1. Introduction
CO
2 has promising prospects as a responsive trigger due to its vital advantages of low cost, environmental friendliness, and wide availability. Amidines and amines are CO
2–responsive molecules [
1,
2,
3,
4] that can react with dissolved CO
2 in water and generate charged amidinium bicarbonate or ammonium salts. These reactions can be reversed by Ar or N
2 bubbling. Because this regulated process involves only CO
2 and other inert gases if no other impurity is introduced [
5], it may have various applications in a wide range of important industries. Many different responsive approaches in which CO
2 is used as a trigger in the form of surfactants [
5], solvents [
6], organic salts [
7], and polymer emulsions [
8] have been reported to achieve this goal.
Aqueous solutions using polymeric surfactants with adjustable surface activities are also attractive. Small–molecule surfactants appeared first and Jessop’s research group successfully prepared a series of intelligent CO
2 switchable surfactants by using CO
2 as a triggering substance to realize CO
2–responsive surface activity [
5,
9]. Our group applied tertiary amines with an ultralong hydrophobic chain and discovered another method wherein CO
2 could be utilized to adjust the surface activities of aqueous solutions [
10,
11,
12]. One of the key advantages of polymeric surfactants over traditional surfactants is the high level of tailoring that can be done with the former to afford optimal stabilization properties. Lu et al. developed CO
2–switchable polymeric surfactants synthesized with 2–(dimethylamino)ethyl methacrylate and the hydrophilic monomer acrylamide [
13]. The polymeric surfactants displayed good performance for emulsifying heavy oil emulsions. Another merit of polymeric surfactants is that their chemical structure, composition, molecular weight, and topology can be easily modified [
14]. We previously reported CO
2–switchable multicompartment micelles with a segregated corona that self–assembles from a linear ABC triblock copolymer. This triblock copolymer aggregates into uniform spherical micelles in the absence of CO
2 and can be switched into micelles with a segregated corona in the presence of CO
2 [
15].
Adjustments in surface activity are based on the molecular structure of surfactants, and topological modification is a powerful approach to construct functional polymers with molecular–level precision [
16]. However, the molecular topology of CO
2–switchable polymeric surfactants has been insufficiently researched, and investigations on block copolymers mainly focus on their molecular weight and dispersity. Developing an alternative method to modify the molecular topologies of CO
2–switchable polymeric surfactants is a challenging topic in polymer chemistry. Polymeric surfactants can be categorized as diblock or triblock copolymers according to the number of their hydrophobic and hydrophilic segments. In nature, cyclic lipids are found in the cell membranes of organisms living in high–temperature environments; some examples of these organisms include thermophilic bacteria in springs and submarine volcanos [
16,
17,
18]. Inspired by this phenomenon, researchers have synthesized an amphiphilic polymer with a cyclic structure that could provide a new approach for obtaining CO
2–switchable polymeric surfactants. However, because the topology of CO
2–switchable copolymeric surfactants is rarely reported, choices for fabricating CO
2–sensitive copolymers are limited.
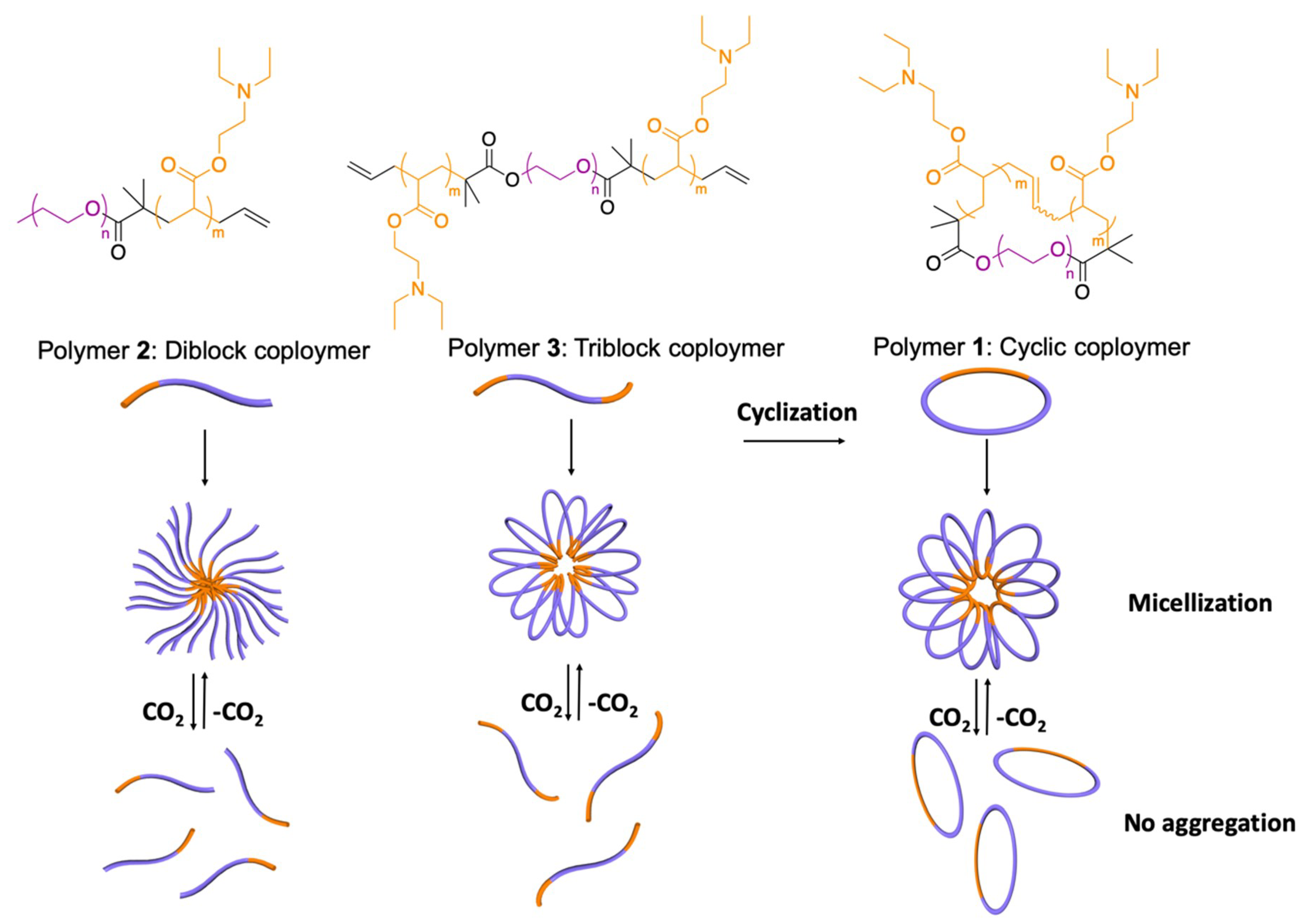
