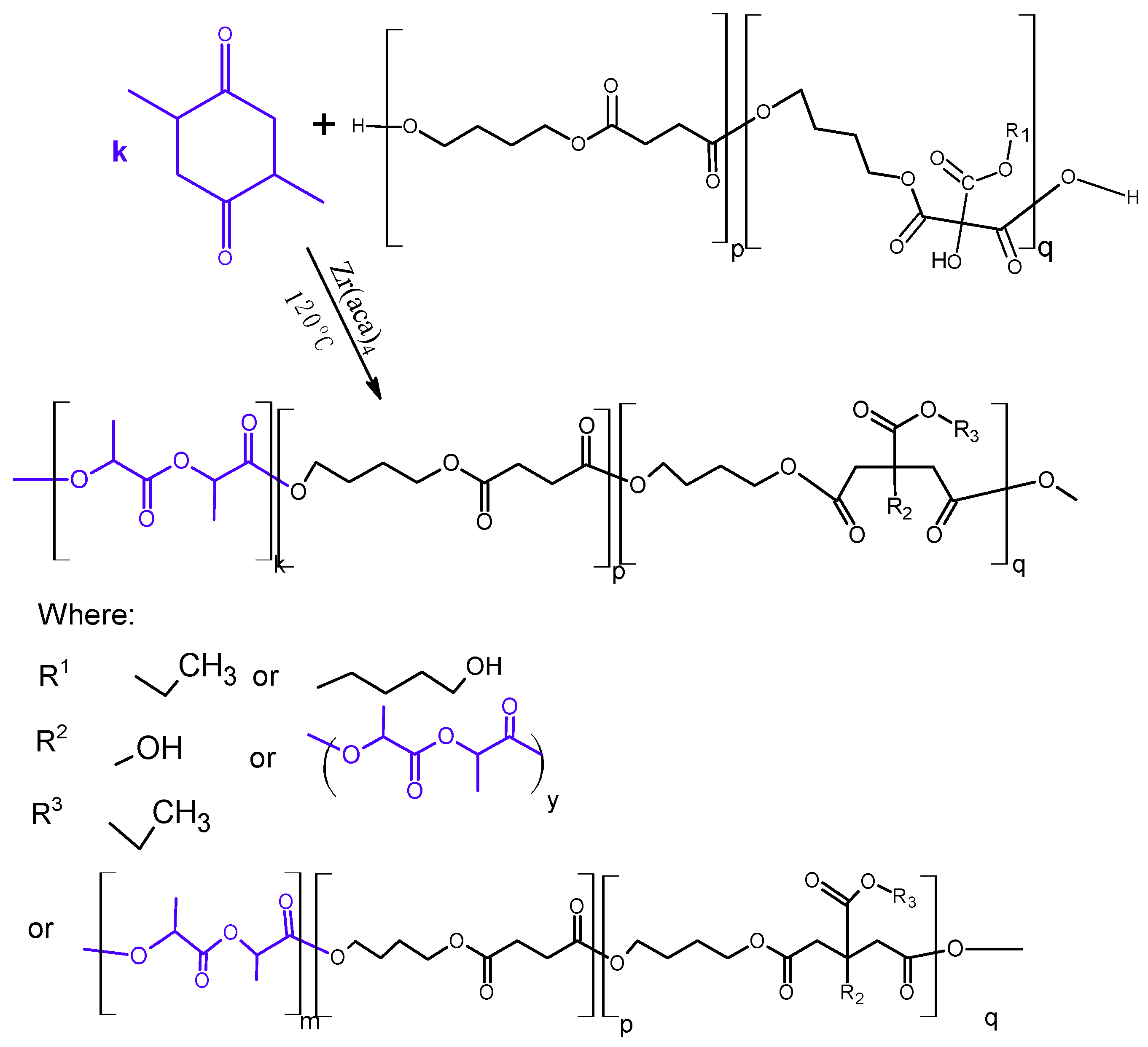
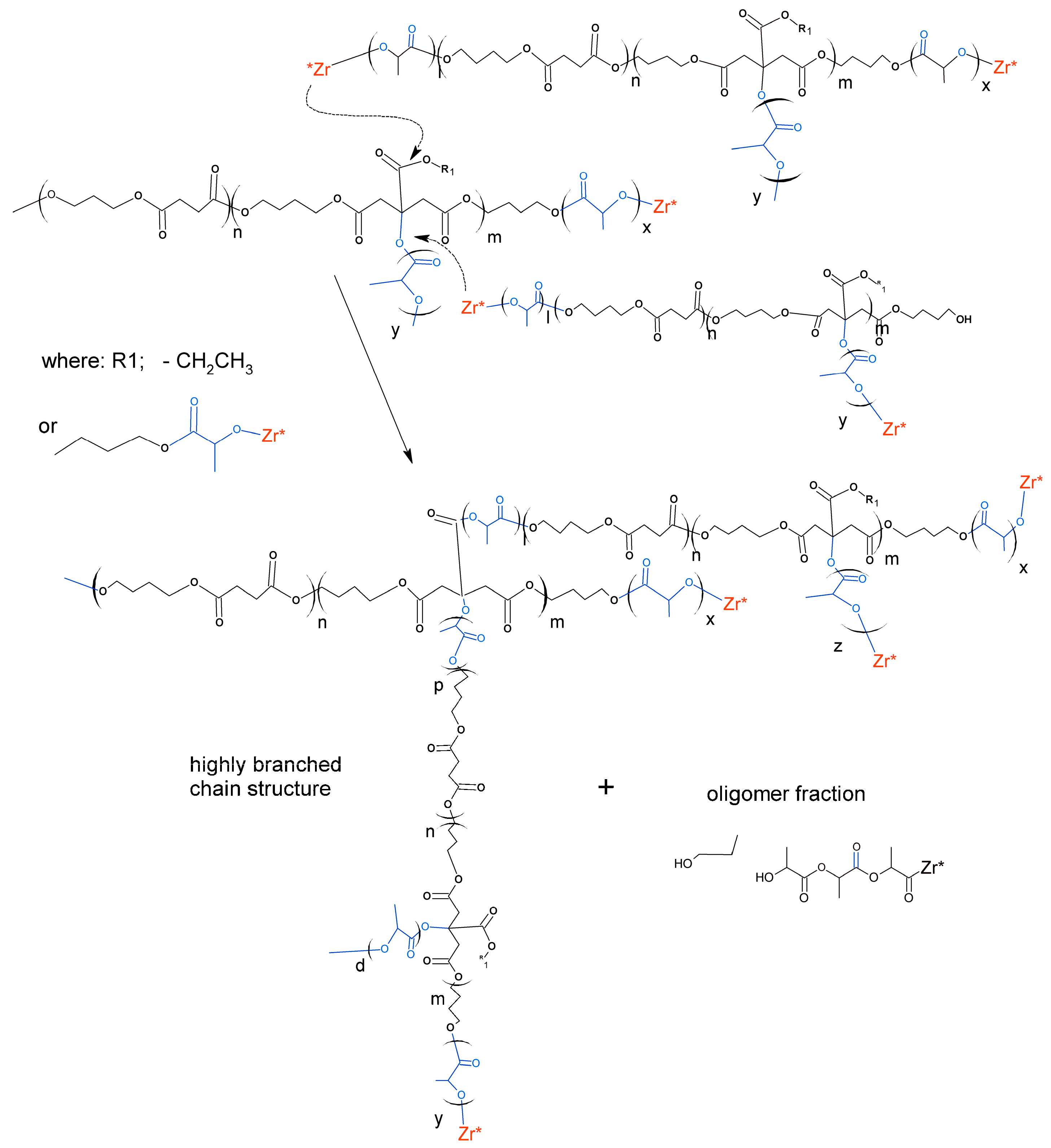
2.3. Synthesis of Poly(l-Lactide–co–Glycolide)–Block–Poly(Butylene Succinate-co-Butylene Citrate)
2.4. Measurements
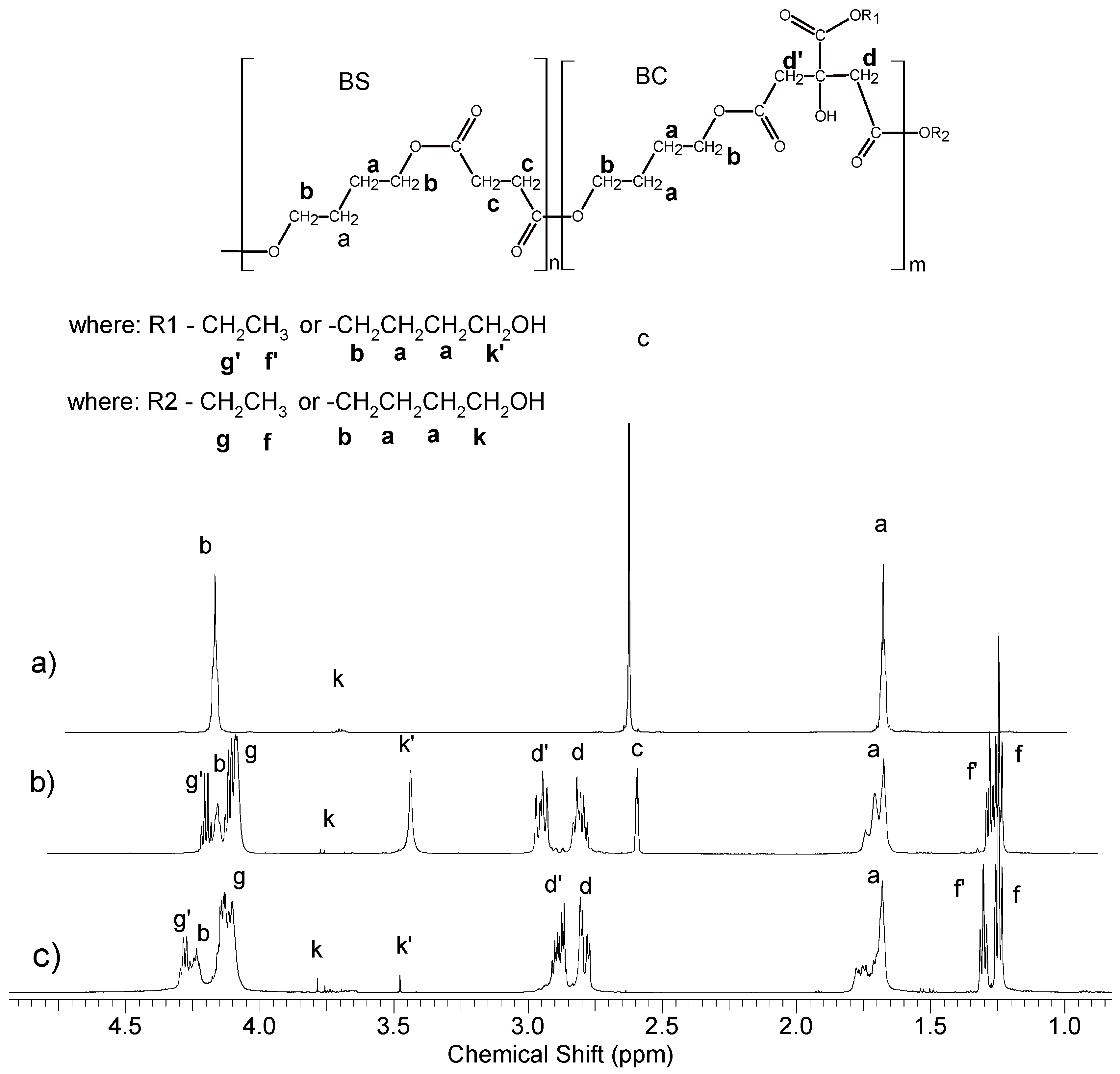
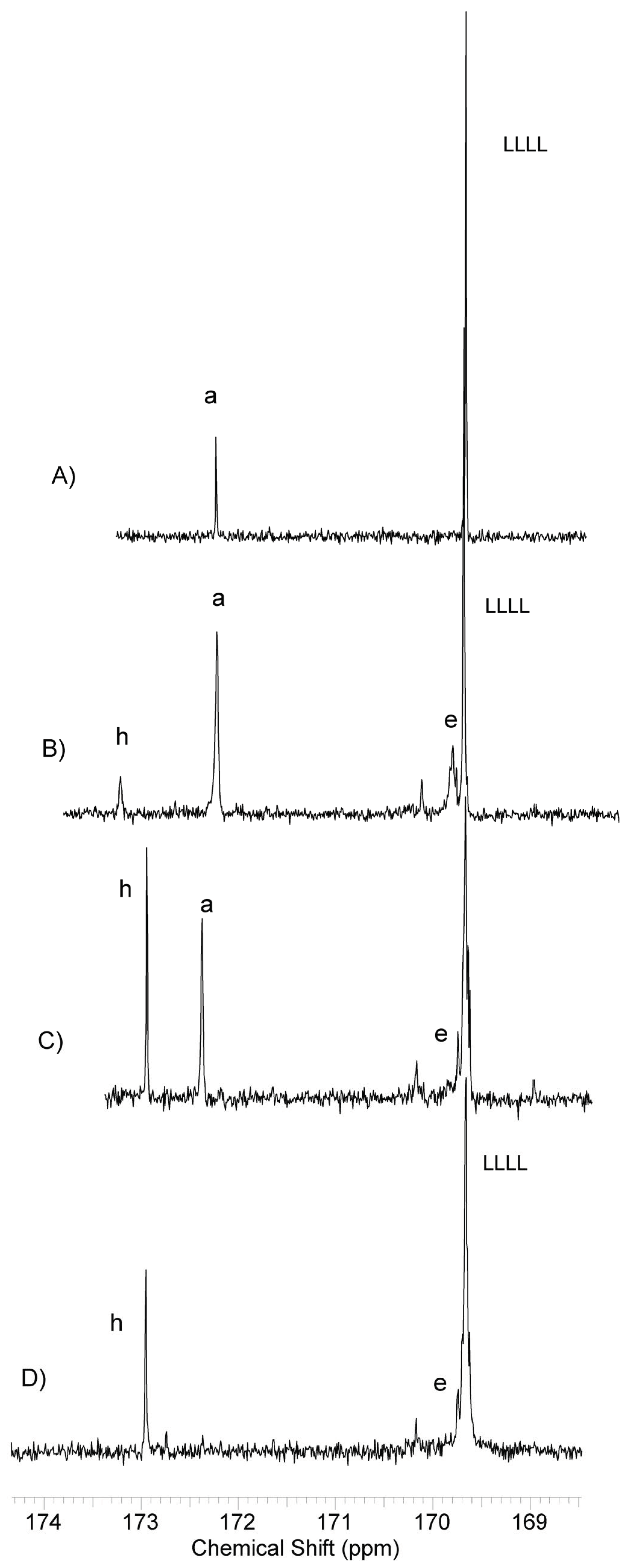
2.4.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.4.2. Thermal Properties
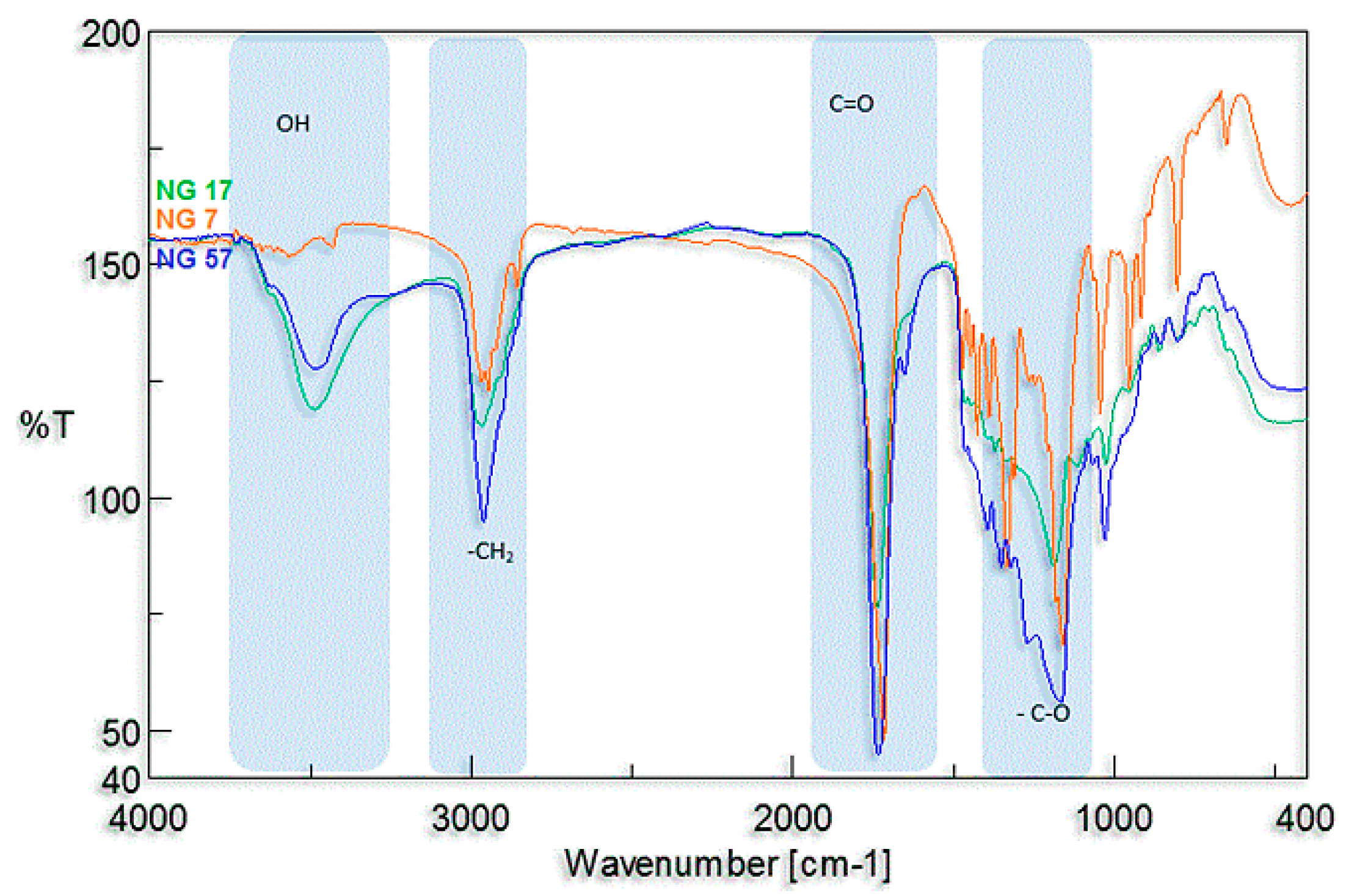
2.4.3. Fourier Transform Infrared (FTIR) Spectroscopy
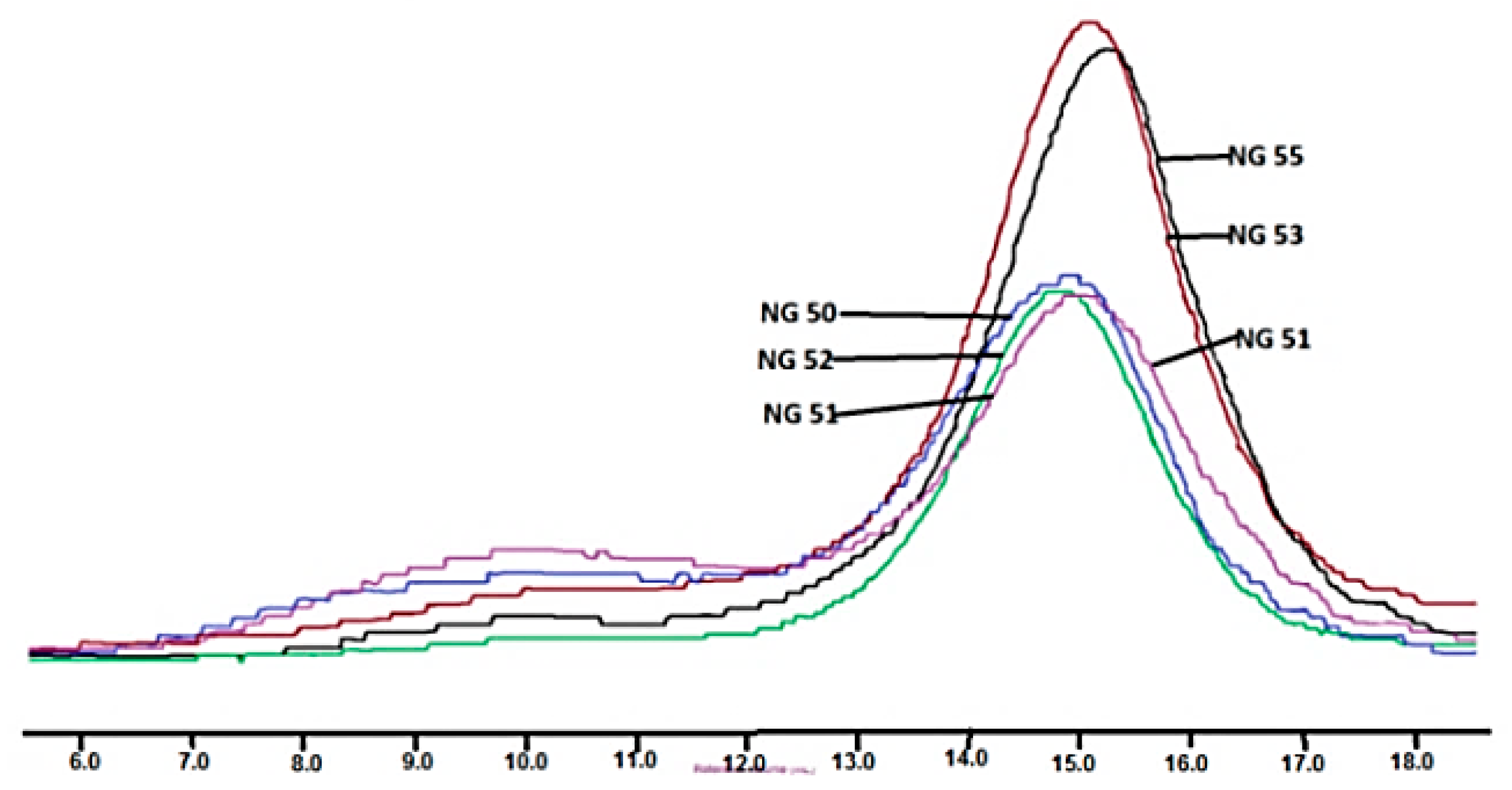
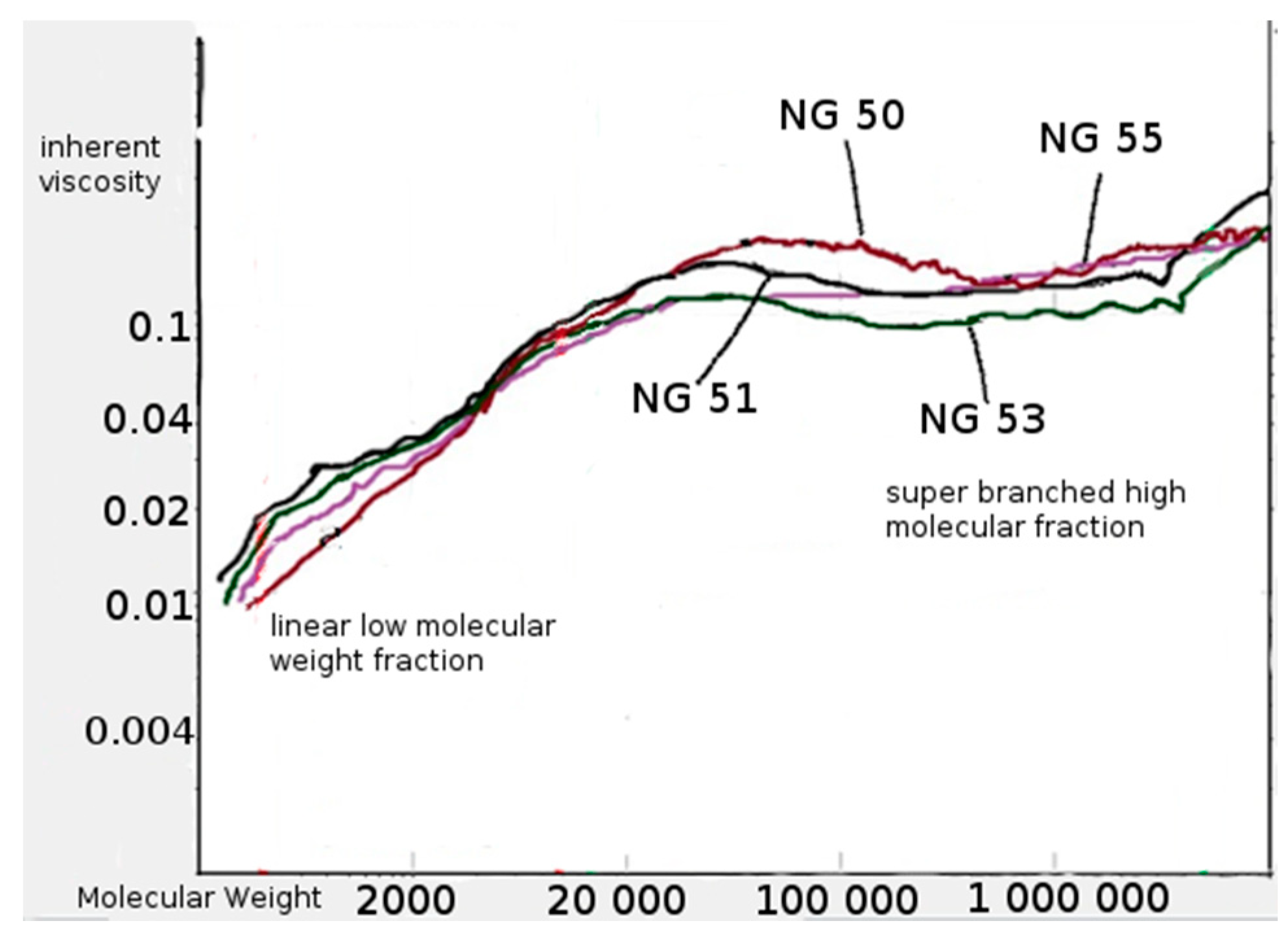
2.4.4. Measurement of Average Molecular Mass and Mass Dispersion
2.4.5. Determination of the Amount of Active Hydroxyl Groups in Obtained Copolymers
2.4.6. Wettability and Surface Free Energy Tests
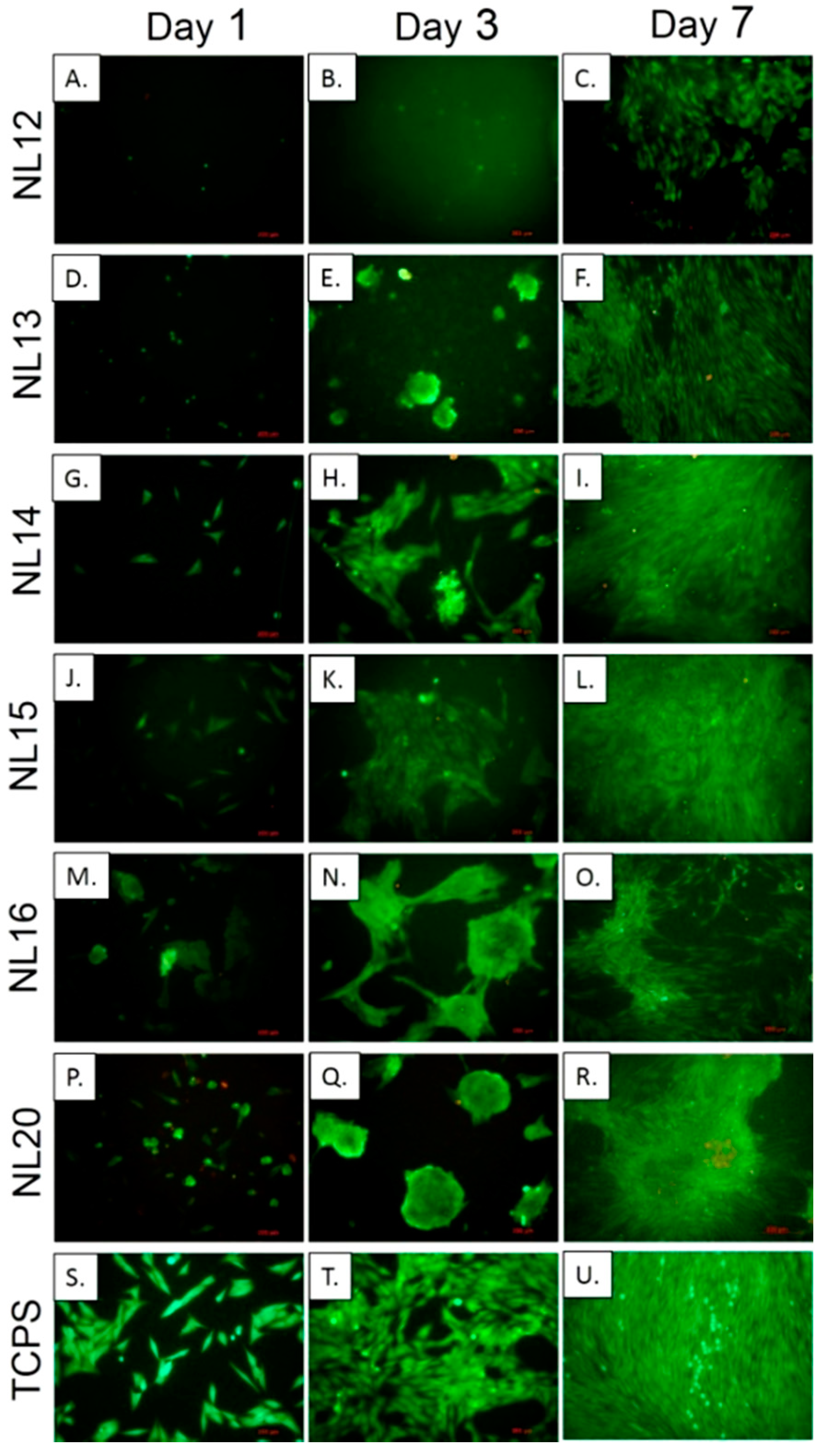
2.4.7. Biological Evaluation
3. Results and Discussion
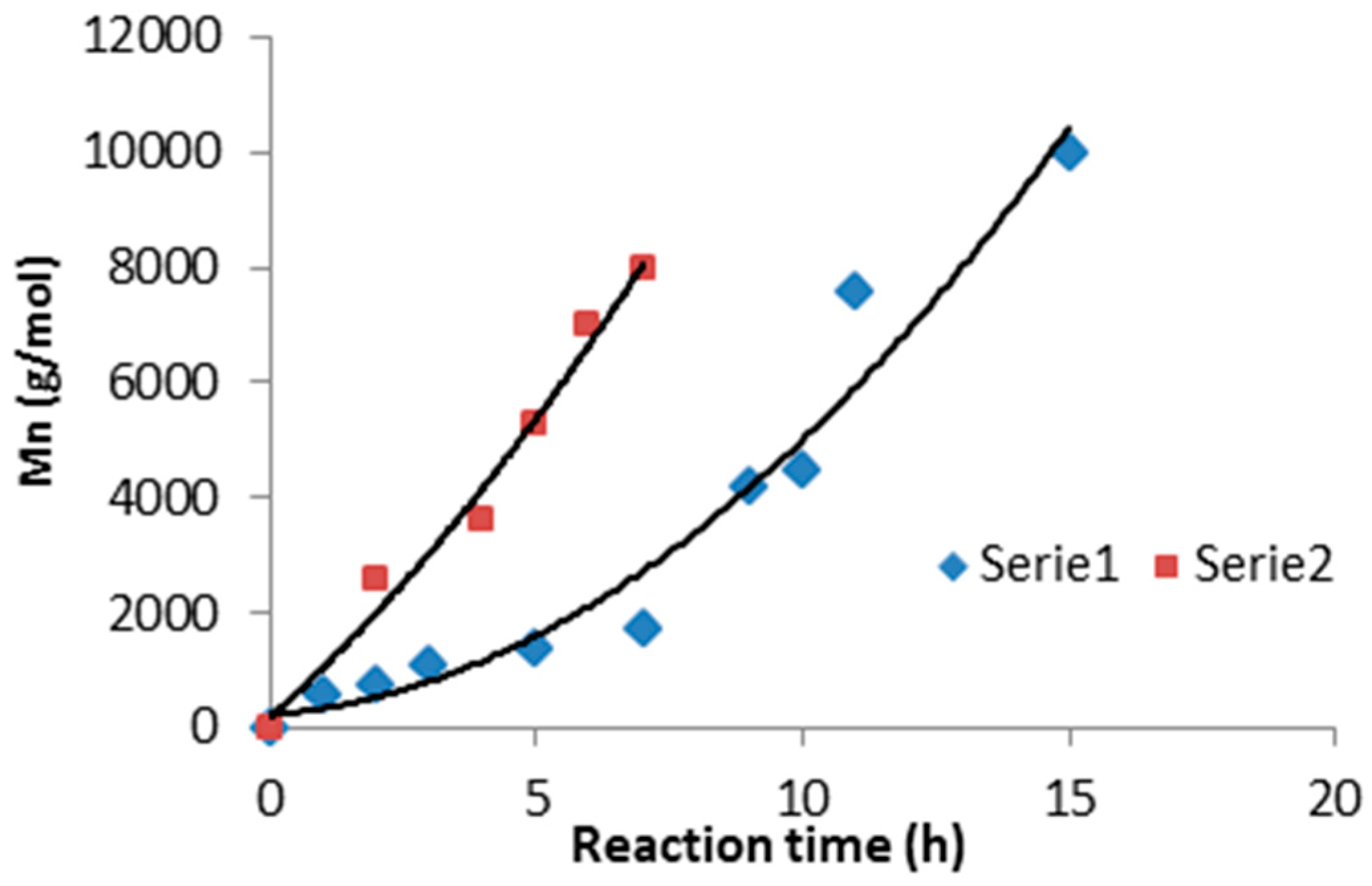
3.1. Synthesis and Characterization of Macro-Initiators—Poly(Butylene Succinate–co–Butylene Citrate)
3.2. Synthesis and Characterization of l-Lactide Copolymers with Butylene Succinate and Butylene Citrate
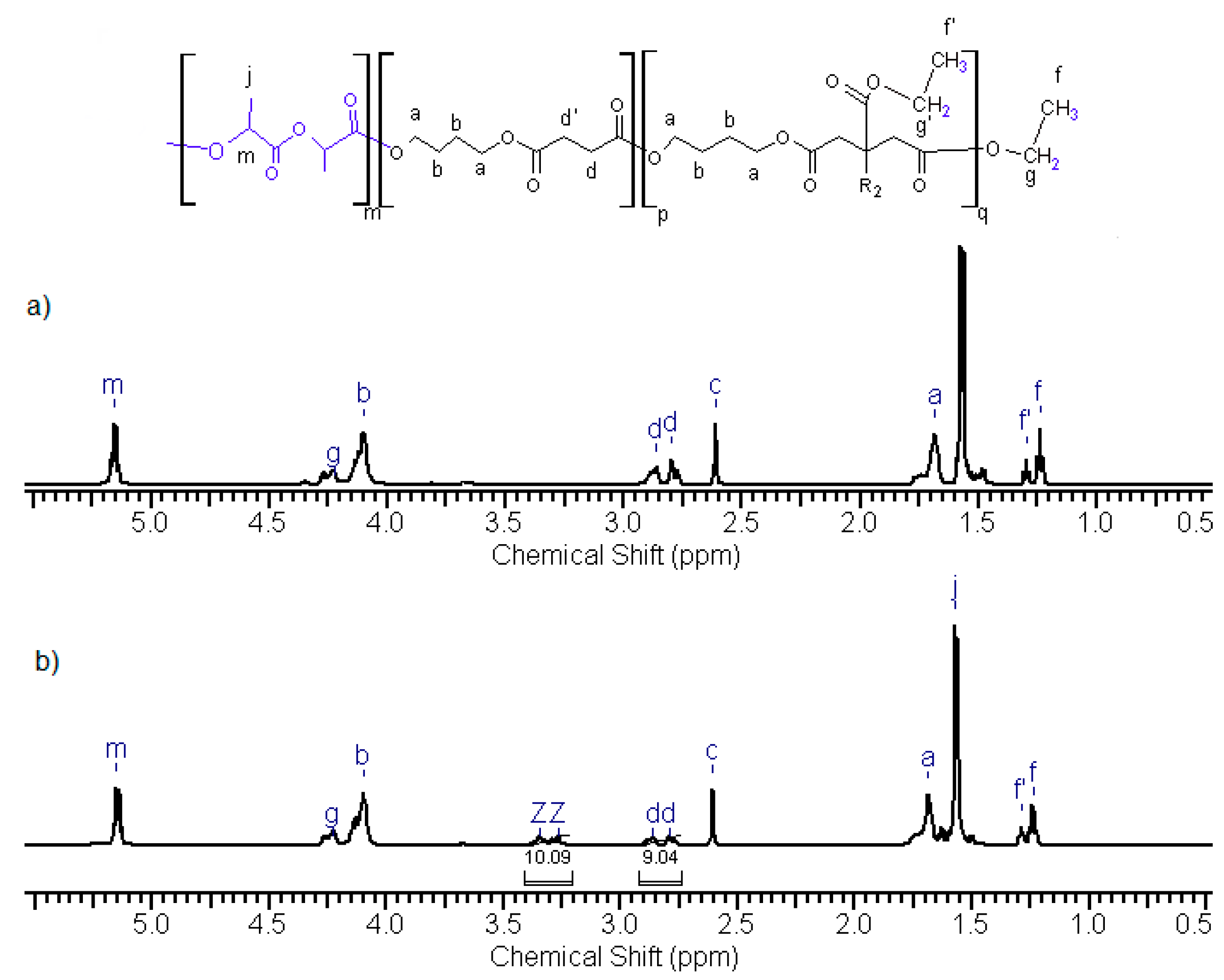
3.3. Synthesis and Characterization of Poly (l-lactide–co–glycolide)–block–poly(butylene succinate–co–butylene citrate)
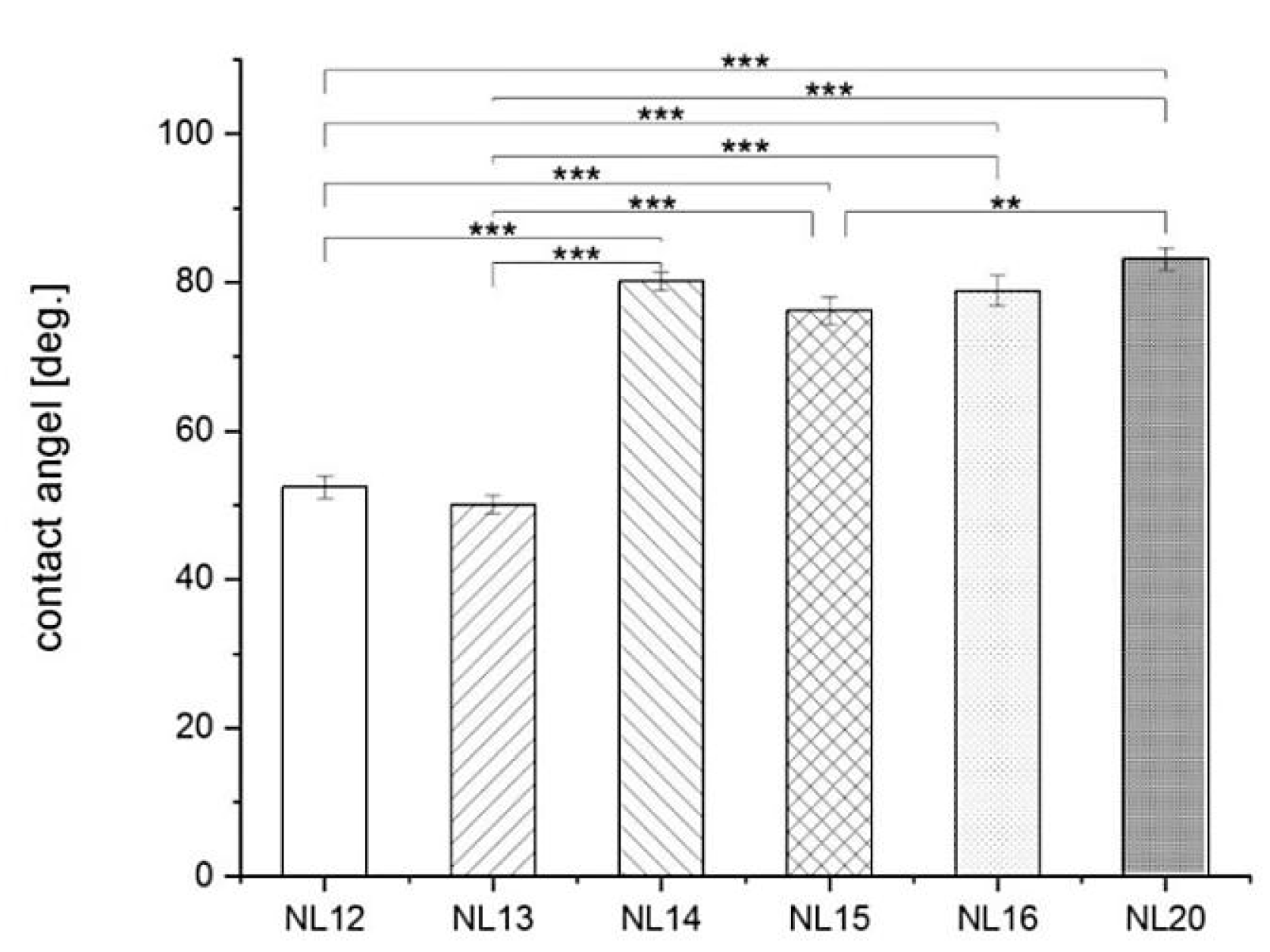
