Review on UV-Induced Cationic Frontal Polymerization of Epoxy Monomers





Abstract
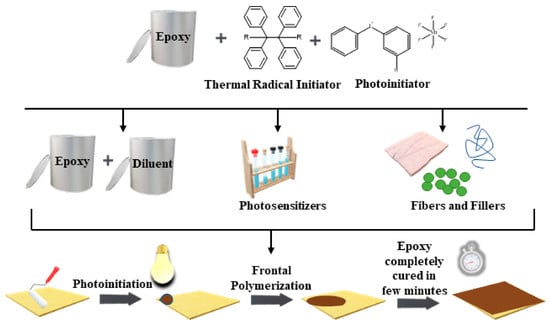
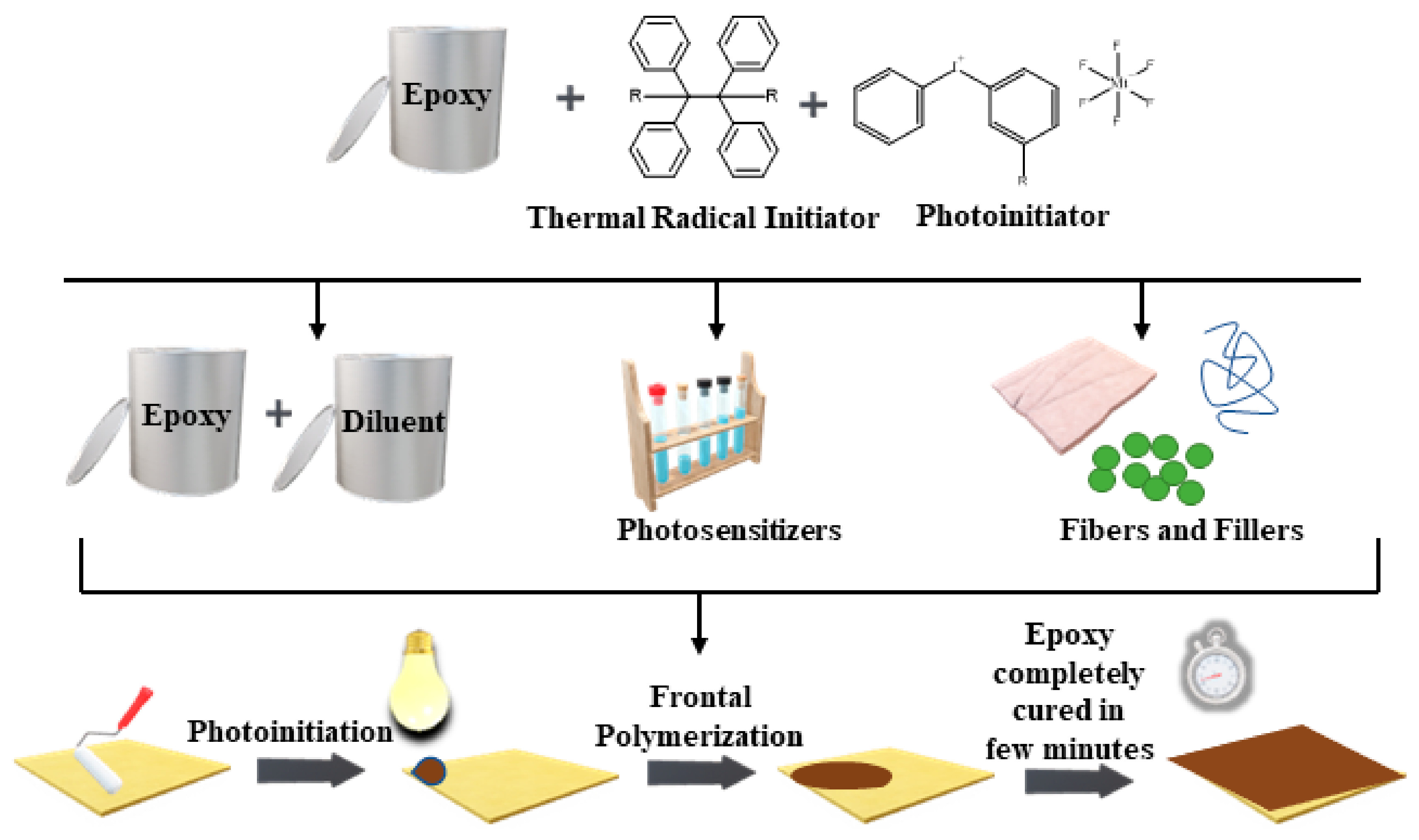