In this study, we present an example of a CO2–switchable polymeric surfactant showing significant topological effects and including diblock, triblock, and cyclic amphiphilic copolymers. The influences of chemical structure and segment composition on the topological effect and tendency of the CO2 response are investigated by evaluating the properties of the self–assembled micelles. Specifically, poly(ethylene oxide) and 2–(diethylamino)ethyl methacrylate (DEAEMA), acting as the hydrophilic and CO2–switchable hydrophobic sections, respectively, are used to synthesize cyclic poly(DEAEMA)–b–poly(ethylene oxide) (copolymer 1), diblock linear polymer poly(DEAEMA)–b–poly(ethylene oxide) (copolymer 2), and triblock linear polymer poly(DEAEMA)–b–poly(ethylene oxide)–b–poly(DEAEMA) (copolymer 3). The relevant properties (i.e., surface activity) of micelles of the three different surfactants are then analyzed and compared.
2. Materials and Methods
2.1. Materials
DEAEMA, 2–bromoisobutyryl bromide, poly(ethylene oxide) (M
n = 2000), CuBr, 4,4′–dinonyl–2,2′–dipyridyl (dNbpy), allyltributyl stannane, and first–generation Grubbs catalyst were purchased from Sigma–Aldrich (St. Louis, MO, USA) and used as received. The poly(ethylene oxide) macroinitiator with 2–bromoisobutyryl terminal groups was prepared through esterification of poly(ethylene oxide) (Mn = 2000) or poly(ethylene glycol) methyl ether (M
n = 2000) with 2–bromoisobutyryl bromide according to a previously reported procedure [
16].
2.2. Synthesis of Amphiphilic Linear Poly(DEAEMA)–b–Poly(Ethylene Oxide)–b–Poly(DEAEMA) (Copolymer 3)
The triblock copolymer was prepared via atom transfer radical polymerization (ATRP) and Keck allylation in accordance with a previously reported procedure [
16]. The poly(ethylene oxide) macroinitiator (400 mg, 0.15 mmol), DEAEMA (420 mg, 23 mmol), dNbpy (30 mg, 73 μmol), and CuBr (10 mg, 70 μmol) were placed in a test tube. The mixture was degassed through freeze–pump–thaw cycling, and the test tube was sealed under vacuum. The resulting suspension was stirred at 110 °C for 1 h, cooled with liquid N
2, and allowed to warm to 20 °C. Allyltributyl stannane (150 mg, 0.45 mmol) was added to the reaction mixture, and the resulting suspension was degassed through freeze–pump–thaw cycling. The test tube was sealed under vacuum, and the suspension was stirred at 110 °C for another 12 h. The reaction mixture was diluted with acetone and filtered through a plug of alumina. The eluent was concentrated and reprecipitated in
n–hexane to allow the isolation of the product as a colorless waxy solid (448 mg) in 71% yield.
2.3. Synthesis of Amphiphilic Cyclic Poly(DEAEMA)–b–Poly(Ethylene Oxide) (Copolymer 1)
First–generation Grubbs catalyst (30 mg, 36 μmol) was added to a CH2Cl2 solution of 3 (0.12 mM, 100 mL), and the resulting solution was refluxed for 48 h. The reaction mixture was concentrated and filtered through a plug of alumina. The eluent was concentrated and reprecipitated in cold n-–hexane to allow the isolation of polymer 1 as a colorless waxy solid (29 mg) in 56% yield.
2.4. Synthesis of Amphiphilic Linear Poly(DEAEMA)–b–Poly(Ethylene Oxide) (Copolymer 2)
Diblock copolymer 2 was prepared via the same procedures applied to obtain polymer 3 except that a different macroinitiator was used. The poly(ethylene oxide) macroinitiator made from poly(ethylene glycol) methyl ether (Mn = 2000) (400 mg, 0.15 mmol), DEAEMA (420 mg, 23 mmol), dNbpy (30 mg, 73 μmol), and CuBr (10 mg, 70 μmol) were placed in a test tube. The mixture was degassed through freeze–pump–thaw cycling, and the test tube was sealed under vacuum. The resulting suspension was stirred at 110 °C for 1 h, cooled with liquid N2, and allowed to warm to 20 °C. Allyltributyl stannane (150 mg, 0.45 mmol) was added to the reaction mixture, and the resulting suspension was degassed through freeze–pump–thaw cycling. The test tube was sealed under vacuum, and the suspension was stirred at 110 °C for another 12 h. The reaction mixture was diluted with acetone and filtered through a plug of alumina. The eluent was concentrated and reprecipitated in n–hexane to allow the isolation of the product as a colorless waxy solid (524 mg) in 78% yield.