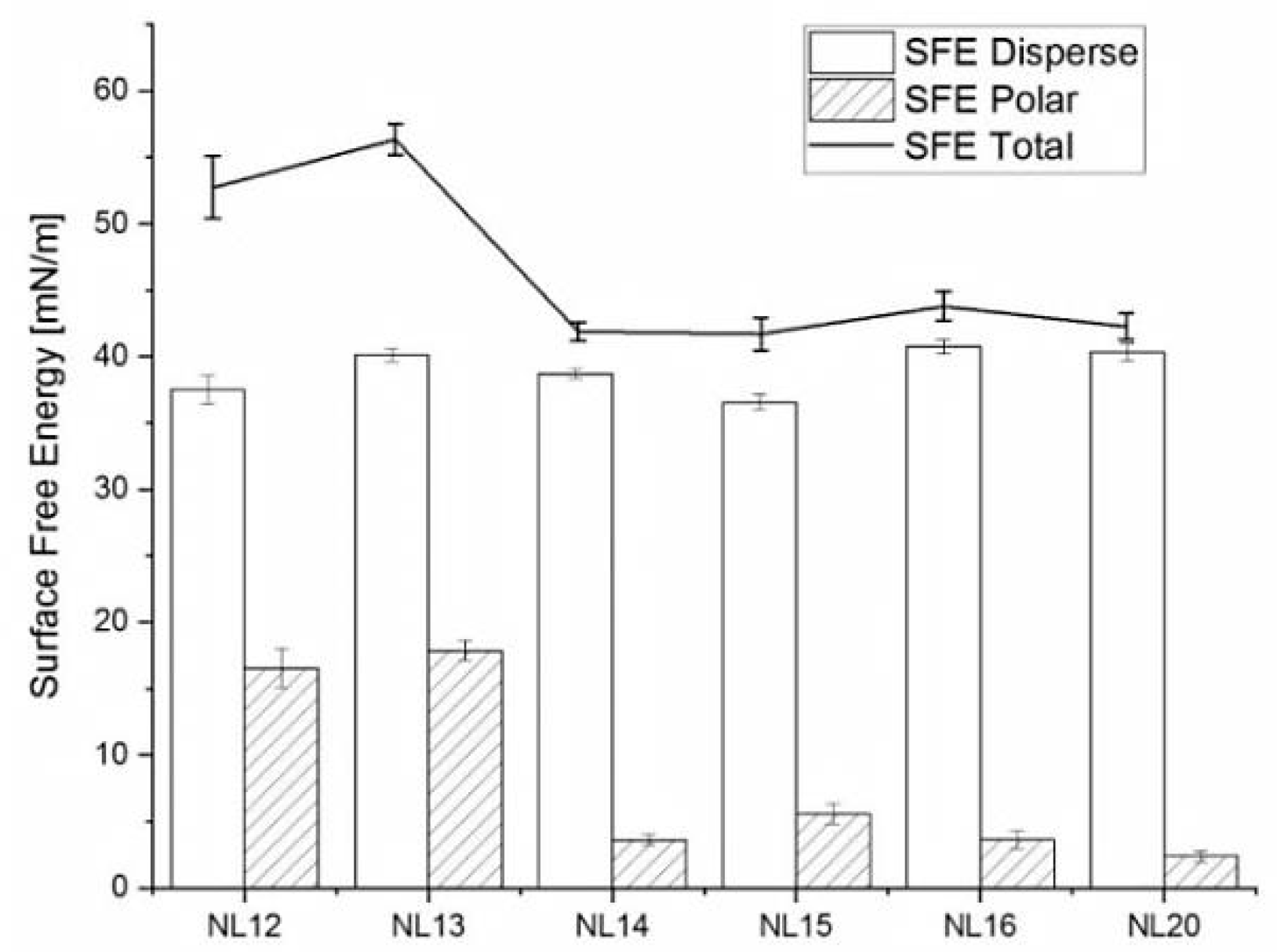
3.4. Wettability and Surface Free Energy
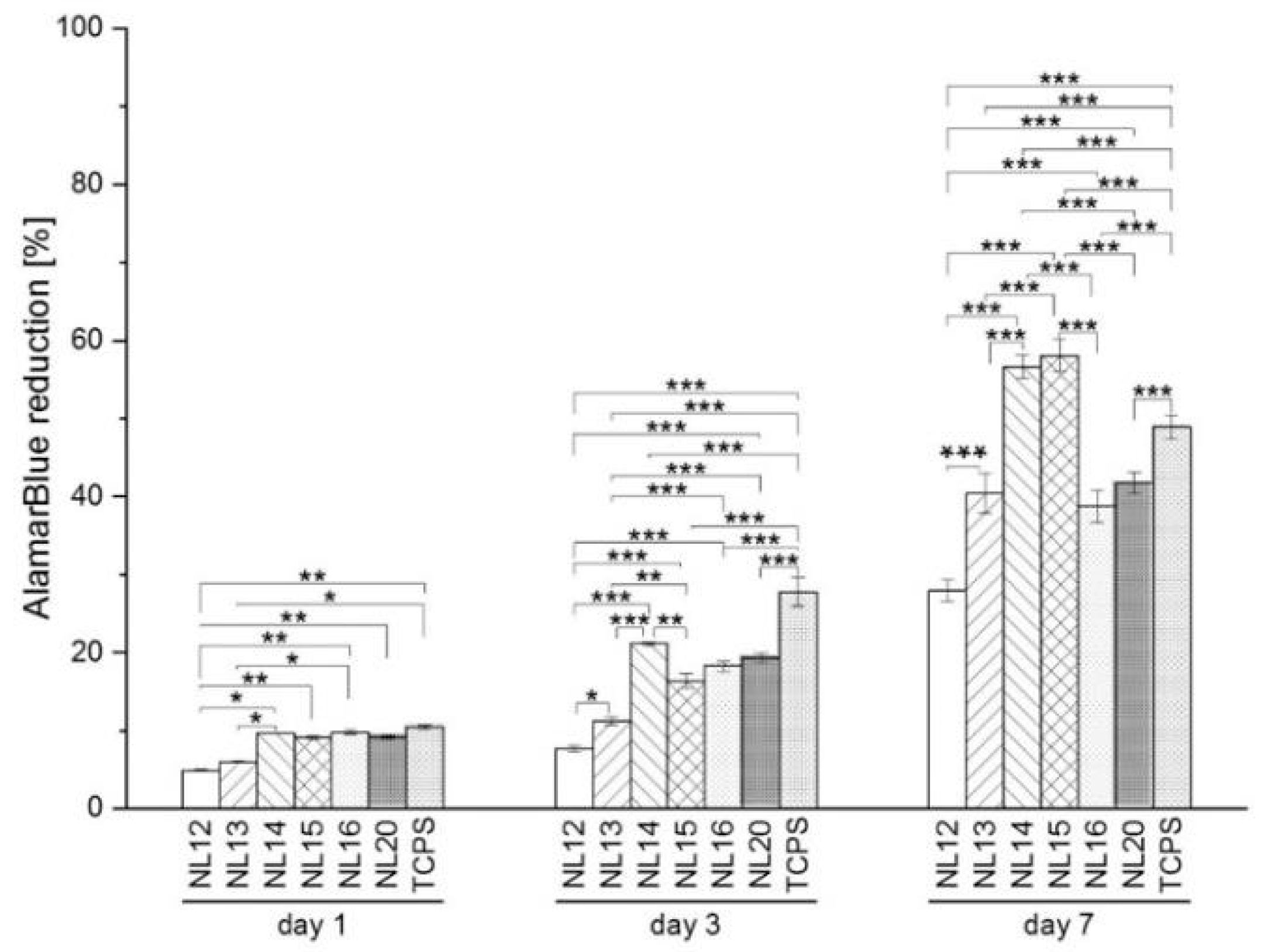
3.5. Cell Metabolic Activity, Viability and Morphology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bikiaris, D.N.; Papageorgiou, G.Z.; Achilias, D.S. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym. Degrad. Stab. 2006, 91, 31–43. [Google Scholar] [CrossRef]
- Corneillie, S.; Smet, M. PLA architectures: The role of branching. Polym. Chem. 2015, 6, 850–867. [Google Scholar] [CrossRef] [Green Version]
- Noordover, B.A.J.; Duchateau, R.; van Benthem, R.A.T.M.; Ming, W.; Koning, C.E. Enhancing the Functionality of Biobased Polyester Coating Resins through Modification with Citric Acid. Biomacromolecules 2007, 8, 3860–3870. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, I.; Choudhury, N.R.; Dutta, N.K.; Kumar, S. Synthesis and characterization of novel citric acid-based polyester elastomers. Polymer 2009, 50, 1682–1691. [Google Scholar] [CrossRef]
- Tran, R.T.; Yang, J.; Ameer, G.A. Citrate-Based Biomaterials and Their Applications in Regenerative Engineering. Annu. Rev. Mater. Res. 2015, 45, 277–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Webb, A.R.; Ameer, G.A. Novel citric acid-based biodegradable elastomers for tissue engineering. Adv. Mater. 2004, 16, 511–516. [Google Scholar] [CrossRef]