:
1. Introduction
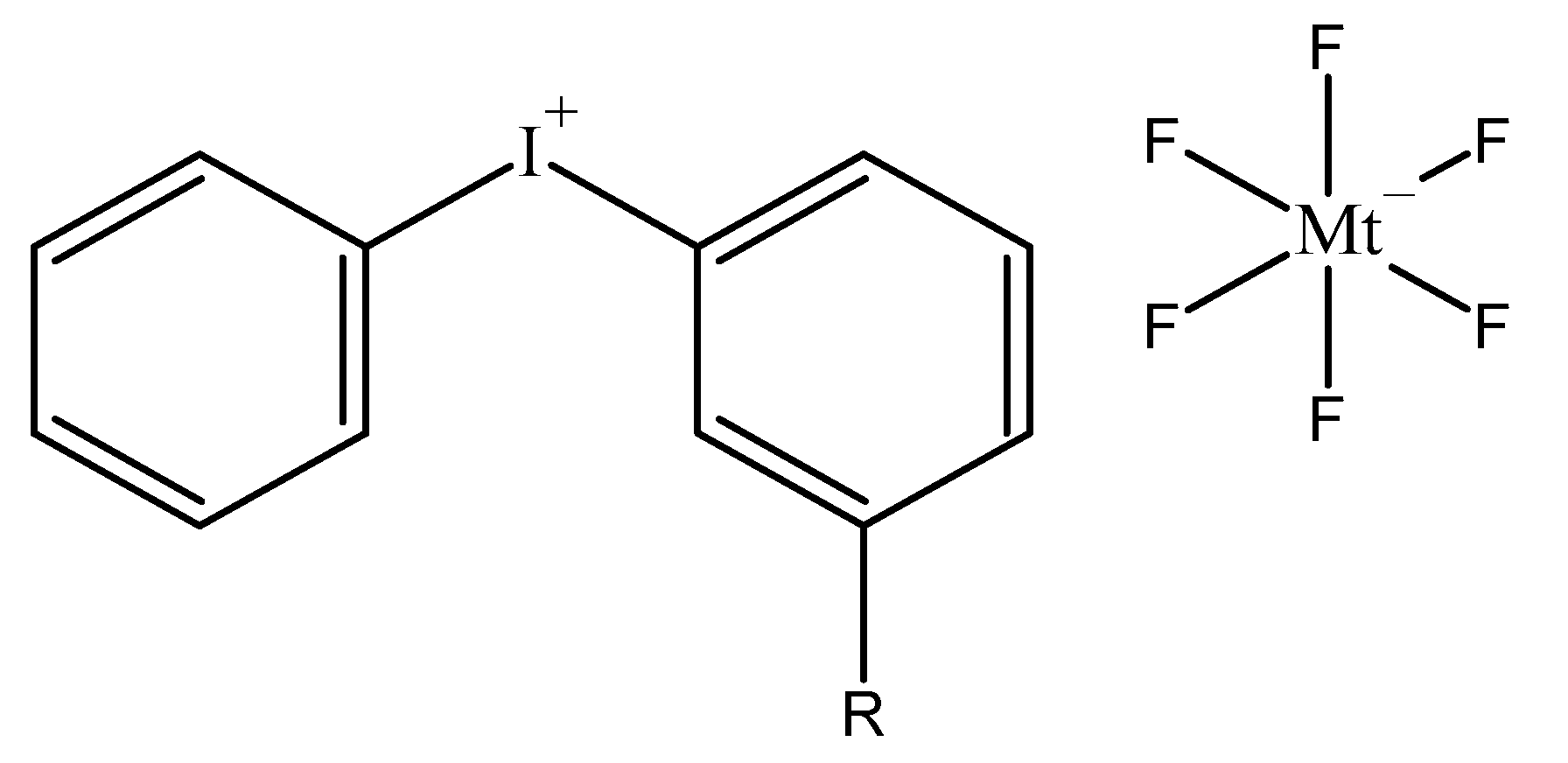
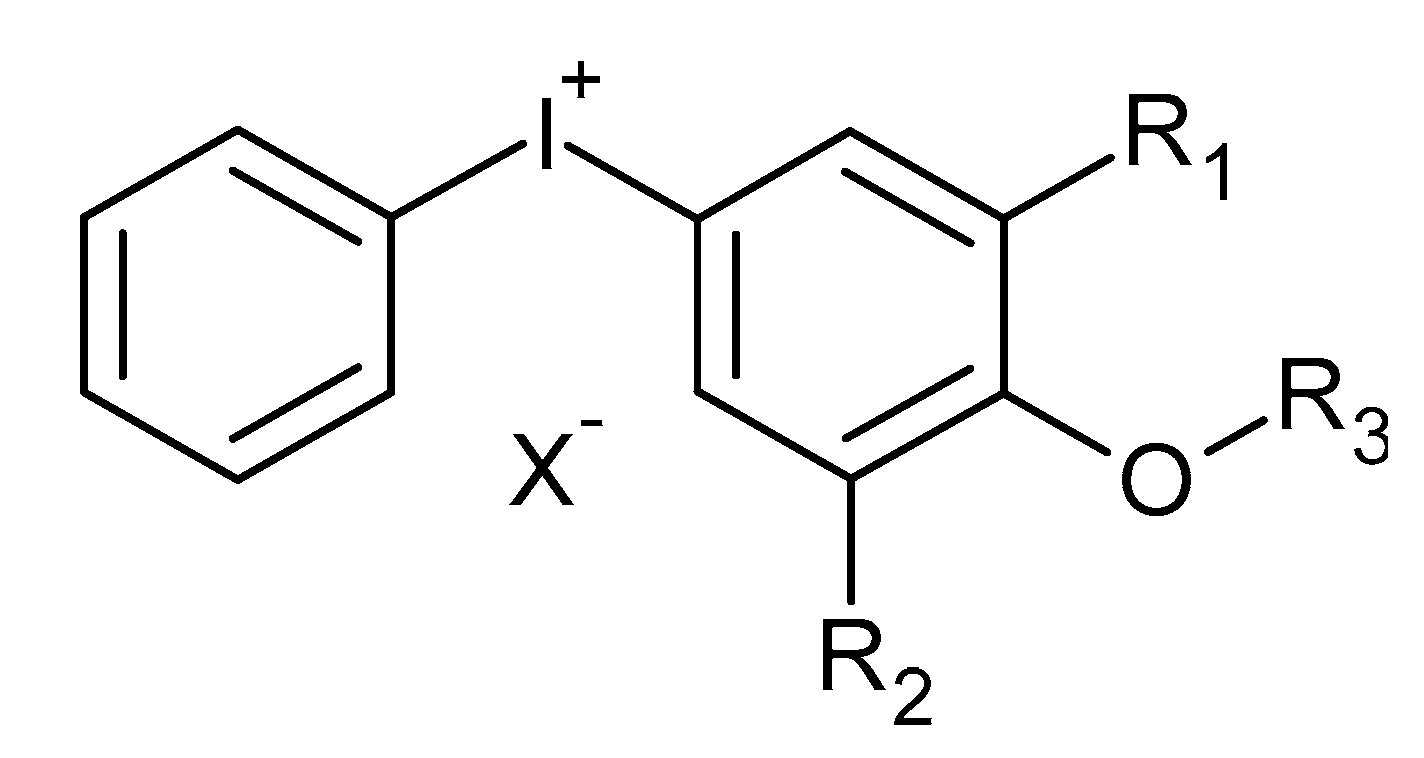
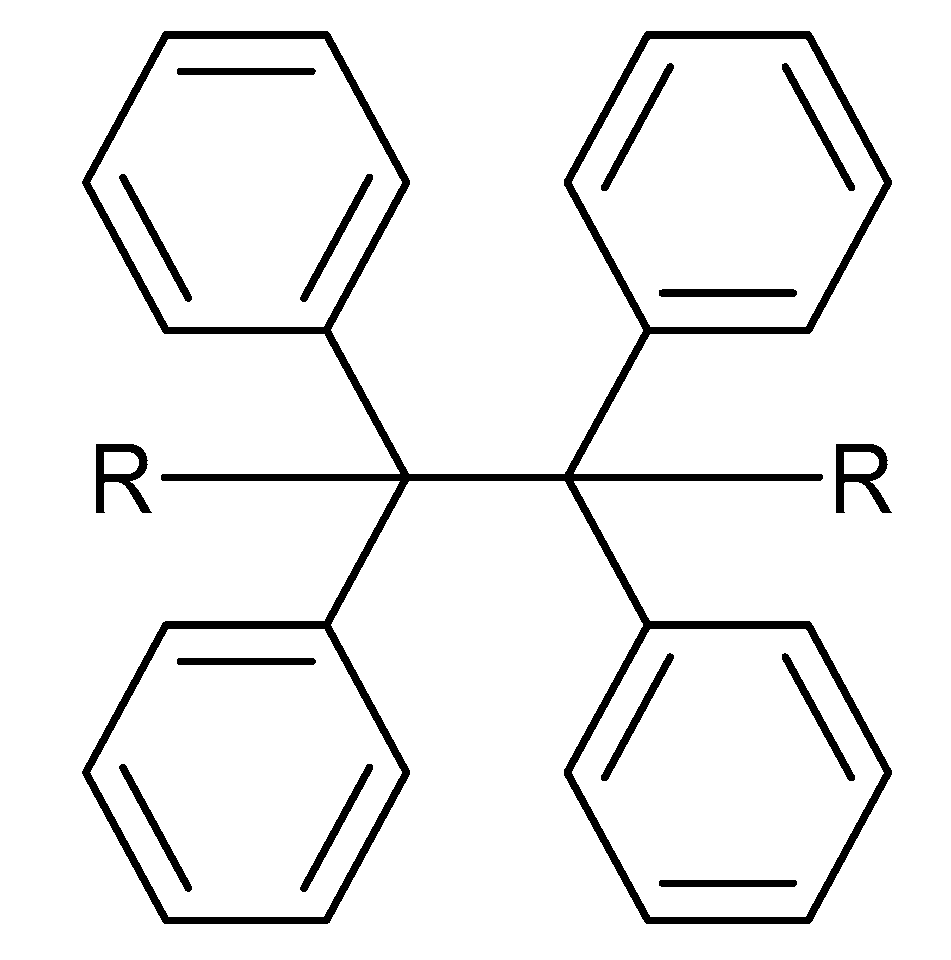