To test the transformation of amine group in the presence of CO2, the protonated copolymer 2 was prepared: copolymer 2 was dissolved in chloroform, then tiny amount of water was added into the solution with the sparging of CO2. After that, precipitation appeared, which was the protonated copolymer 2.
2.5. Characterization
Nuclear magnetic resonance (NMR) spectroscopy: 1H NMR spectra were obtained on a Bruker Avance–II 400 MHz NMR spectrometer (Billerica, MA, USA).
Gel permeation chromatography (GPC): GPC measurements (Shodex 3000 GPC system with an HPLC pump, Santa Clara, CA, USA) were conducted at 40 °C to measure the molecular weight of samples. The GPC system was equipped with a Waters separation module, including four polystyrene columns with pore sizes of 100, 500, 103, and 104 Å, and a differential refraction detector. Tetrahydrofuran was used as the eluent with a flowrate of 0.3 mL/min. Molecular weights were determined by using polystyrene standards in the range of 870,000−875,000 g/mol.
Surface tension: Surface tension at different concentrations was measured and recorded using a Krüss K100 full–automatic surface tension analyzer (Hamburg, Germany). When the standard deviation of five successive measuring points was less than 0.05 mN/m, the tension was defined as the equilibrium surface tension of the measured solution.
Hydrodynamic size: The hydrodynamic sizes of the copolymers were determined with a Malvern Zetasizer Nano ZS instrument (Malvern, UK). Impurities in the tested samples were removed with a 0.8 μm probe filter, the experimental temperature was set to 25 °C, and each sample was tested at least three times.
Conductivity: Conductivity was measured using a DDS–11A conductometer with a DJS–1C platinum electrode (Shanghai, China).
TEM: Transmission electron microscopy (TEM) visualization was conducted on a Hitachi H600 electron microscope (Tokyo, Japan).
Transmittance: A UV–Vis spectrophotometer (Mapada UV–6100, Shanghai, China) equipped with dual channels for detecting the reference deionized water and sample solutions was utilized for all measurements at 25 °C. Each sample solution was measured in triplicate to obtain average values.
Viscosity: Viscosity was measured on an MCR 302 (Anton Paar, Graz, Austria) rotational rheometer with a Searle–type concentric cylinder geometry (CC27), and the shear rate was set to 10/s. Prior to the measurements, sample solutions were equilibrated at 25 °C for 10 min with a Peltier temperature control device.
3. Results and Discussion
Linear triblock amphiphilic polymers containing different blocks, as well as the corresponding cyclic copolymer
1 (
Scheme 1), were synthesized and characterized according to previously reported methods [
16,
19]. Copolymer 2 and copolymer 3 were prepared through ATRP [
20] and Keck allylation [
21]. Using copolymer 3 as a precursor, copolymer 1 was synthesized via cyclization triggered by metathesis polymerization with a second–generation Hoveyda–Grubbs catalyst [
22] in DMSO.
1H NMR (
Figures S1–S4) confirmed that propylene units were converted into internal olefin units through metathesis polymer cyclization. GPC revealed a decrease in hydrodynamic volume (
Figure 1). The molecular weight of polymer 1 was lower than that of its precursor polymer 3, as shown in
Table 1, thus indicating that the former presentrf a cyclic structure. After treatment of water and CO
2, it was found that copolymer 2 had bicarbonate, proving that the polymer can be protonated by CO
2 in the presence of water.
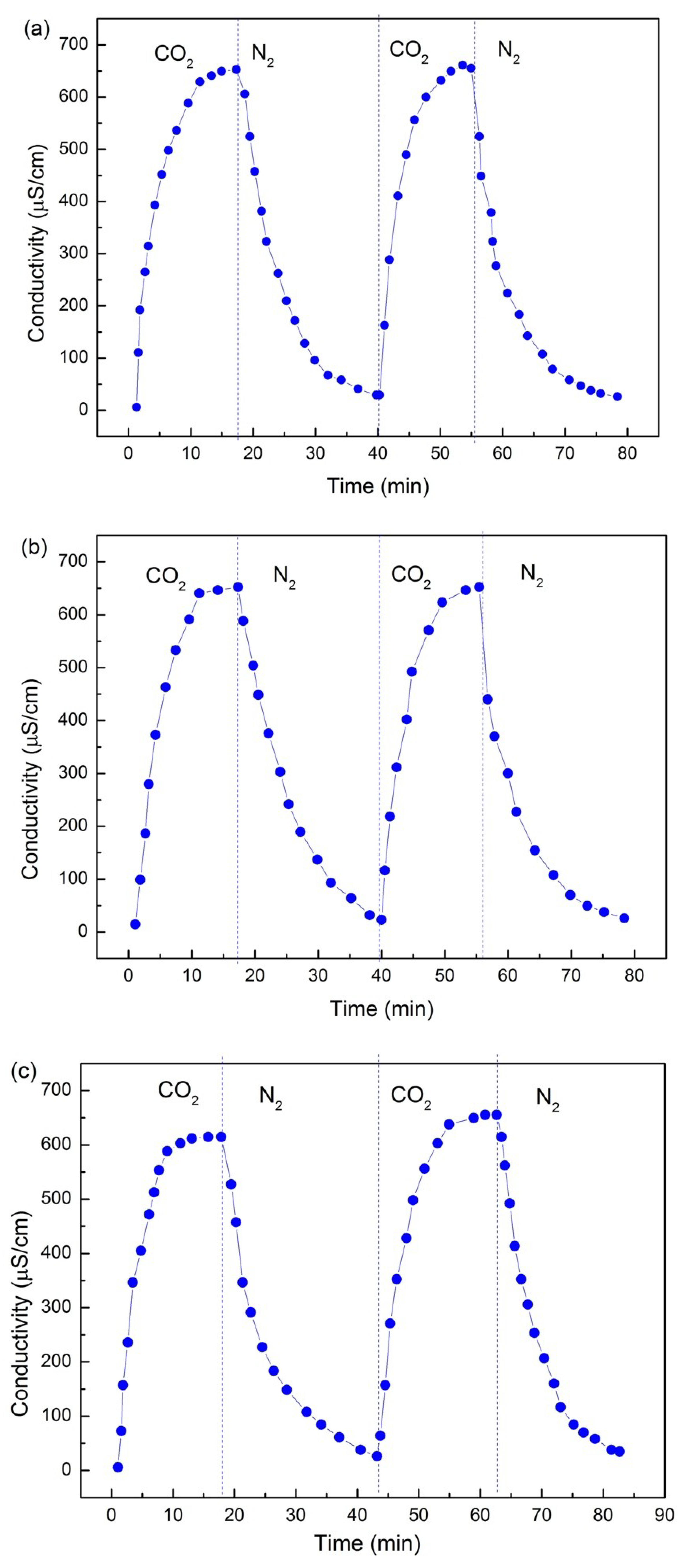
3.1. CO2 Switchability
Given that copolymers 1, 2, and 3 had a tertiary amine as their switchable hydrophobic segment and PEO as their permanent hydrophilic component, the three block polymers presented switchable behaviors that could be controlled by CO
2. Determining changes in conductivity ratio is the most classic and effective method to verify the reversibility and repeatability of CO
2/N
2 switching (
Figure 2). When polymers remained in neutral form in the absence of CO
2 in an aqueous solution, the conductivity ratio of the solution was nearly zero. By contrast, when polymers reacted with CO
2 and form cations in water, a large number of bicarbonate ions were distributed in the solution and the conductivity ratio increased drastically.
