Oxidation–Responsive Emulsions Stabilized with Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide)

Abstract
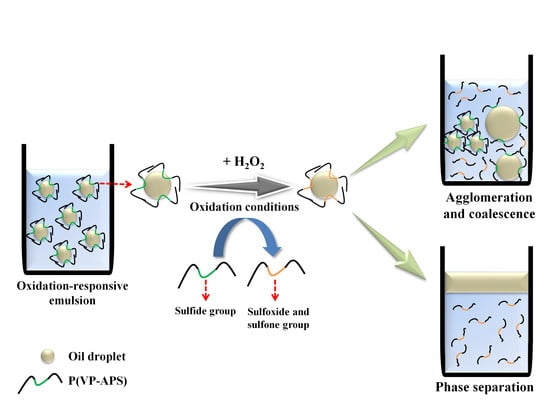
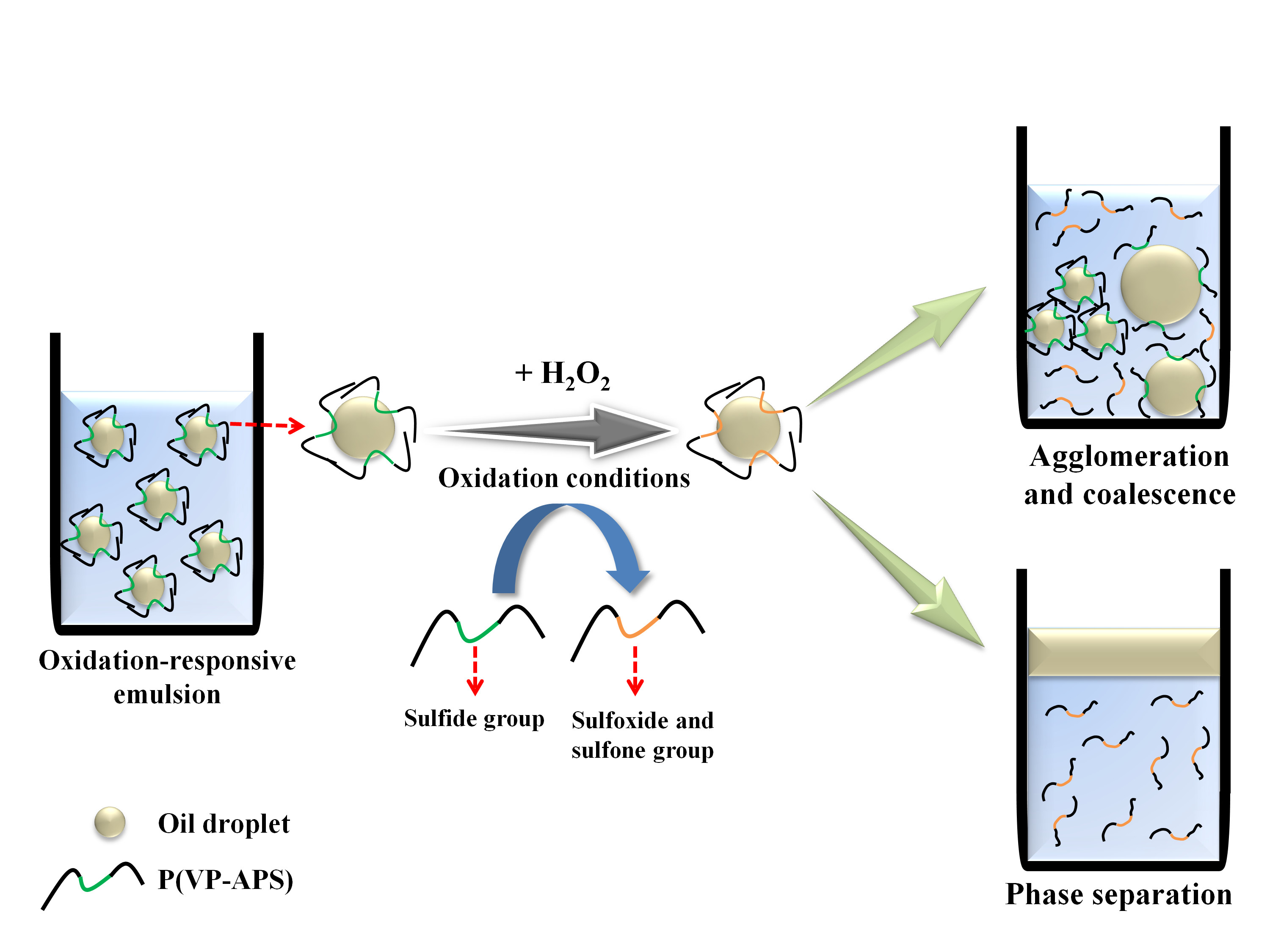
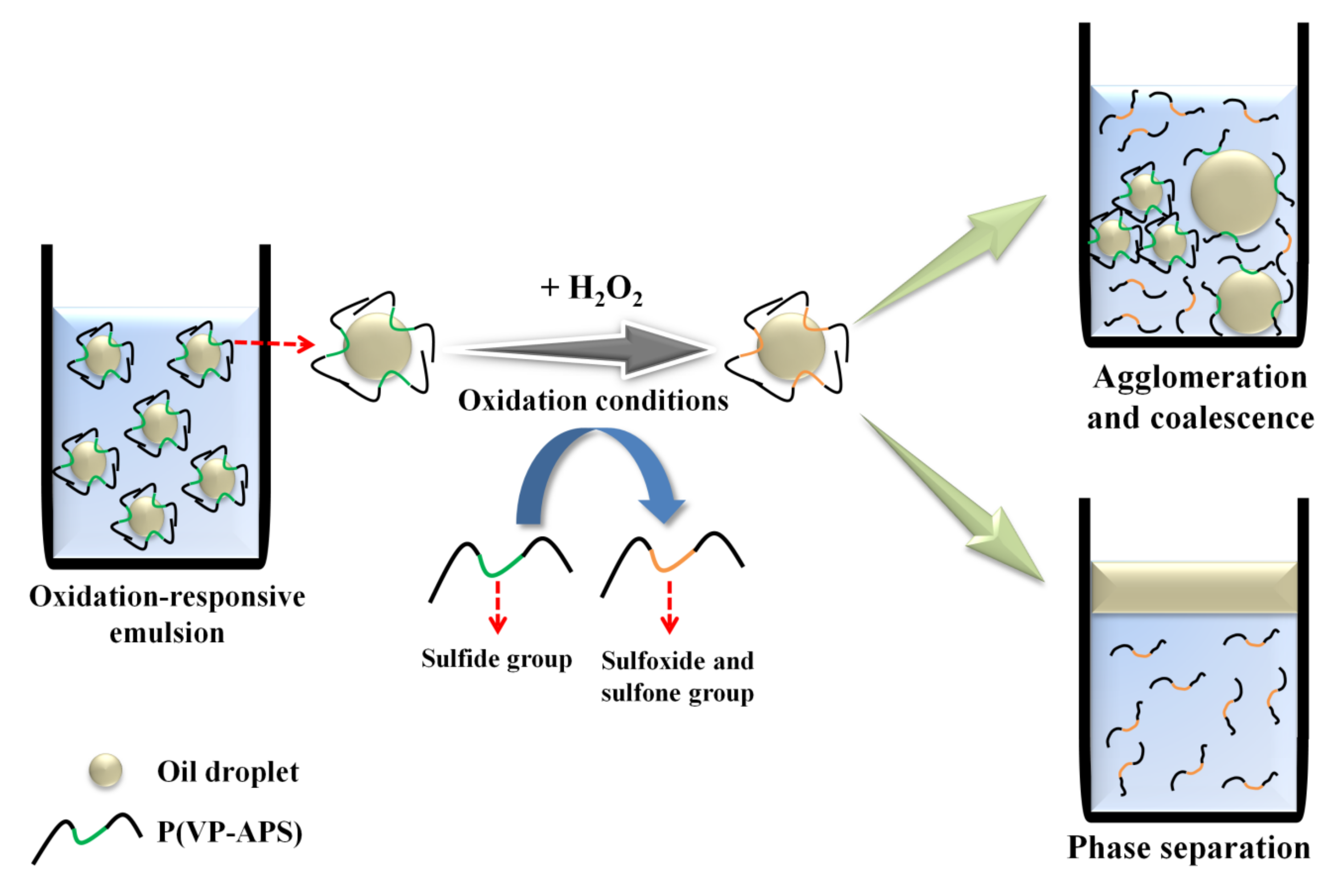
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide)
2.3. Oxidizing Treatment of Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide)
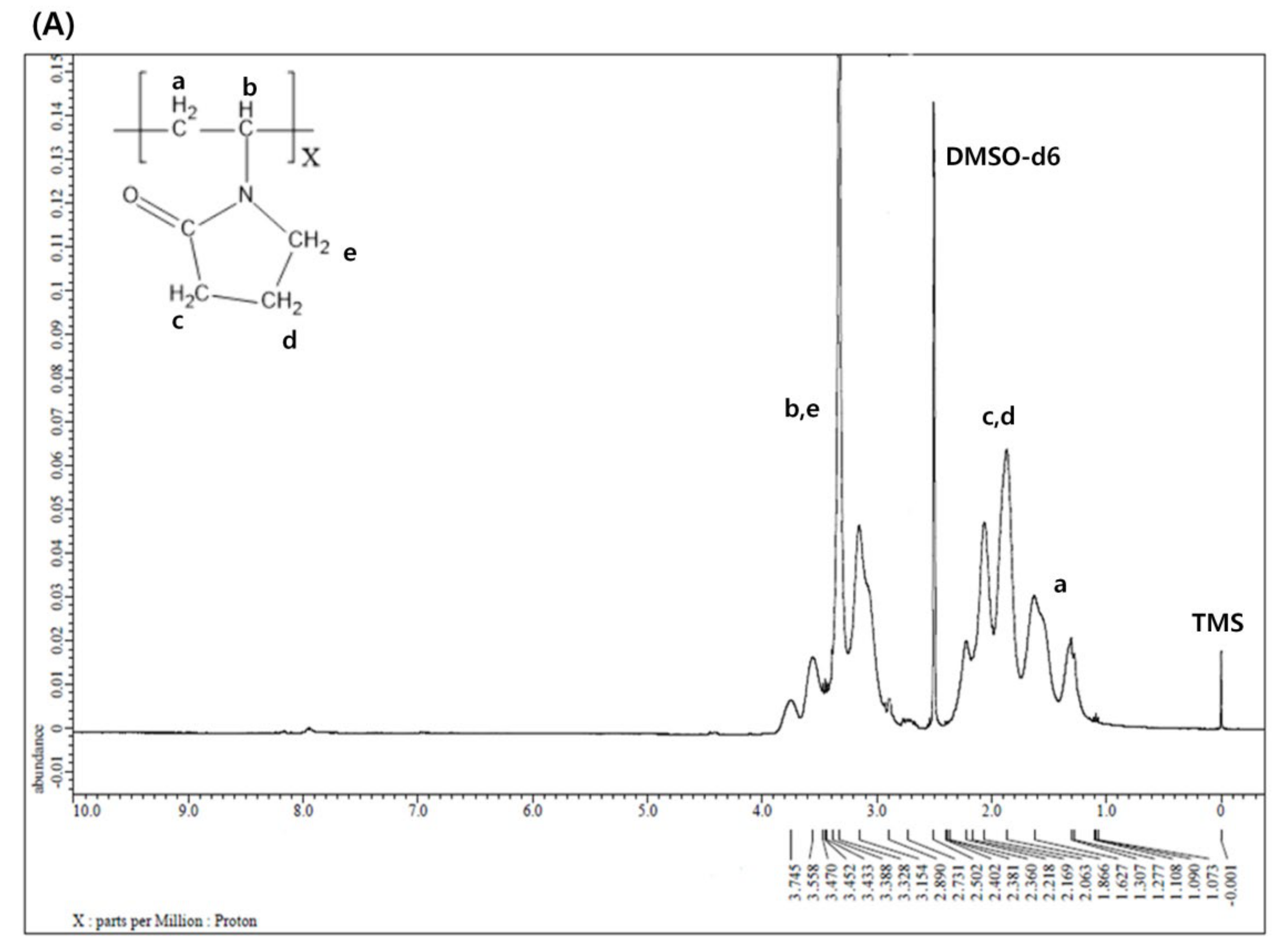
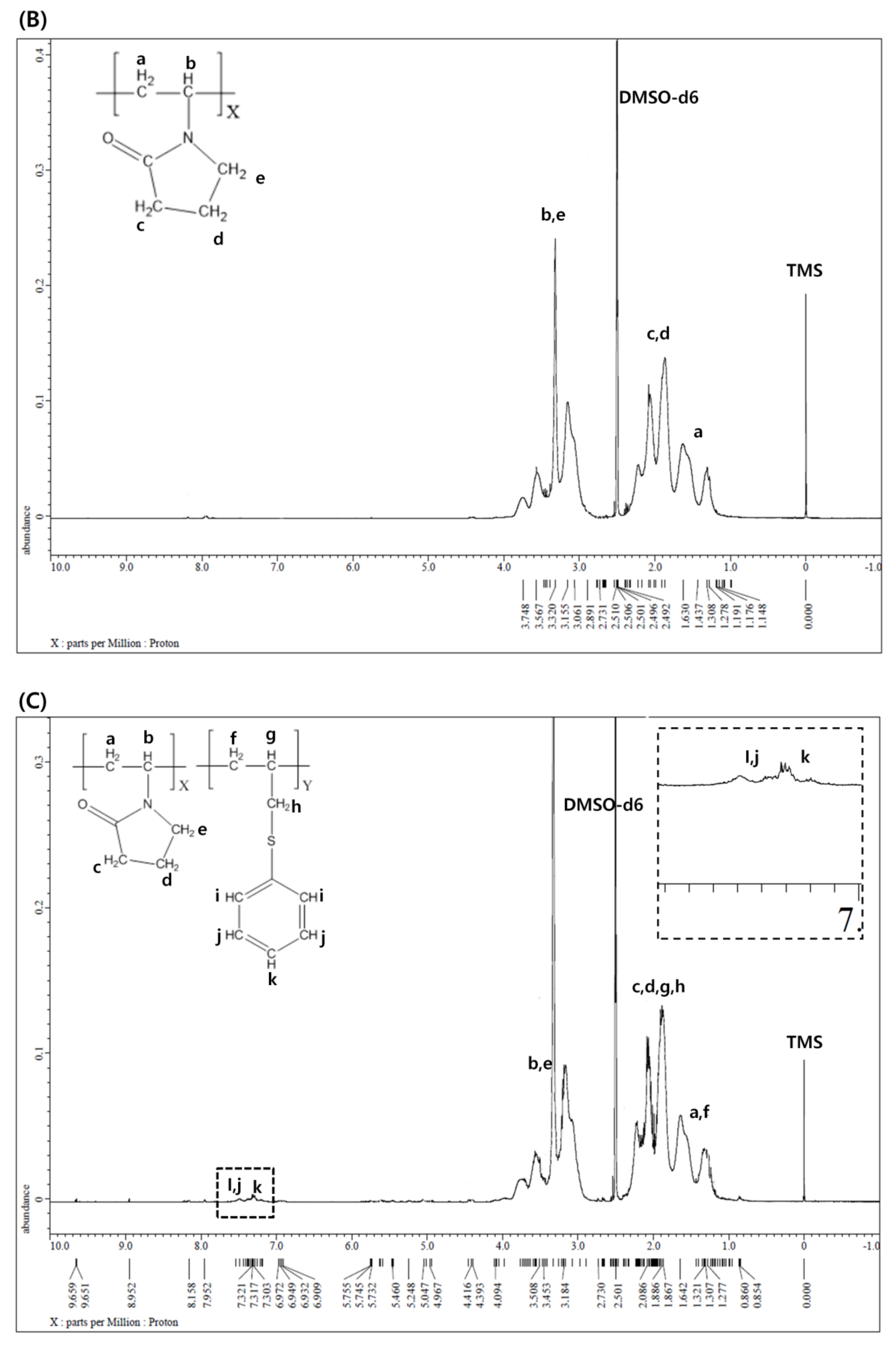
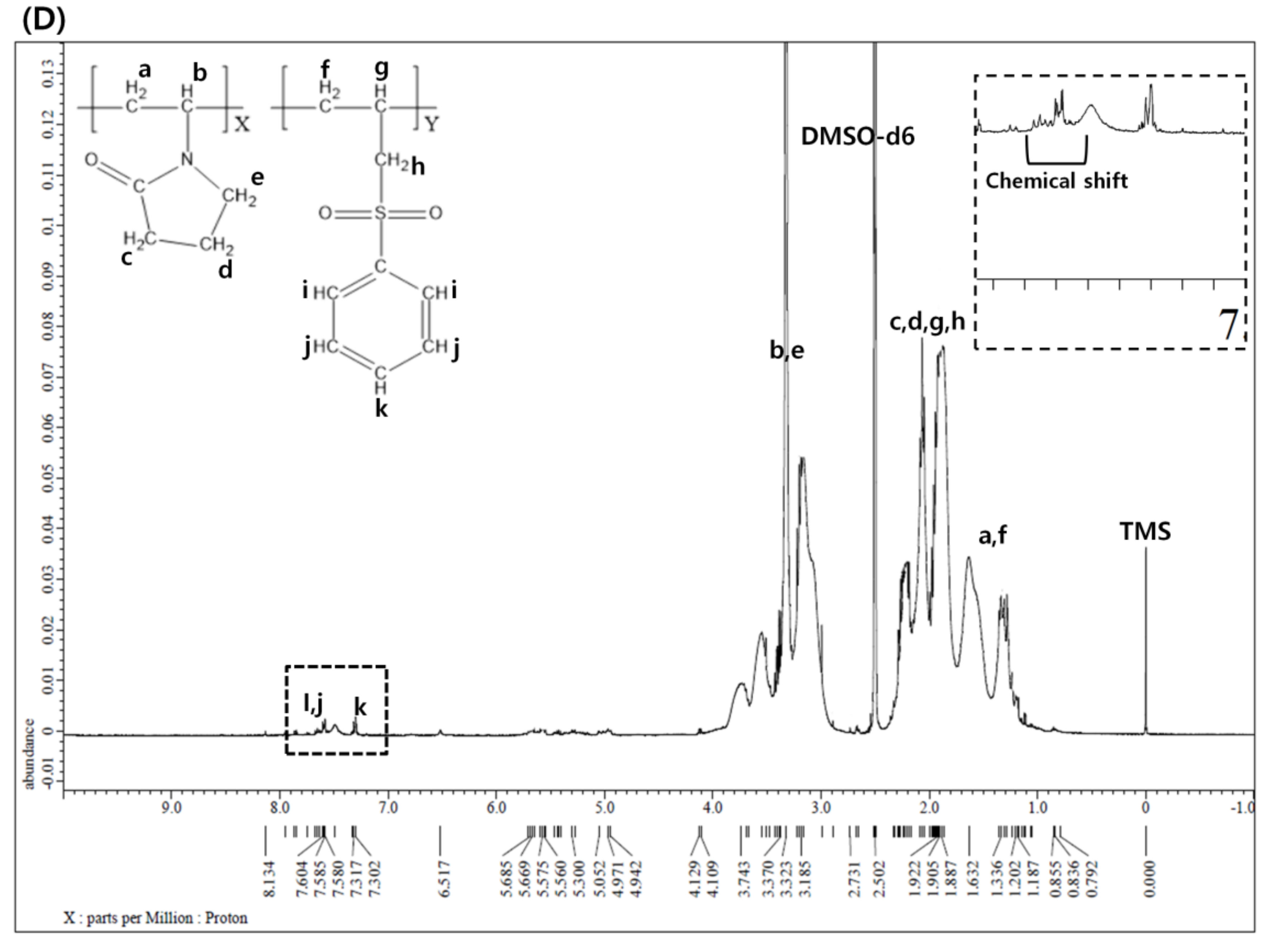
2.4. 1H NMR Spectroscopy
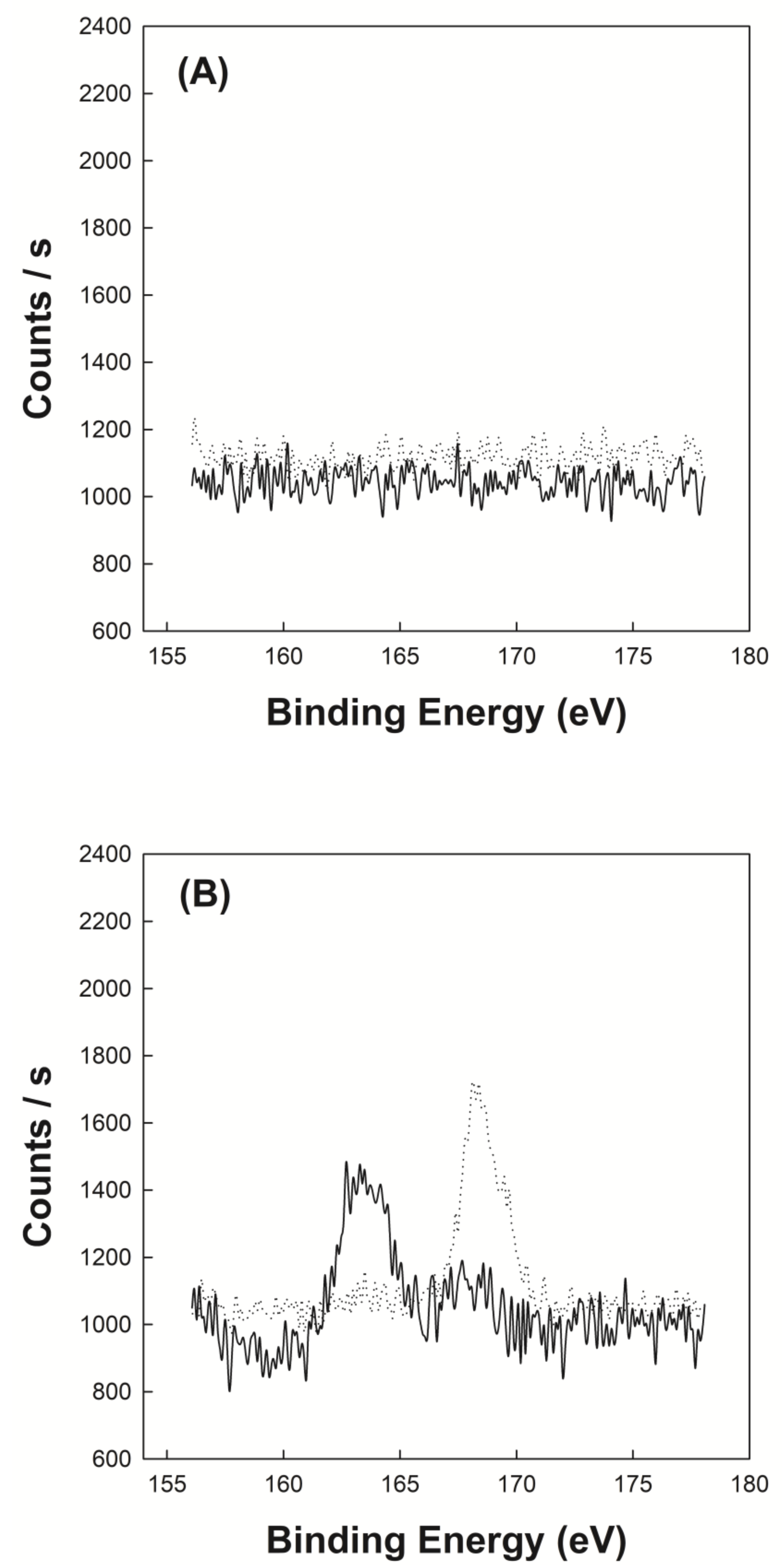
2.5. X-ray Photoelectron Spectroscopy (XPS)
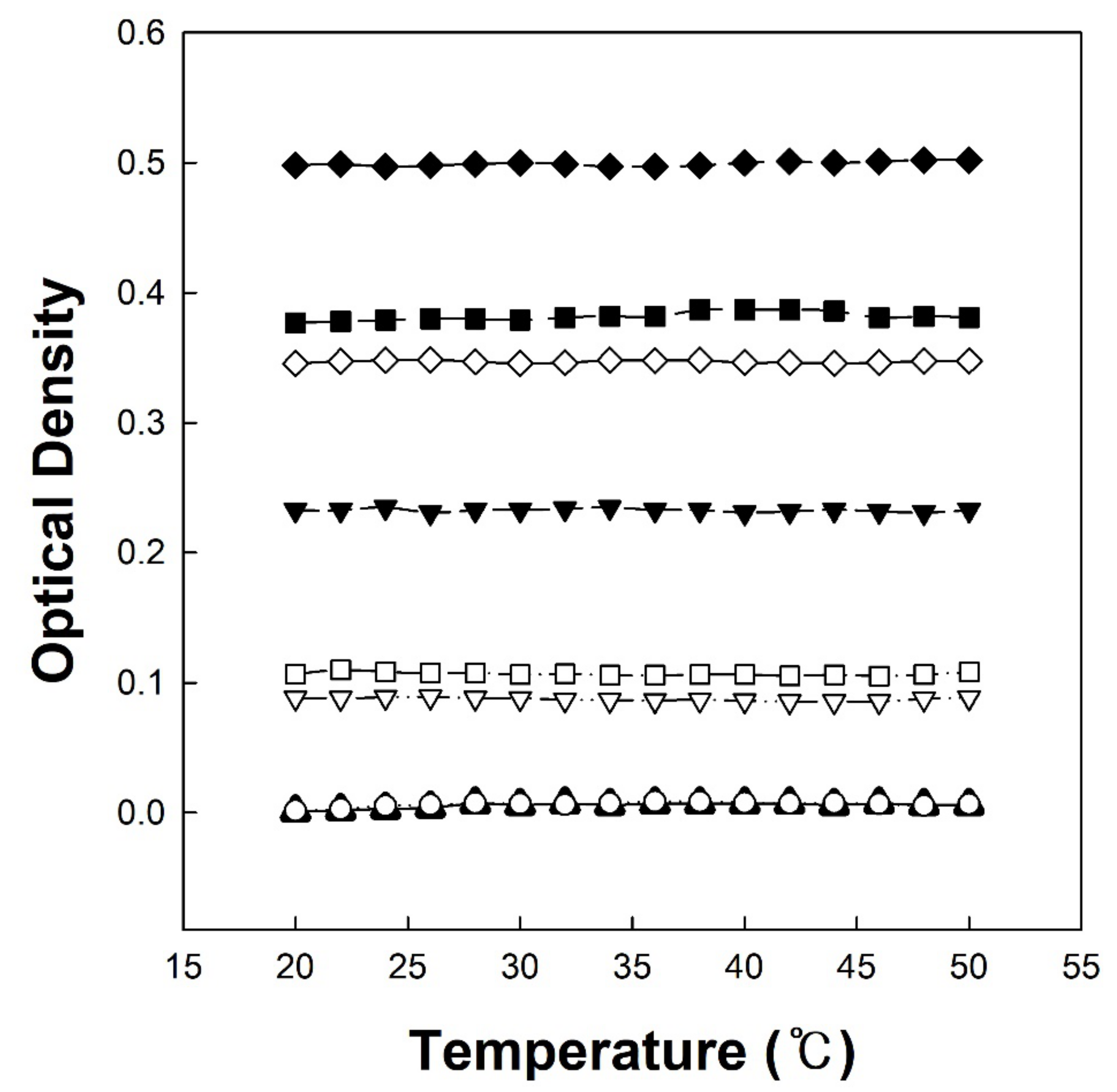
2.6. Temperature-Dependent Optical Density Change of Copolymer Solutions
2.7. Measurements of Air/Water Interfacial Tensions