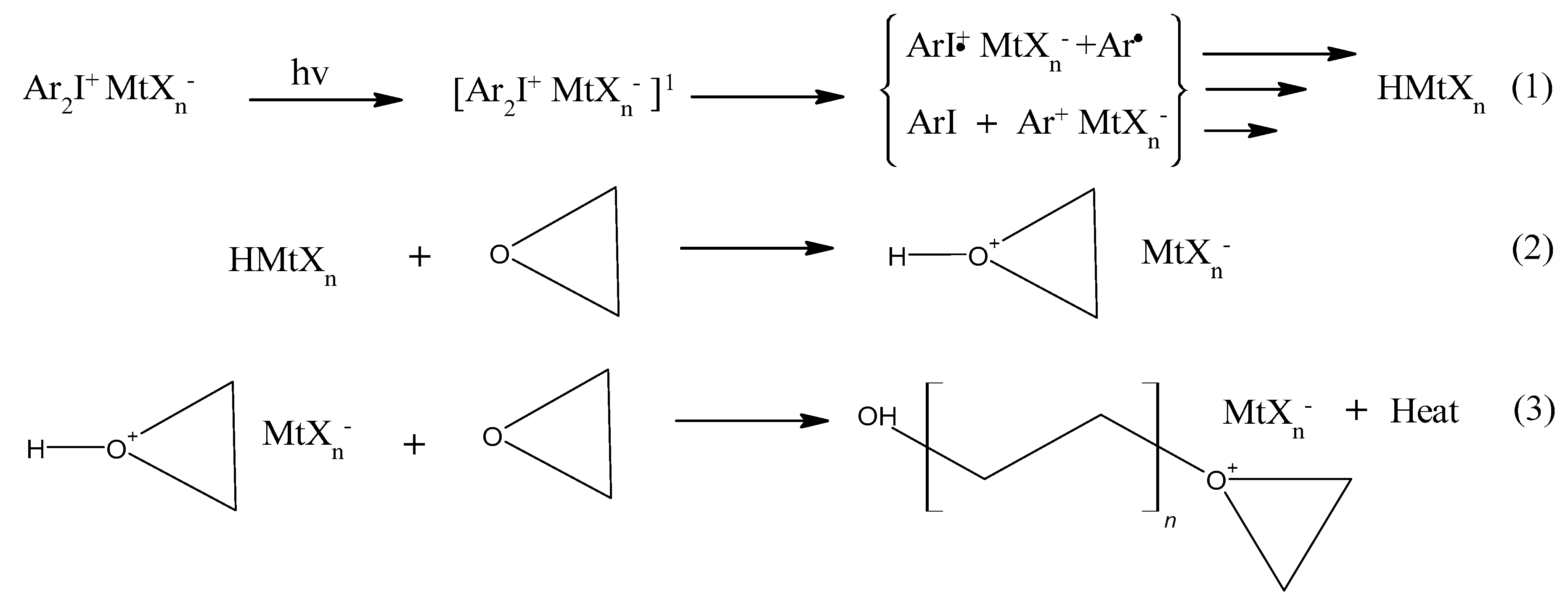
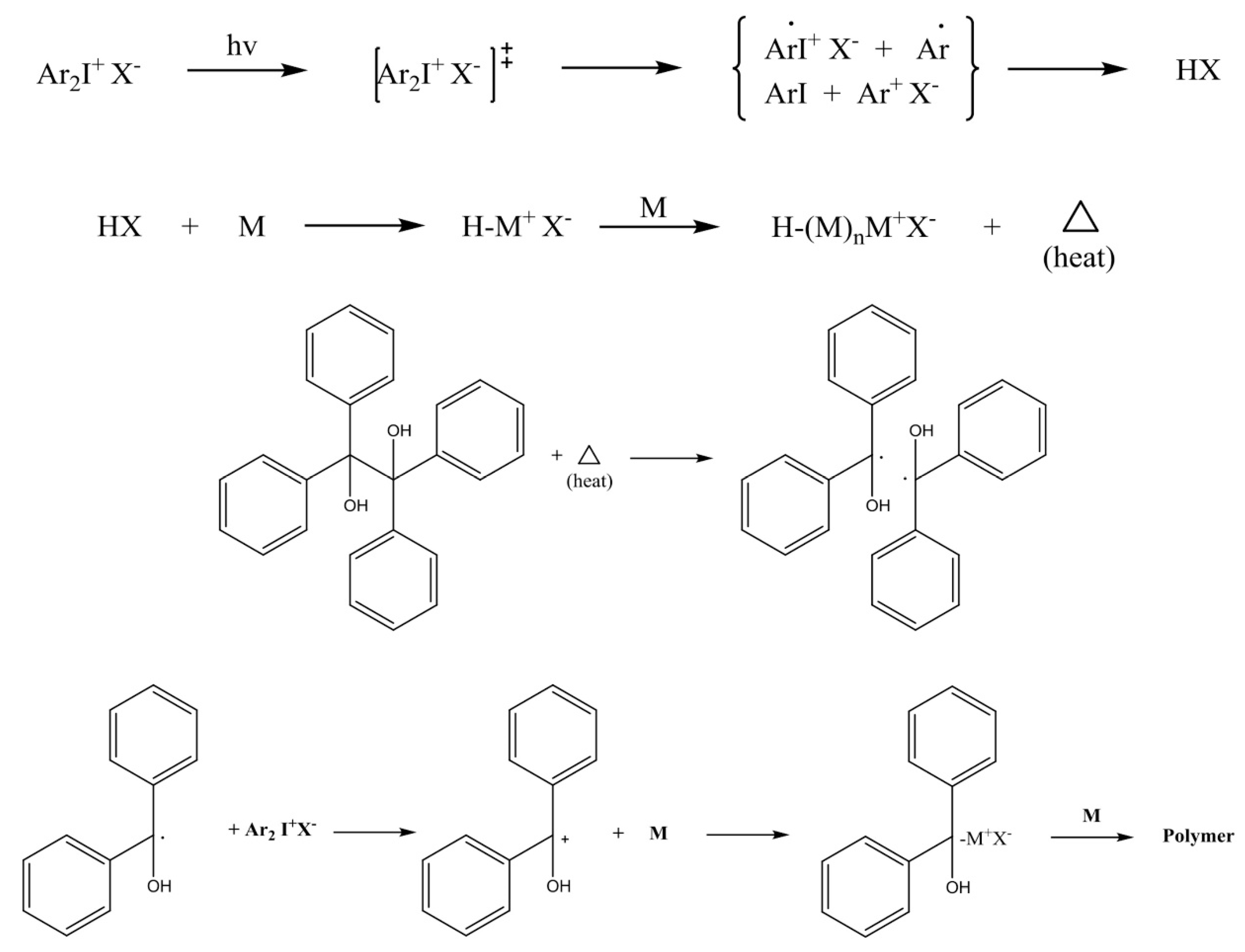
2. Chemistry of Cationic Photoinitiators Based on Diaryliodonium Salts