The experiments showed that the initial form of the polymer in the solution was a neutral nonionic compound; thus, its conductivity ratio is consistent with that of pure water. However, the conductivity of the solution in its initial state was slightly higher than zero, likely because of the presence of tertiary amines in the copolymers. Continuous insertion of CO
2 led to the reaction of the polymers with CO
2 and subsequent formation of quaternary ammonium bicarbonates, thus resulting in a rapid increase in conductivity [
4]. When conversion was completed, the increase rate of the conductivity ratio gradually decreased and stabilized. As N
2 bubbling continued, the bicarbonate salt gradually decomposed into a neutral nonionic compound, resulting in a gradual loss of conductivity. The conductivity of the solution eventually decreased to the initial value and gradually stabilized. The final conductivity measured was slightly higher than the initial value because of the incomplete reversible conversion of oxycarbonate. The conductivity experiments confirm the CO
2/N
2 responsiveness of the polymers and the repeatability of their switching behavior.
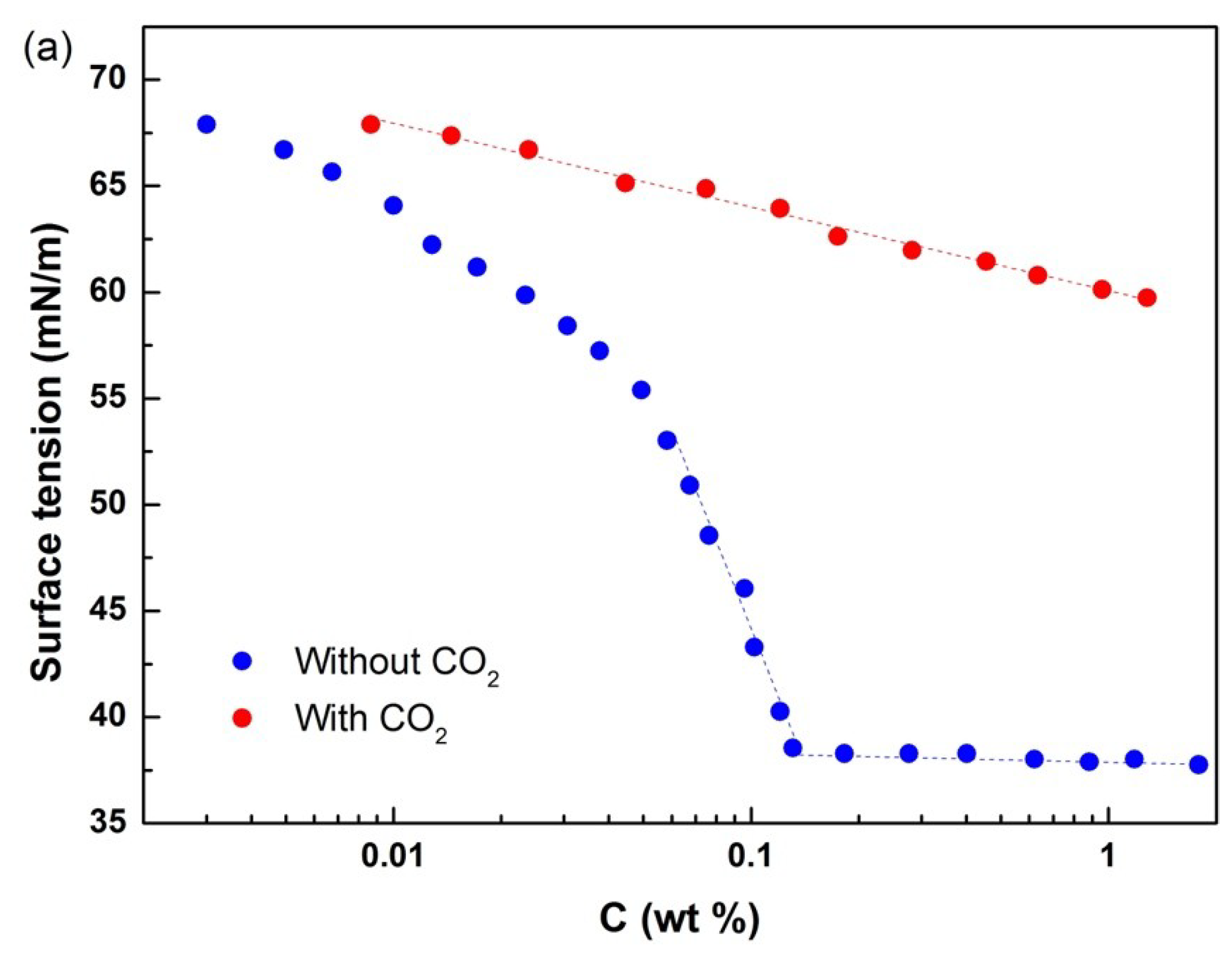
3.2. Surface Tension
The conductivity test showed that the poly–DEAEMA segment of block polymers in the aqueous solution could be reversibly modified by using CO
2 and that copolymers 1, 2, and 3 could exhibit reversible surface activity under the control of CO
2.