- Yang, J.; Webb, A.R.; Pickerill, S.J.; Hageman, G.; Ameer, G.A. Synthesis and evaluation of poly(diolcitrate) biodegradable elastomers. Biomaterials 2006, 27, 1889–1898. [Google Scholar] [CrossRef]
- Su, L.C.; Xie, Z.; Zhang, Y.; Nguyen, K.T.; Yang, J. Study on the antimicrobial properties of citrate-based biodegradable polymers. Front. Bioeng. Biotechnol. 2014, 2, 23. [Google Scholar] [CrossRef]
- Naeini, A.T.; Adeli, M.; Vossoughi, M. Poly (citric acid)-block-poly (ethylene glycol) copolymers—New biocompatible hybrid materials for nanomedicine. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 556–562. [Google Scholar] [CrossRef]
- Jaworska, J.; Kawalec, M.; Pastusiak, M.; Reczynska, K.; Janeczek, H.; Lewicka, K.; Pamula, E.; Dobrzynski, P. Biodegradable Polycarbonates Containing Side Carboxyl Groups—Synthesis, Properties, and Degradation Study. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2756–2769. [Google Scholar] [CrossRef]
- Djordjevic, I.; Chothury, R.N.; Dutta, N.K.; Sunil, K. Poly[octanediol–co–(citric acid)–co–(sebacic acid)] elastomers: Novel bio-elastomers for tissue engineering. Polym. Int. 2011, 60, 333–343. [Google Scholar] [CrossRef]