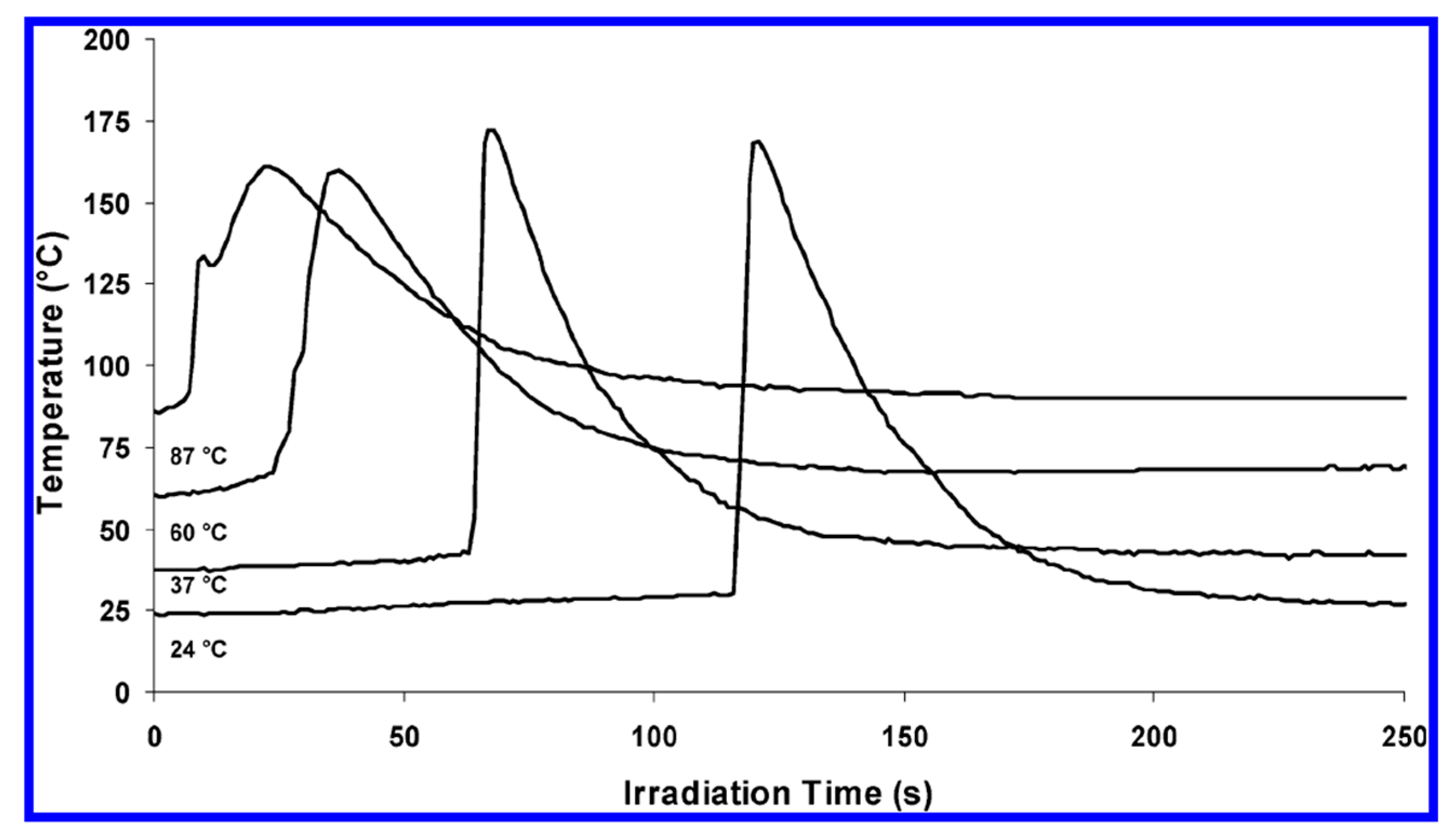
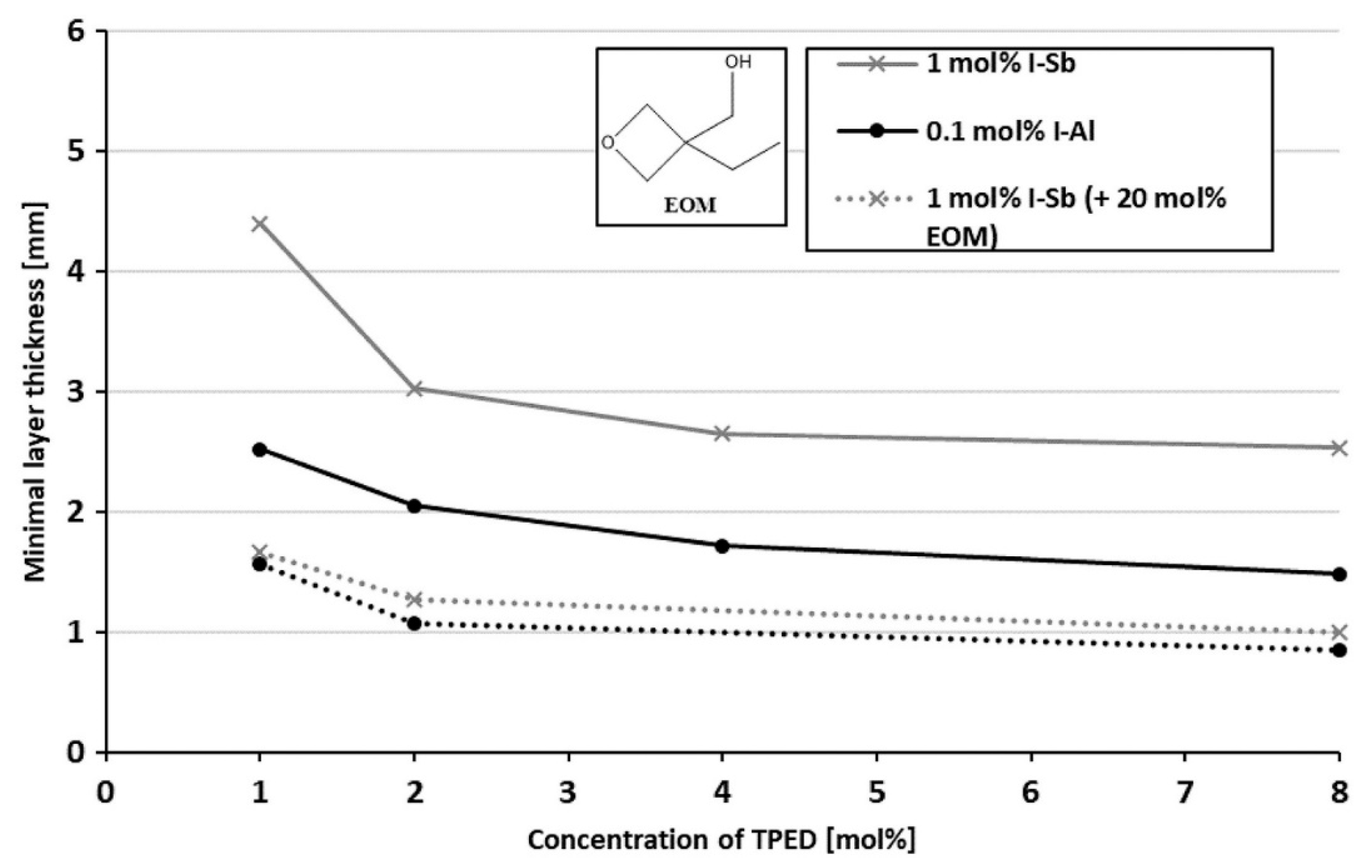
3. Onium Salt Photoinitiators for Different Epoxy Monomers