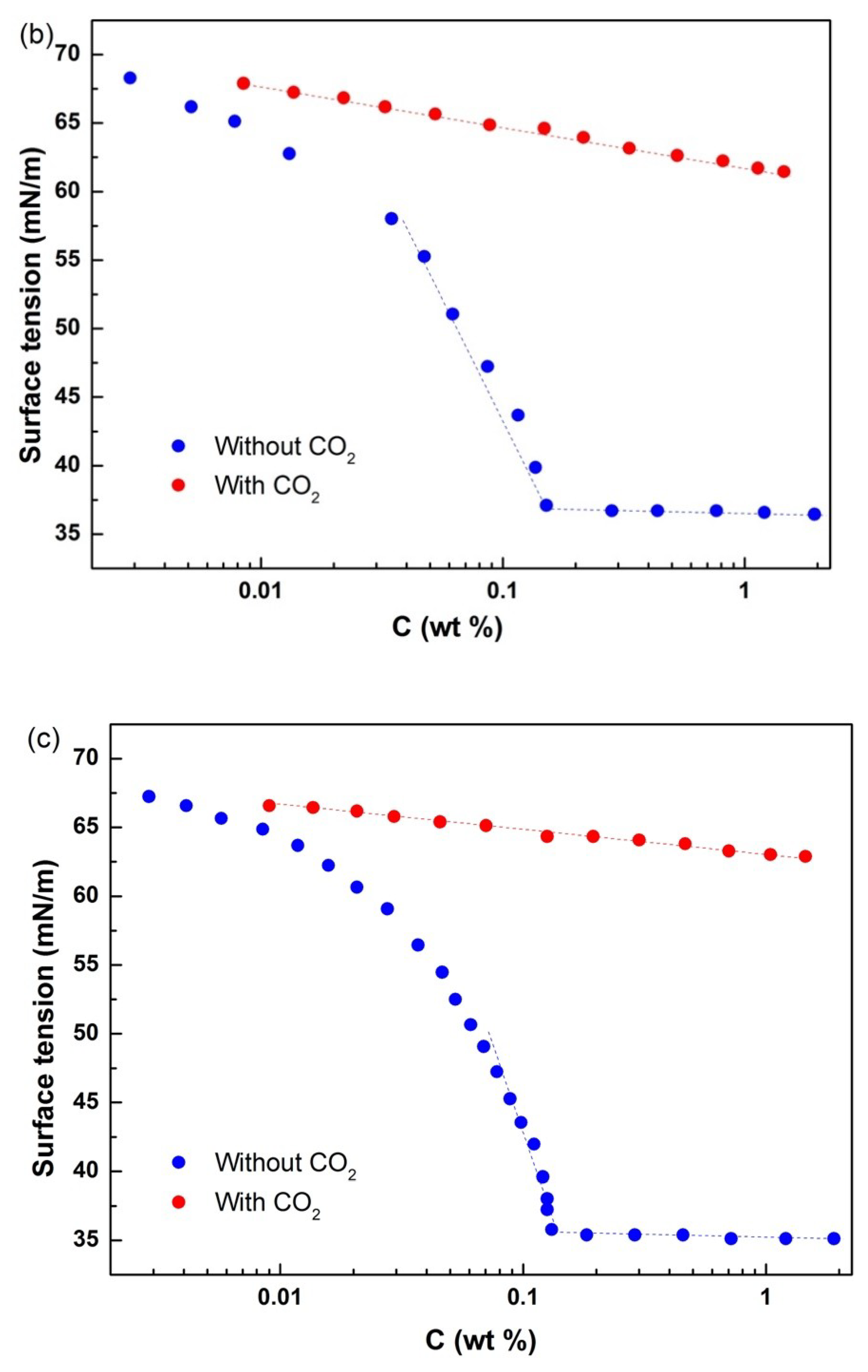
Figure 3 presents the surface tension and critical micellar concentration (CMC) of the copolymers. When the three copolymers were dispersed in aqueous solution without CO
2, they behaved as normal surfactants. After CO
2 bubbling, the whole polymers became hydrophilic due to protonation of tertiary amines in their hydrophobic segments. Therefore, the polymers lacked a CMC in the presence of CO
2. When CO
2 was expelled from the solution by using N
2, the original properties of the polymers were restored, the micelles were regenerated, and surface activity returned to its original level with CMC. As the charged copolymers were converted into a neutral form, the ionic segment of DEAEMA became neutral. Therefore, the aqueous solution containing polymer 1 exhibited switchable surface tension behavior under the influence of CO
2. The surface tension of the cyclic diblock copolymer was compared with those of the linear diblock and triblock copolymers, as shown in
Table 1. The results showed that the three copolymers have similar surface tension data, thereby indicating that they had similar micelle–formation and assembly behaviors on water/air surfaces.