2.8. Preparation of Emulsion Stabilized with of Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide)
2.9. Investigation of Emulsion Stability
3. Results and Discussion
3.1. 1H NMR Spectroscopy
3.2. X-ray Photoelectron Spectroscopy
3.3. Temperature-Dependent Optical Density Change of Copolymer Solutions
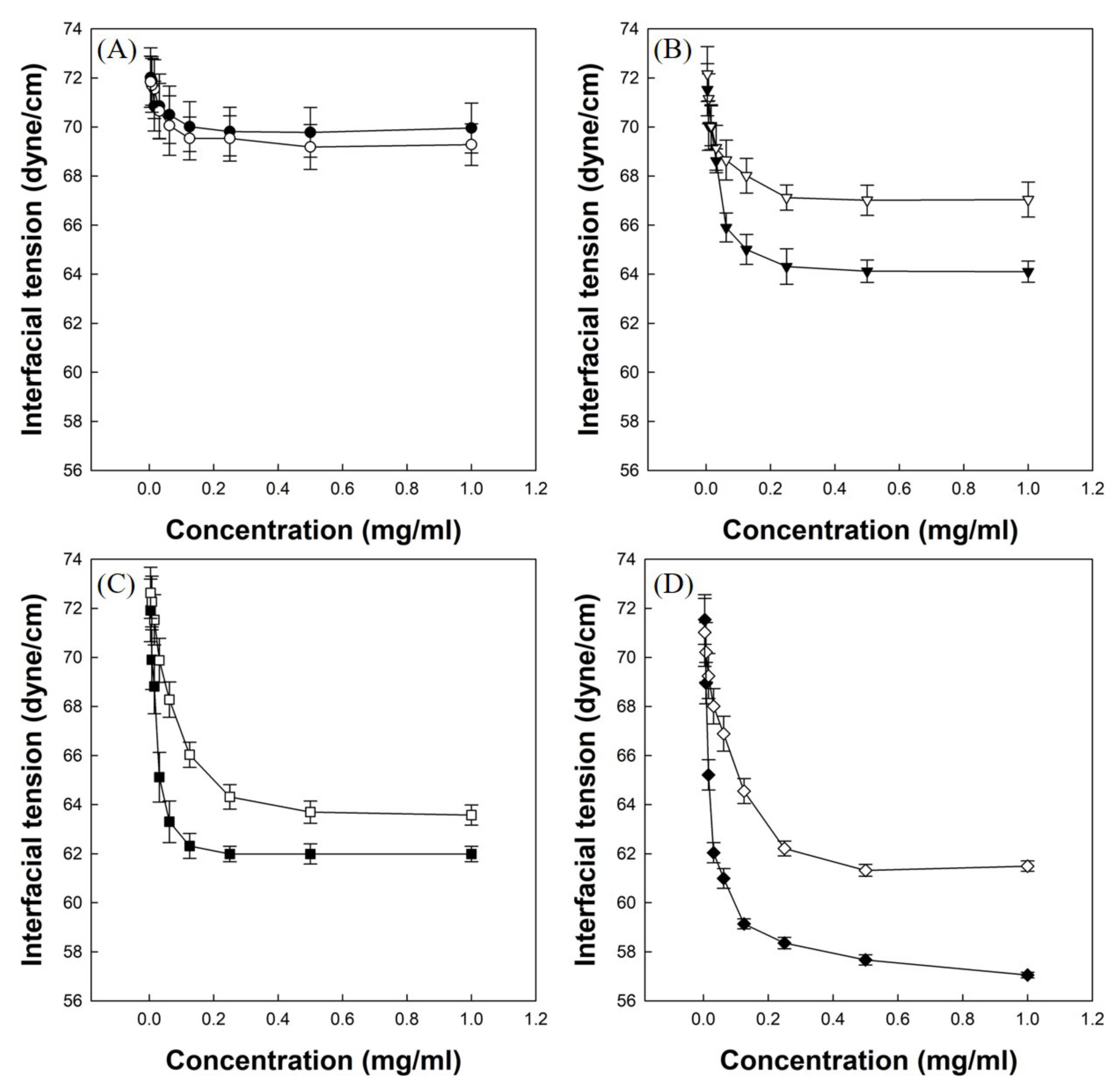
3.4. Measurements of Air/Water Interfacial Tensions
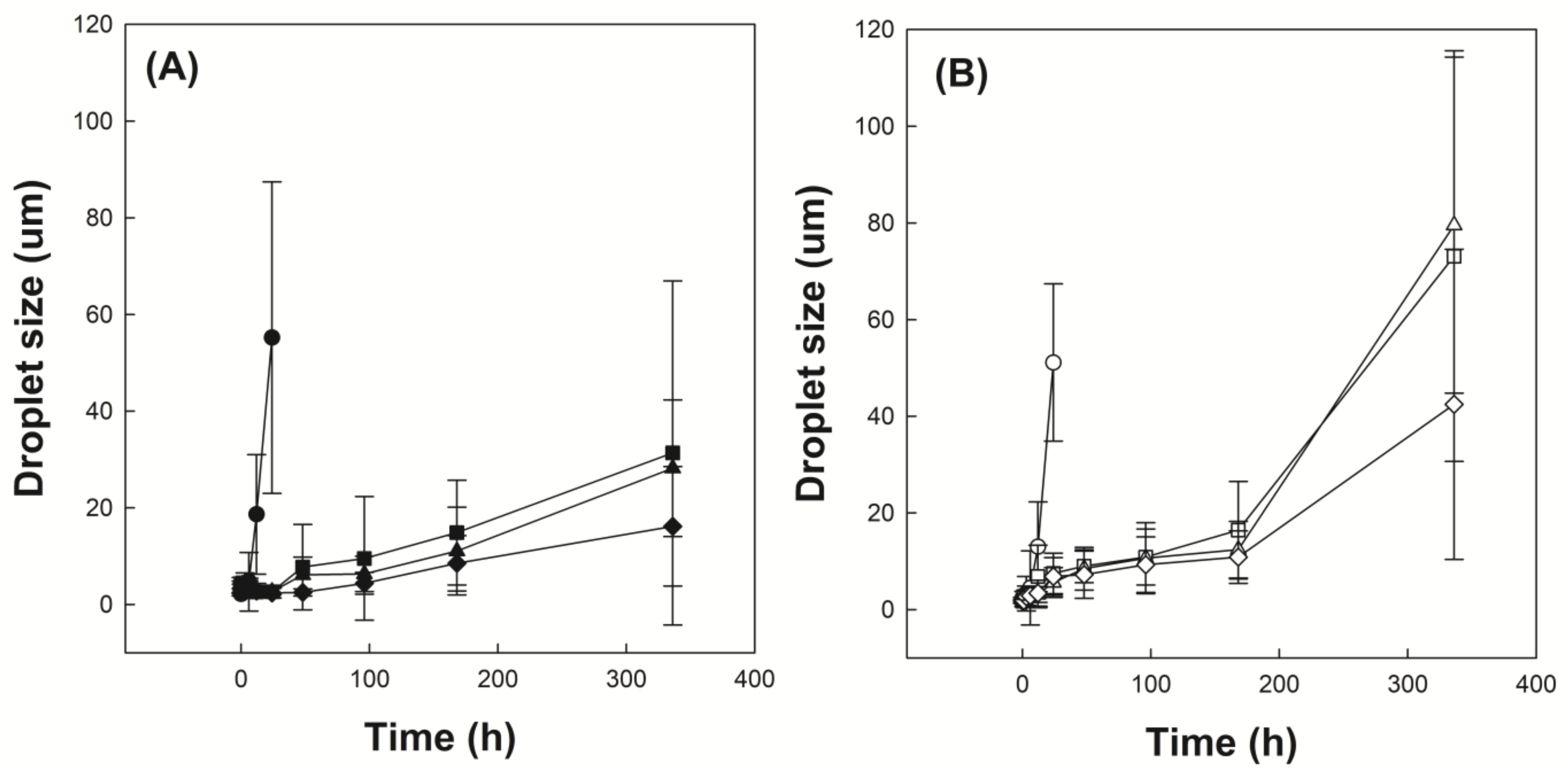
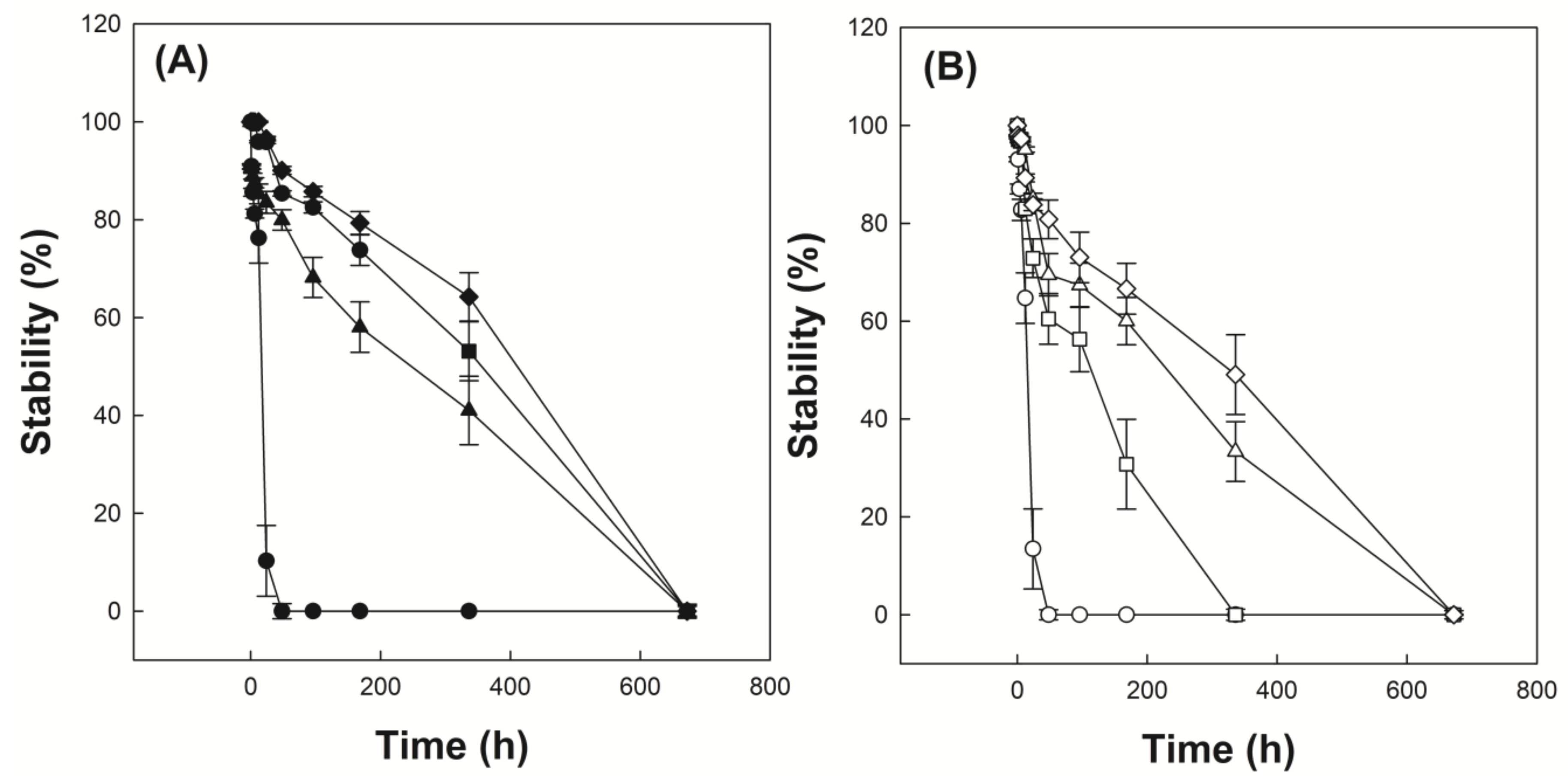
3.5. Investigation of Emulsion Stability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brugger, B.; Richtering, W. Magnetic, Thermosensitive Microgels as Stimuli-Responsive Emulsifiers Allowing for Remote Control of Separability and Stability of Oil in Water-Emulsions. Adv. Mater. 2007, 19, 2973–2978. [Google Scholar] [CrossRef]