Effect of Sensitizers
4. Chemistry of Thermal Radical Initiators
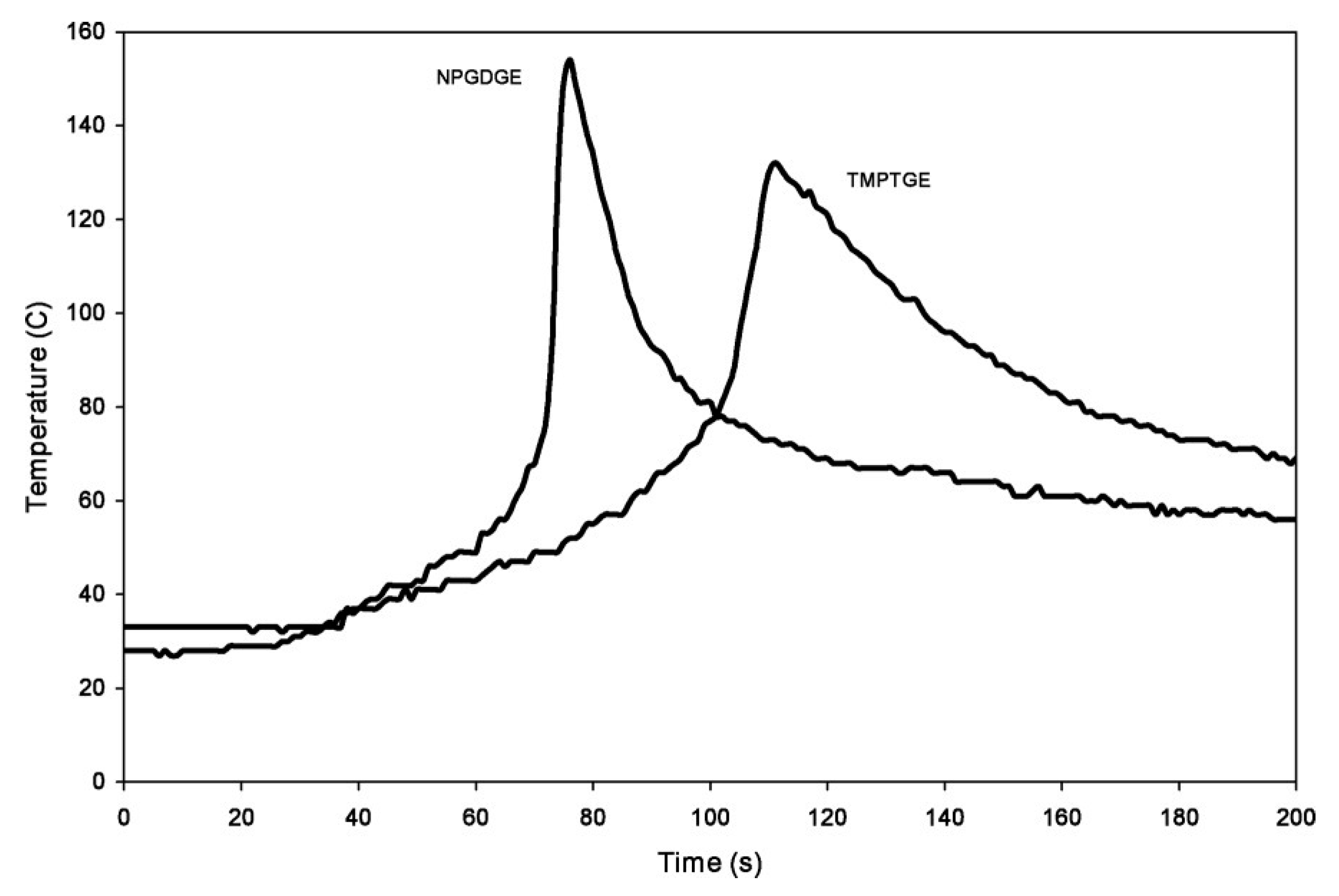
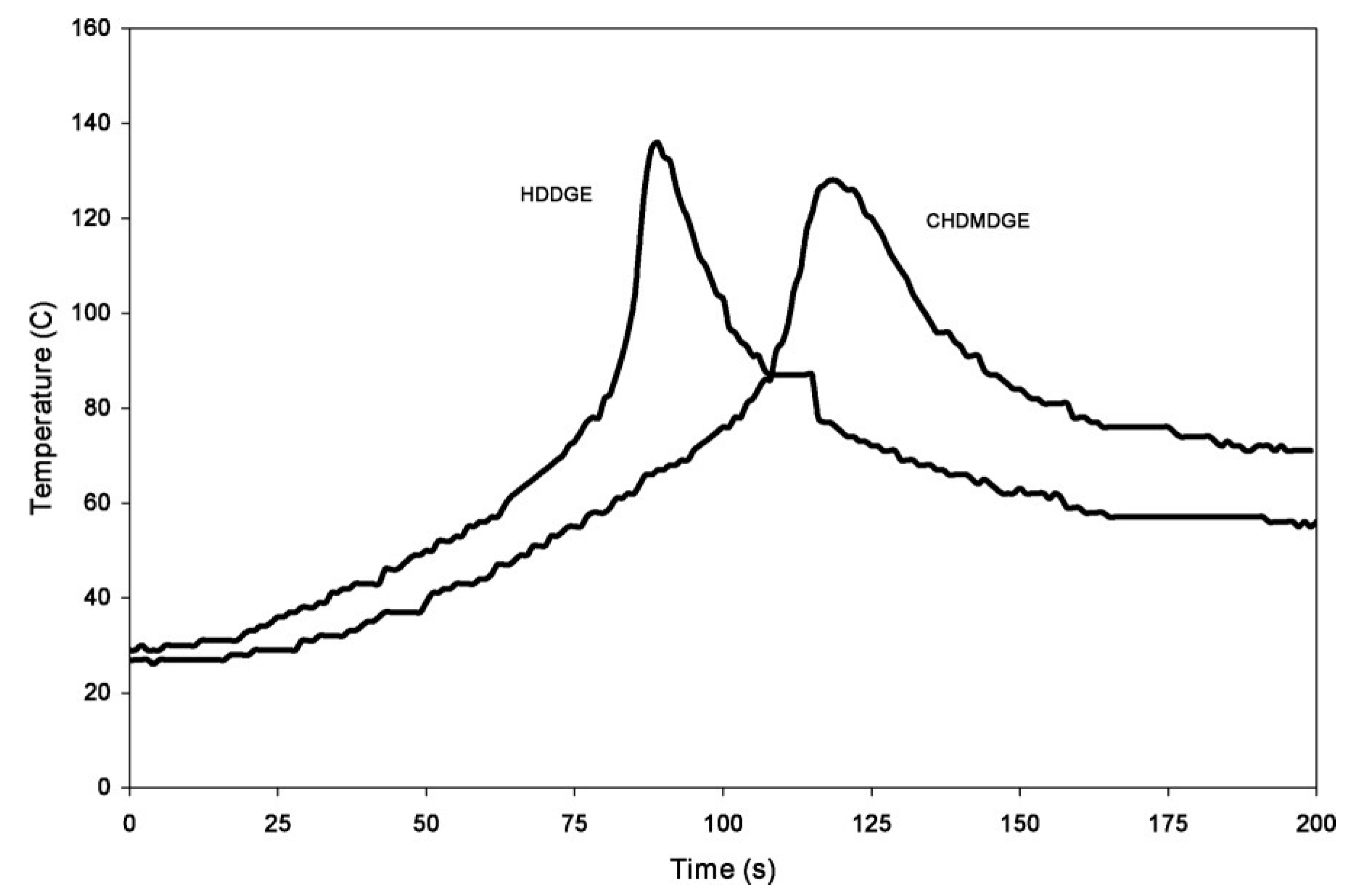