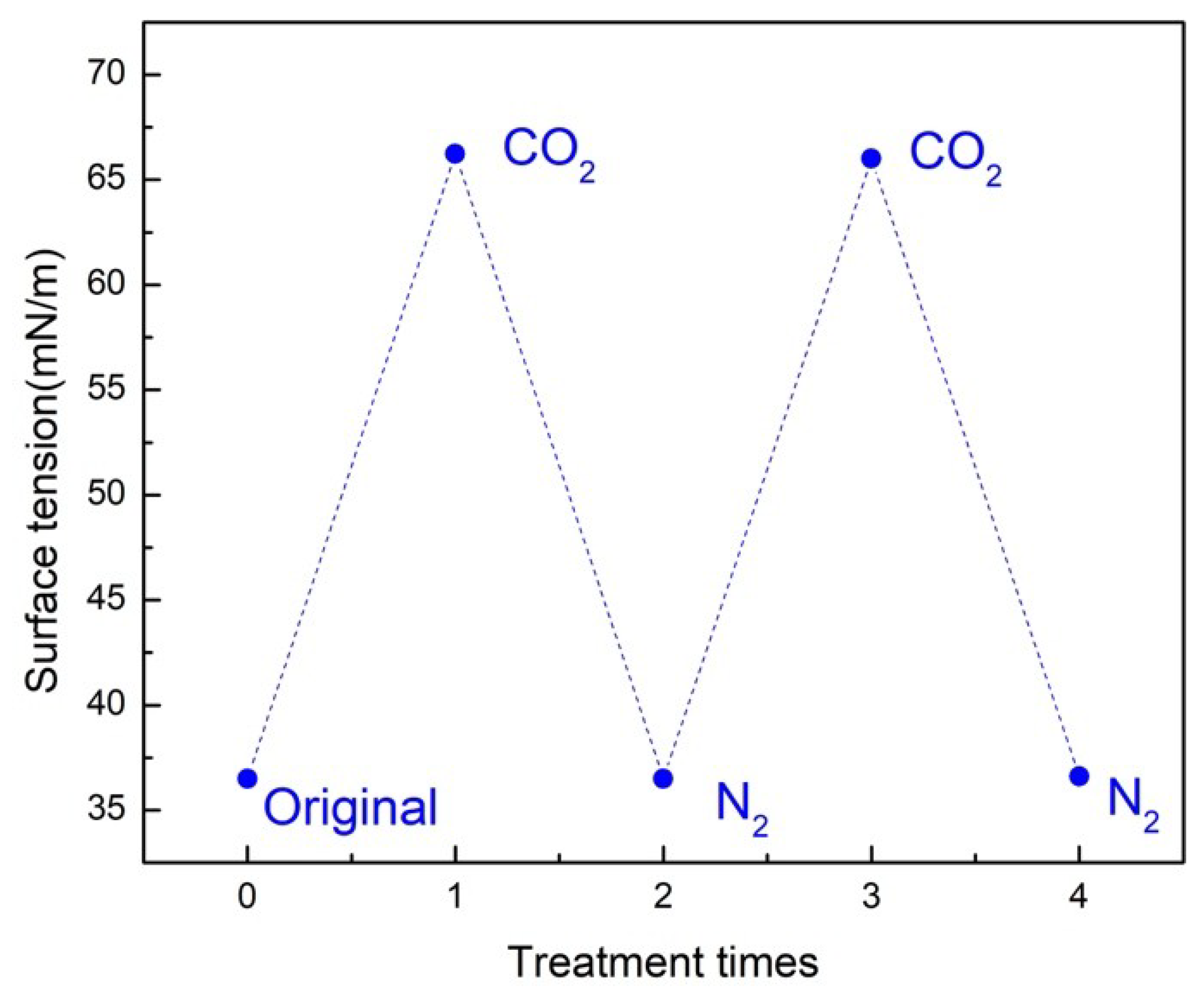
CO
2 can be a good trigger to change the surface tension of aqueous solutions with the three copolymers. In addition, the response to surface tension is reversible. As an example, the surface tension of an aqueous solution of polymer 1 during two cycles of alternating CO
2/N
2 bubbling was measured at 25 ± 0.5 °C (
Figure 4). N
2 bubbling was required to remove CO
2 from the aqueous solution and subsequently neutralize carbamate.
Figure 3 displays fluctuations in surface tension and pH of the aqueous solution containing copolymer 1 (0.2 wt%) during two cycles of alternating CO
2/N
2 bubbling. Surface tension decreased from 34.9 mN/m to 31.0 mN/m after CO
2 bubbling for 20 min. pH fluctuated from 7.2 to 5.6 during alternating CO
2/N
2 bubbling. The initial value was reached after bubbling N
2 into the aqueous solution.
Changes in effective diameter confirm the switchability of copolymer 1 under a CO
2/N
2 atmosphere. The concentration of copolymer 1 was determined to be 0.2 wt%. The results indicated that the diameters of the surfactants are affected by the atmosphere. Prior to CO
2 sparging, polymer molecules tended to aggregate, forming micellar structures with large hydrodynamic sizes. During bubbling with CO
2, the micellar structure was diminished because of the effect of electrostatic repulsion, resulting in small hydrodynamic sizes (
Figure 5, right plot). The observed results prove that the obtained copolymer 1, a cyclic amphiphilic copolymer, could be reversibly converted into micelles via self–assembly in water. In the presence of CO
2, copolymers 1, 2 and 3 did not form micelles; In the absence of CO
2, the particle size (546 nm) of 1 was the largest among the three polymers. The possible reason is that cyclic structure of copolymer 1 promotes the formation of micelles and the increases the aggregate number of micelles.
3.3. Switchable Emulsion
Emulsification is an effective approach to reduce the viscosity of crude oil. If CO2 can trigger the reversible conversion of crude oil emulsions, the problems encountered in the process of crude oil transport through pipelines may be addressed. Specifically, crude oil emulsions with low viscosity can be prepared and water can be separated from crude oil after removing CO2 from or adding CO2 to crude oil emulsions after transportation.