- Díaz, A.; Katsarava, R.; Puiggalí, J. Synthesis, Properties and Applications of Biodegradable Polymers Derived from Diols and Dicarboxylic Acids: From Polyesters to Poly (ester amide) s. Int. J. Mol. Sci. 2014, 15, 7064–7123. [Google Scholar] [CrossRef] [Green Version]
- Puchalski, M.; Szparaga, G.; Biela, T.; Gutowska, A.; Sztajnowski, S.; Krucińska, I. Molecular and Supramolecular Changes in Polybutylene Succinate (PBS) and Polybutylene Succinate Adipate (PBSA) Copolymer during Degradation in Various Environmental Conditions. Polymers 2018, 10, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yang, J.; Liu, H.; Chang, J.; Cao, A. Synthesisi and Characterization of Poly (butylene succinate–co–butylene malate): A New Biodegradable Copolyester Bearing Hydroxyl Pendant Groups. Biomacromolecules 2003, 4, 437–445. [Google Scholar] [CrossRef]
- Yang, J.; Hao, Q.; Liu, X.; Ba, C.; Coa, A. Novel biodegradable aliphatic poly(butylene succinate–co–cyclic carbonate)s with functionalizable carbonate building blocks, 1. Chemical synthesis and their structural and physical characterization. Biomacromolecules 2004, 5, 209–218. [Google Scholar] [CrossRef]
- Gheybi, H.; Entezami, A.A. Polymeric Micelles Based on Poly(citric acid)-b-poly(l-lactide)-b-poly(citric acid) Copolymer: Synthesis and Characterization. Polym. Plast. Technol. Eng. 2014, 53, 19–29. [Google Scholar] [CrossRef]