- Tang, J.; Quinlan, P.J.; Tam, K.C. Stimuli-responsive Pickering emulsions: Recent advances and potential applications. Soft Matter 2015, 11, 3512–3529. [Google Scholar] [CrossRef]
- Brugger, B.; Rosen, B.A.; Richtering, W. Microgels as Stimuli-Responsive Stabilizers for Emulsions. Langmuir 2008, 24, 12202–12208. [Google Scholar] [CrossRef]
- Besnard, L.; Marchal, F.; Paredes, J.F.; Daillant, J.; Pantoustier, N.; Perrin, P.; Guenoun, P. Multiple Emulsions Controlled by Stimuli-Responsive Polymers. Adv. Mater. 2013, 25, 2844–2848. [Google Scholar] [CrossRef]
- Khoukh, S.; Perrin, P.; Bes de Berc, F.; Tribet, C. Reversible Light-Triggered Control of Emulsion Type and Stability. Chem. Phys. Chem. 2005, 6, 2009–2012. [Google Scholar] [CrossRef]
- Li, Z.; Ngai, T. Stimuli-responsive gel emulsions stabilized by microgel particles. Colloid. Polym. Sci. 2011, 289, 489–496. [Google Scholar] [CrossRef]
- Seo, S.R.; Lee, H.Y.; Kim, J.C. Thermo- and pH-Responsiveness of Emulsions Stabilized with Acidic Thermosentive Polymers. J. Dispers. Sci. Technol. 2013, 34, 1280–1285. [Google Scholar] [CrossRef]
- Seo, S.R.; Kim, J.C. Emulsions Stabilized with poly(Hydroxyethyl Acrylate-co-Coumaryl Acrylate-co-2-Ethylhexyl acrylate). J. Macromol. Sci. A 2013, 50, 855–860. [Google Scholar] [CrossRef]
- Kwon, K.; Kim, J.C. Emulsion stabilized with disulfide proteinoid and its stability in reducing condition. J. Disper. Sci. Technol. 2018, 39, 333–340. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, J.C. Oil-in-gold nanoparticle solution emulsion stabilized with amphiphilic polymers and its stability under NIR irradiation. J. Dispers. Sci. Technol. 2018, 39, 961–969. [Google Scholar] [CrossRef]