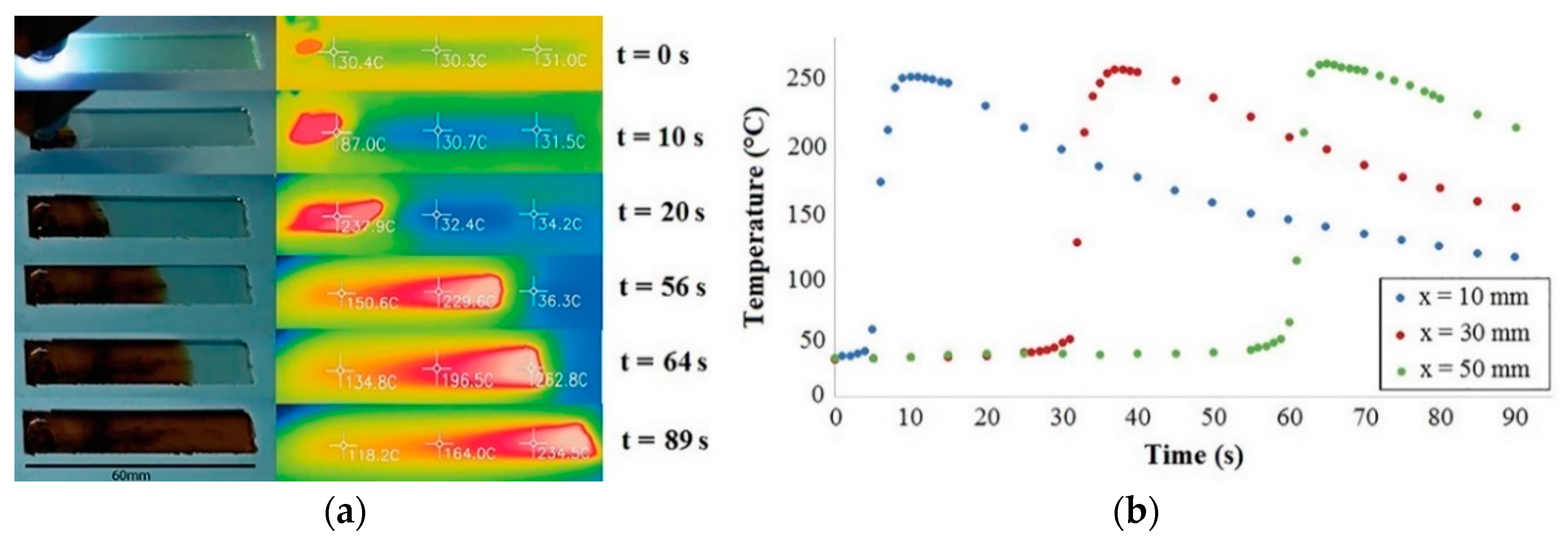
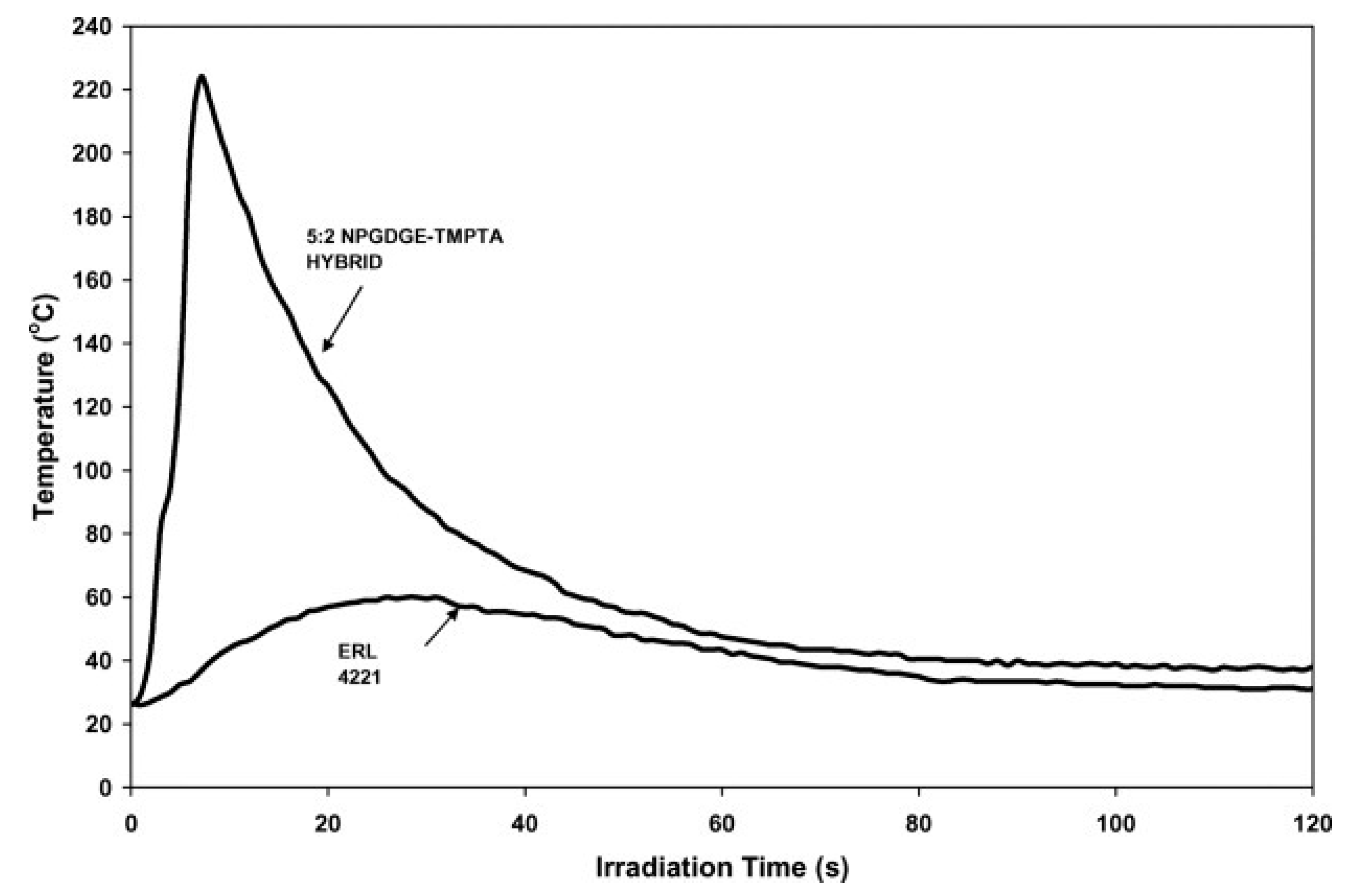
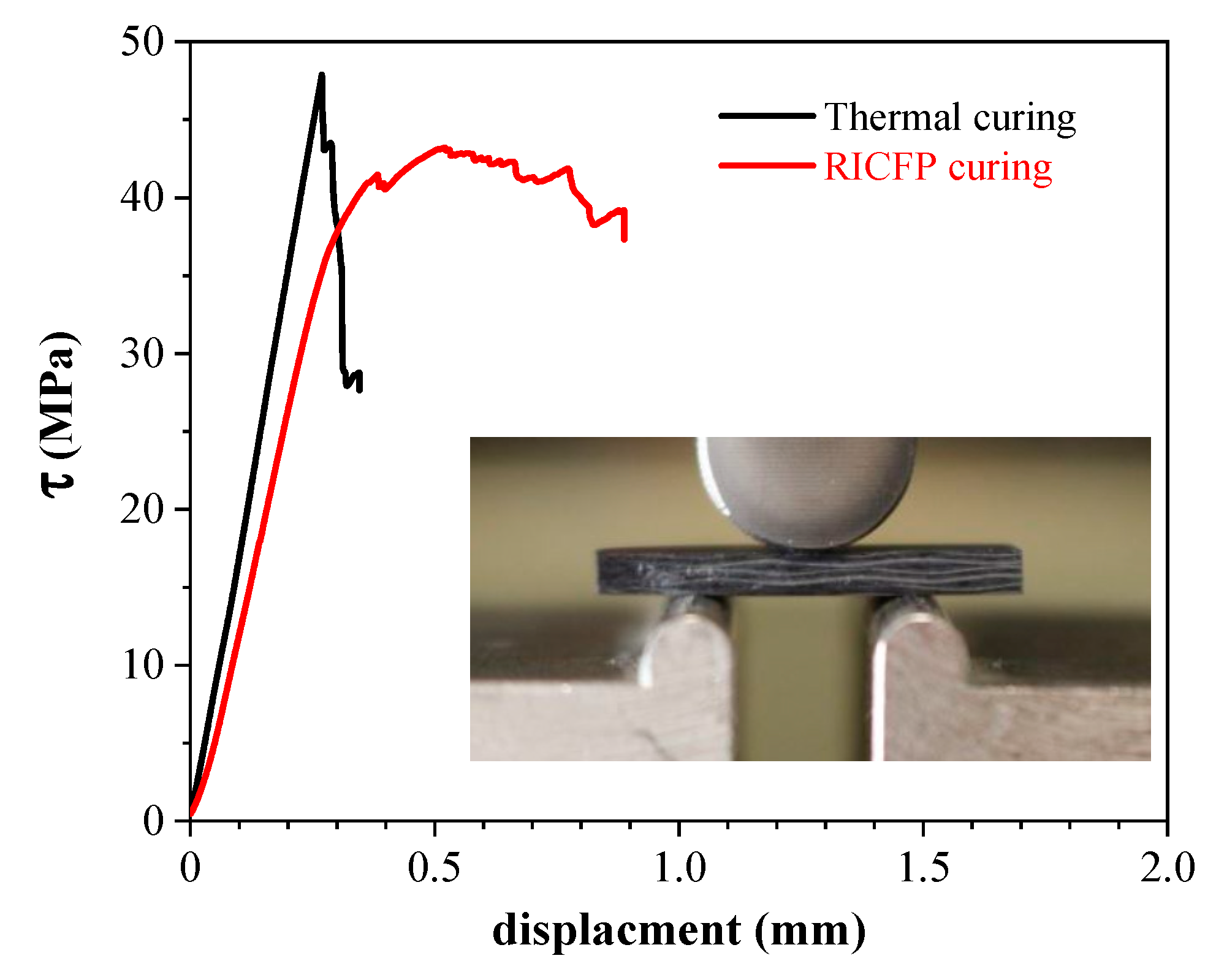