- Nikolic, M.S.; Djonlagic, J. Synthesis and characterization of biodegradable poly(butylene succinate–co–butylene adipate)s. Polym. Degrad. Stab. 2001, 74, 263–270. [Google Scholar] [CrossRef]
- Dobrzynski, P.; Kasperczyk, J.; Janeczek, H.; Bero, M. Synthesis of Biodegradable Copolymers with the Use of Low Toxic Zirconium Compounds. 1. Copolymerization of Glycolide with l-lactide Initiated by Zr(Acac)4. Macromolecules 2001, 34, 5090–5098. [Google Scholar] [CrossRef]
- Dobrzynski, P. Mechanism of ε-caprolactone polymerization and ε-caprolactone/trimethylene carbonate copolymerization carried out with Zr(Acac)4. Polymer 2007, 48, 2263–2279. [Google Scholar] [CrossRef]
- Dobrzynski, P.; Pastusiak, M.; Jaworska, J.; Kaczmarczyk, B.; Kwiecien, M.; Kawalec, M. Zirconium (IV) Acetylacetonate: Ring-Opening Initiator Mediating One-Step Synthesis of Biodegradable Polyacids. Adv. Polym. Technol. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Striegel, A.M. Viscometric Detection in Size-Exclusion Chromatography: Principles and Select Applications. Chromatographia 2016, 79, 945–960. [Google Scholar] [CrossRef]