Investigation of the CO
2 responsiveness of the three copolymers indicated that the modification approach is also feasible for emulsions containing the copolymers. A crude oil–in–water emulsion (weight ratio, 0.8:1) was prepared using the copolymers (0.4 wt%) to confirm the effect of the copolymers on emulsion stability. Under this condition, the neutral copolymers 1, 2, and 3 behaved as impressive emulsifiers (
Figure 6). Given the diffused electric double layer surrounding the milky droplets, an inner layer consisting of polymer 1 adsorbed on the surfaces of the droplets and the outer layer involved Na ions, which are attracted by surface charges. The neutral polymer 1 had no adverse effect on the electric double layer in the absence of CO
2.
Figure 4 shows that crude oil stably existed in the aqueous emulsion for more than 36 h after 20 min of CO
2 bubbling. However, in the presence of CO
2, tertiary amines in polymer 1 reacted with CO
2 and generated polymeric bicarbonate salts. Therefore, polymer 1 lost its surfactant capability. Without the assistance of a surfactant, the stability of the emulsion drastically decreased as the emulsion droplets coalesced within 20 min after introducing CO
2 to polymer 1 (
Figure 4). The two phases became completely apparent after another 20 min. However, when N
2 was sprayed onto a mixture containing crude oil/water phases to remove CO
2, the tertiary amine of polymer 1 was converted back into its neutral form. Then, the mixture was stirred to prepare a stable emulsion. Alternating CO
2/N
2 bubbling could be used to convert emulsions into two phases. This conversion process triggers the behavior of the emulsion phase in the reservoir via the injection of CO
2. Therefore, the emulsification of transferable crude oil by adding and removing CO
2 to decrease or improve the stability of the emulsion of polymer 1 is possible. The results showed no differences among the performances of the three copolymers for reversibly emulsifying crude oil under the control of CO
2.
3.4. Switchable Viscosity
The hydrophobic component of copolymers 1, 2, and 3 could be reversibly switched so that the materials become hydrophilic by CO
2 addition, and using CO
2 to control viscosity would be possible [
23].
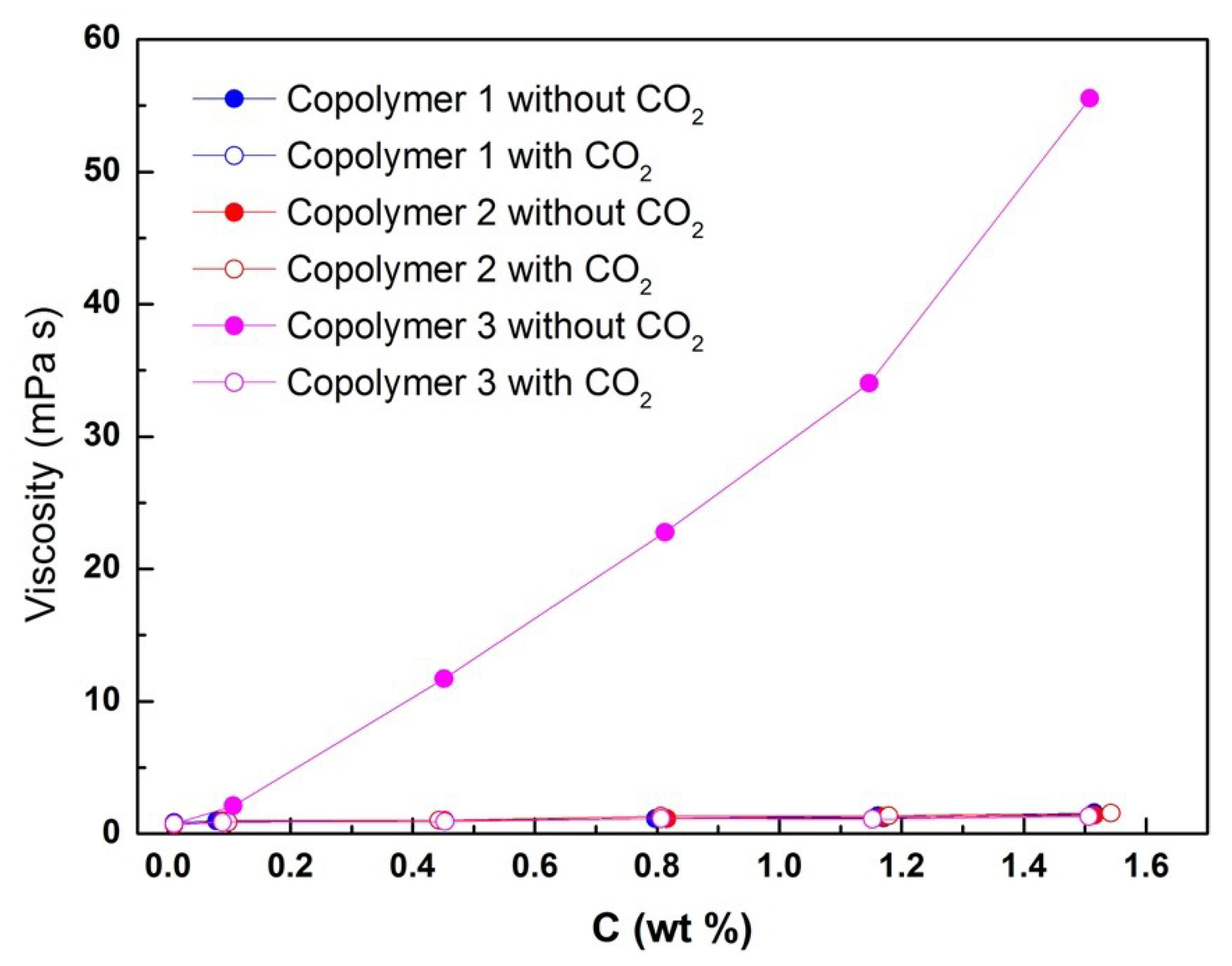
Figure 7 shows the viscosities of aqueous solutions containing copolymers 1, 2, and 3 at different concentrations in the absence or presence of CO
2. The results showed that copolymer 3 had the highest viscosity among the samples in the absence of CO
2. The viscosity of copolymer 3 likely increased because this copolymer, as a triblock polymer, can achieve cross–micellar connections through its two hydrophobic segments, thus forming a network structure at the microscopic level. The viscosity of copolymer 3 was CO
2–responsive and diminished by addition of CO
2. The viscosity of copolymer 3 is only slightly higher than that of pure water at 25 °C. Viscosity decreased because the conversion of the hydrophobic chain segments into hydrophilic cationic chains with bicarbonate counterions results in the breakup of hydrophobic associations. Viscosity measurements were subsequently carried out after bubbling N
2 through the solution to remove CO
2 and confirm that the switch was reversible. After 50 min, the viscosity recovered to its initial value.