5. UV-Induced Cationic Frontal Polymerization of Different Epoxies
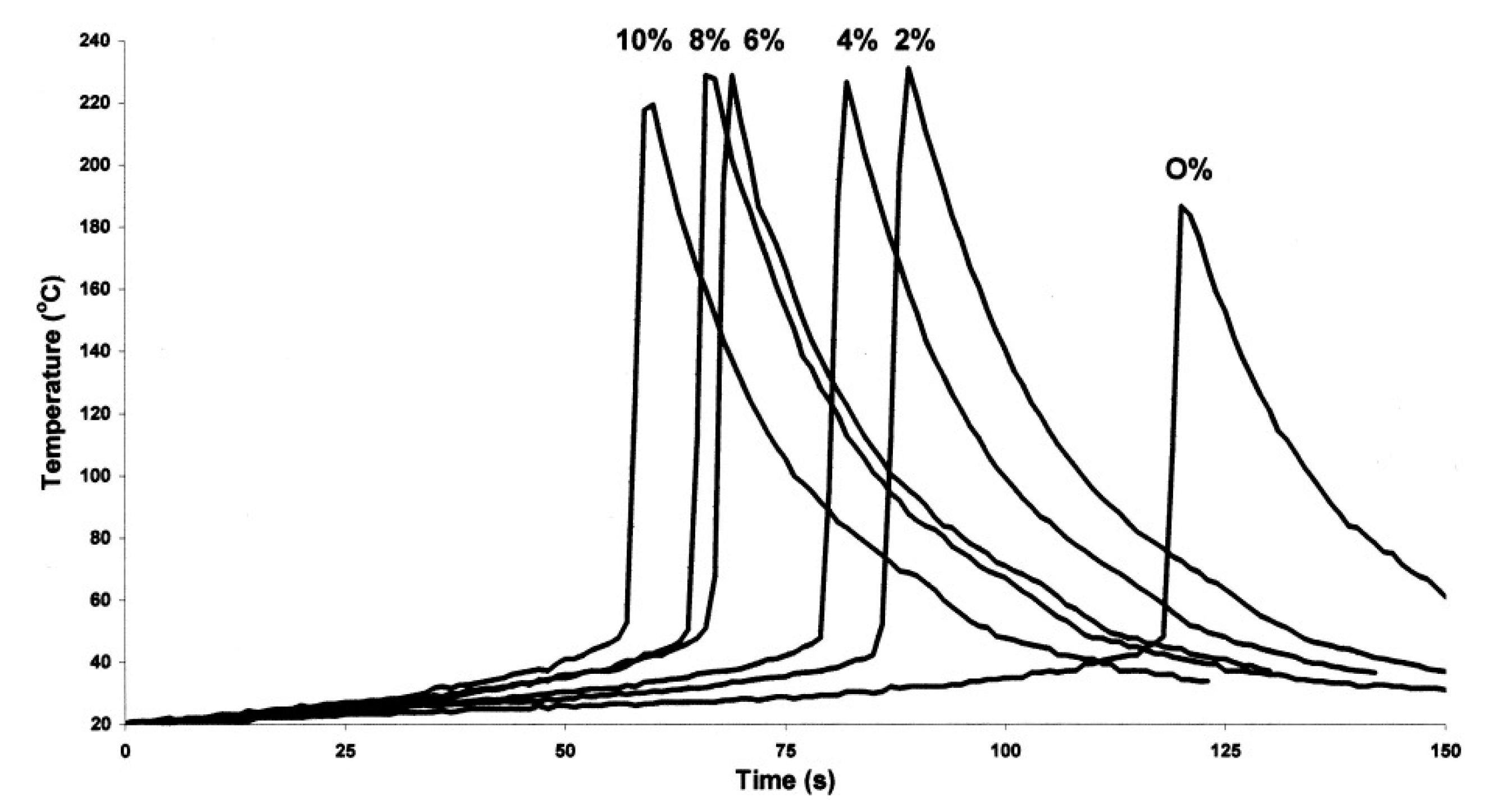
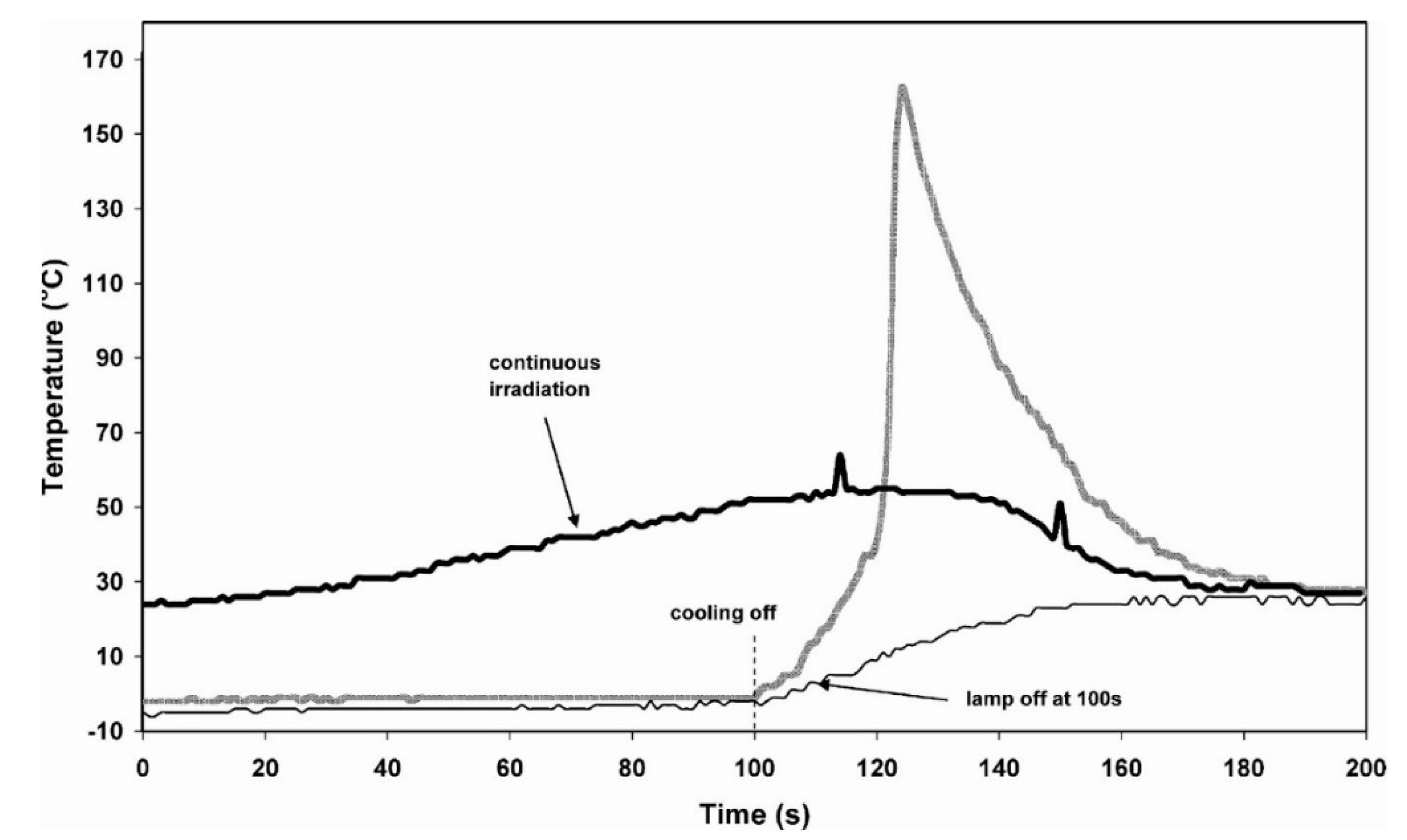