- Chung, S.L.; Ferrier, L.K. pH and Sodium Chloride Effects on Emulsifying Properties of Egg Yolk Phosvitin. J. Food Sci. 1992, 57, 40–42. [Google Scholar] [CrossRef]
- Zorba, Ö. The effects of the amount of emulsified oil on the emulsion stability and viscosity of myofibrillar proteins. Food Hydrocoll. 2006, 20, 698–702. [Google Scholar] [CrossRef]
- Liu, Y.; Bhatnagar, A.; Ji, Q.; Riffle, J.S.; McGrath, J.E.; Geibel, J.F.; Kashiwagi, T. Influence of polymerization conditions on the molecular structure stability and physical behavior of poly(phenylene sulfide sulfone) homopolymers. Polymer 2000, 41, 5137–5146. [Google Scholar] [CrossRef]
- Zhunuspayev, D.E.; Mun, G.A.; Khutoryanskiy, V.V. Temperature-Responsive Properties and Drug Solubilization Capacity of Amphiphilic Copolymers Based on N-Vinylpyrrolidone and Vinyl Propyl Ether. Langmuir 2010, 26, 7590–7597. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Wennerstroem, H. Entropic forces between amphiphilic surfaces in liquids. J. Phys. Chem. 1992, 96, 520–531. [Google Scholar] [CrossRef]
- Zilman, A.; Tlusty, T.; Safran, S.A. Entropic networks in colloidal, polymeric and amphiphilic systems. J. Phys. Condens. Matter 2002, 15, S57–S64. [Google Scholar] [CrossRef]
- Nie, Z.; Fava, D.; Kumacheva, E.; Zou, S.; Walker, G.C.; Rubinstein, M. Self-assembly of metal–polymer analogues of amphiphilic triblock copolymers. Nat. Mater. 2007, 6, 609–614. [Google Scholar] [CrossRef]