3.5. Thermal Stability
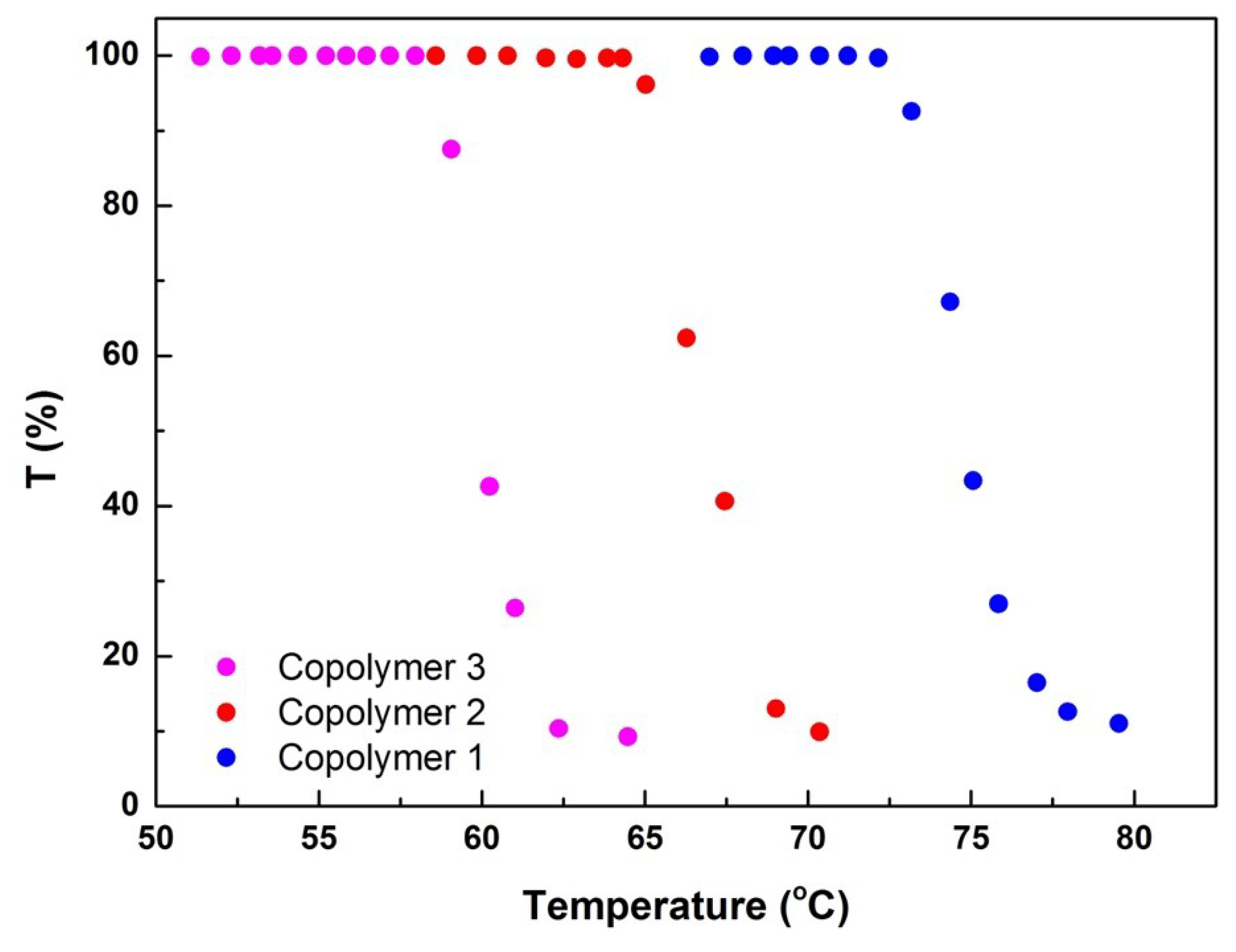
Given that all three copolymers contained PEO strands, they were expected to have cloud points, which means their aqueous solutions should have become cloudy when the temperature was increased to a certain extent. Aqueous solutions (1.0 wt%) of the copolymers were tested for light transmission at different temperatures to study the effects of topological structures on the thermal stability of the materials.
Figure 8 reveals that the cloud point of copolymer 3 was 60 °C and that of copolymer 2 was 66 °C. Among the samples, cyclic copolymer 1 had the highest cloud point of 75 °C. The ring–shaped and linear assemblies exhibited significantly different thermal stabilities. The lower thermal stability of the linear triblock composition is attributed to intermicellar bridging caused by the combination of a suspended polymer chain and dehydration, which results in reunion at a low temperature [
16]. Light transmission was not measured under CO
2 because CO
2 does not stably exist in aqueous solutions at temperatures above 60 °C [
6,
7].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}