- Pastusiak, M.; Jaworska, J.; Kawalec, M.; Kasperczyk, J.; Dobrzynski, P. Obtaining Aliphatic Branched Polycarbonates via Simple Copolymerization of Trimethylene Carbonate with Cyclic Carbonate Containing Pendant Ester Groups. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 808–819. [Google Scholar] [CrossRef]
- Rychter, P.; Lewicka, K.; Pastusiak, M.; Domanski, M.; Dobrzynski, P. PLGA-PEG terpolymers as a carriers of bioactive agents, influence of PEG blocks content on degradation and release of herbicides into soil. Polym. Degrad. Stab. 2019, 161, 95–107. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SM:CE:BD | BS:BC (% mol) | Mn (g/mol) | Đ | NOH | Tg (°C) | Tm (°C) | ΔH (J/g) |
|---|---|---|---|---|---|---|---|---|
| NG 6 | 1:0:1.07 | 100:0 | 7000 | 2.2 | ≈2 | −18 | 114 | 81 |
| NG 17 | 7:3:10.7 | 68:32 | 7000 | 4.0 | 10 | −33 | 77 | 52 |
| NG 23 | 5:5:10.7 | 52:48 | 8000 | 8.7 | 17 | −29 | 55 | 14 |
| NG 49 | 3:7:10.7 | 27:73 | 7000 | 9.2 | 20 | −29 | - | - |
| NG 48 | 0:1:1.07 | 0:100 | 7500 | 10.5 | 24 | −31 | - | - |
| Sample | C (%) | M/I | (LA:PBS)0 (wt %) | LA:BS (wt %) | LA:BS (%mol.) | Mw (g/mol) | TMw (g/mol) | Đ | Tg (°C) | Tm (°C) | ΔH (J/g) | ηinh (dL/g) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NG7 | 99 | 190/1 | 75:25 | 75:25 | 80:20 | 56; 200 | 68; 720 | 2.0 | 38 | 165 | 43 | 1.1 |