- Busico, V.; Ferraro, A.; Vacatello, M. Thirmotropic Smectic Liquid Crystals of Ionic Amphiphilic Compounds: A General Discussion. Mol. Cryst. Liquid Cryst. 1985, 128, 243–261. [Google Scholar] [CrossRef]
- Mondal, J.; Yethiraj, A. Driving Force for the Association of Amphiphilic Molecules. J. Phys. Chem. Lett. 2011, 2, 2391–2395. [Google Scholar] [CrossRef]
- Wesslén, B.; Kober, M.; Freij-Larsson, C.; Ljungh, Å.; Paulsson, M. Protein adsorption of poly(ether methane) surfaces modified by amphiphilic and hydrophilic polymers. Biomaterials 1994, 15, 278–284. [Google Scholar] [CrossRef]
- Maltesh, C.; Xu, Q.; Somasundaran, P.; Benton, W.J.; Hung, N. Aggregation behavior of and surface tension reduction by comblike amphiphilic polymers. Langmuir 1992, 8, 1511–1513. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.H.; Kim, J.-C. Oxidation–Responsive Emulsions Stabilized with Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide). Polymers 2020, 12, 498. https://doi.org/10.3390/polym12020498
Park SH, Kim J-C. Oxidation–Responsive Emulsions Stabilized with Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide). Polymers. 2020; 12(2):498. https://doi.org/10.3390/polym12020498
Chicago/Turabian StylePark, Seok Ho, and Jin-Chul Kim. 2020. "Oxidation–Responsive Emulsions Stabilized with Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide)" Polymers 12, no. 2: 498. https://doi.org/10.3390/polym12020498
APA StylePark, S. H., & Kim, J. -C. (2020). Oxidation–Responsive Emulsions Stabilized with Poly(Vinyl Pyrrolidone-co-allyl Phenyl Sulfide). Polymers, 12(2), 498. https://doi.org/10.3390/polym12020498