| NG8 | 96 | 75/1 | 60:40 | 59:41 | 65:35 | 38; 100 | 44; 500 | 2.5 | 7 | 108; 150 | 29; 23 | 0.8 |
| NG9 | 95 | 33/1 | 40:60 | 38:62 | 45:55 | 44; 500 | 43; 480 | 3.7 | −16 | 108 | 50 | 0.8 |
| Sample | Initiator | C (%) | M/I | (LA:BS:BC)0 (%mol.) | LA:BS:BC (%mol.) | Mw (g/mol) | Đ | Tg (C°) | Tm (C°) | ΔH (J/g) | ηinh (dL/g) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NG 20 | NG 17 | 99 | 60/1 | 55:30:15 | 55:29:16 | 24; 200 | 2.2 | 11 | 40; 115 | 38; 6.1 | 0.3 |
| NG 51 | NG 49 | 88 | 60/1 | 55:12:23 | 49:14:27 | 190; 000 | ≈100 | −1 | 126 | 16 | 1.3 |
| NG 53 | NG 48 | 96 | 75/1 | 59:0:41 | 60:0:40 | 203; 000 | ≈57 | −15 | - | - | 1.7 |
| NG 55 | NG 48 | 98 | 12/1 | 19:0:81 | 19:0:81 | 148; 000 | ≈58 | −26 | - | - | 1.6 |
| Sample | Initiator | C (%) | M/I | (LA:GL:BS:BC)0 (% mol) | La:GL:BS:BC (% mol) | Mw (g/mol) | Đ | Tg (°C) | Tm (°C) | ΔH (J/g) | ηinh (dL/g) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NL 14 | NL7 | 98 | 120/1 | 67:11:7:15 | 66:10:9:15 | 42; 500 | 3.5 | 32 | 78; 131 | 3.9, 2.0 | 0.45 |
| NL 15 | NL3 | 97 | 120/1 | 67:11:11:11 | 64:12:12:12 | 34; 300 | 3.7 | 33 | 83; 120 | 5.4, 1.5 | 0.41 |
| NL20 | NL7 | 97 | 60/1 | 55:9:11:26 | 57:10:11:22 | 28; 100 | 3.8 | 21 | 73; 117 | 14.2, 2.8 | 0.37 |
| NL16 | NL3 | 98 | 60/1 | 55:9:18:18 | 56:10:17:17 | 29; 400 | 3.8 | 23 | 77; 113 | 13.7, 0,9 | 0.35 |
| NL12 | NL7 | 91 | 40/1 | 46:7:14:33 | 47:8:13:32 | 15; 300 | 4.0 | 5 | 95; 139 | 15.4, 0,9 | 0.24 |
| NL13 | NL3 | 94 | 40/1 | 46:7:23:24 | 48:8:22:22 | 19; 700 | 3.9 | 6 | 105 | 17.0 | 0.28 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śmigiel-Gac, N.; Pamuła, E.; Krok-Borkowicz, M.; Smola-Dmochowska, A.; Dobrzyński, P. Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate. Polymers 2020, 12, 214. https://doi.org/10.3390/polym12010214
Śmigiel-Gac N, Pamuła E, Krok-Borkowicz M, Smola-Dmochowska A, Dobrzyński P. Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate. Polymers. 2020; 12(1):214. https://doi.org/10.3390/polym12010214
Chicago/Turabian StyleŚmigiel-Gac, Natalia, Elżbieta Pamuła, Małgorzata Krok-Borkowicz, Anna Smola-Dmochowska, and Piotr Dobrzyński. 2020. "Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate" Polymers 12, no. 1: 214. https://doi.org/10.3390/polym12010214
APA StyleŚmigiel-Gac, N., Pamuła, E., Krok-Borkowicz, M., Smola-Dmochowska, A., & Dobrzyński, P. (2020). Synthesis and Properties of Bioresorbable Block Copolymers of l-Lactide, Glycolide, Butyl Succinate and Butyl Citrate. Polymers, 12(1), 214. https://doi.org/10.3390/polym12010214