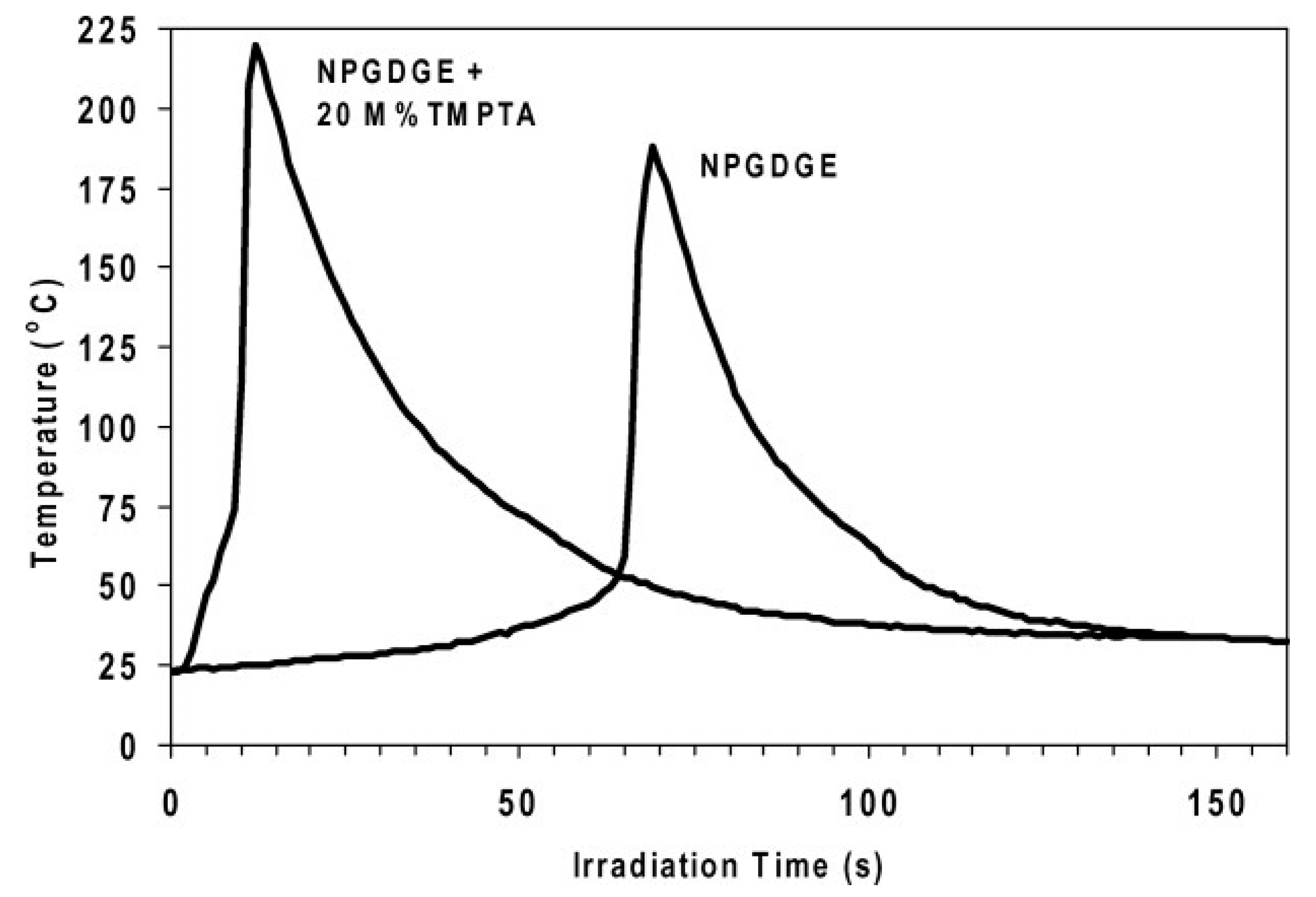
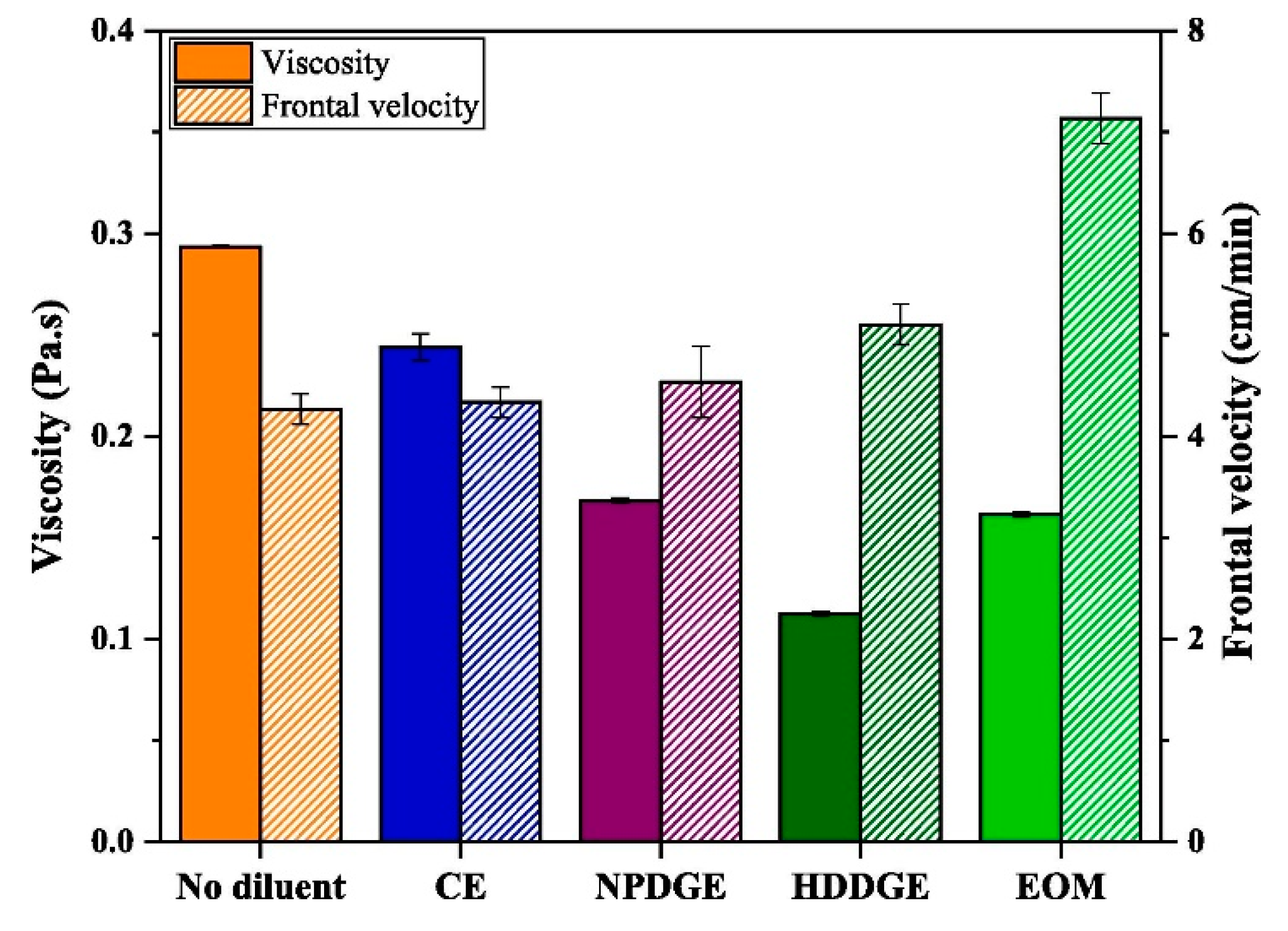
Effect of Diluents in UV-Induced Cationic Frontal Polymerization of Epoxide Monomers
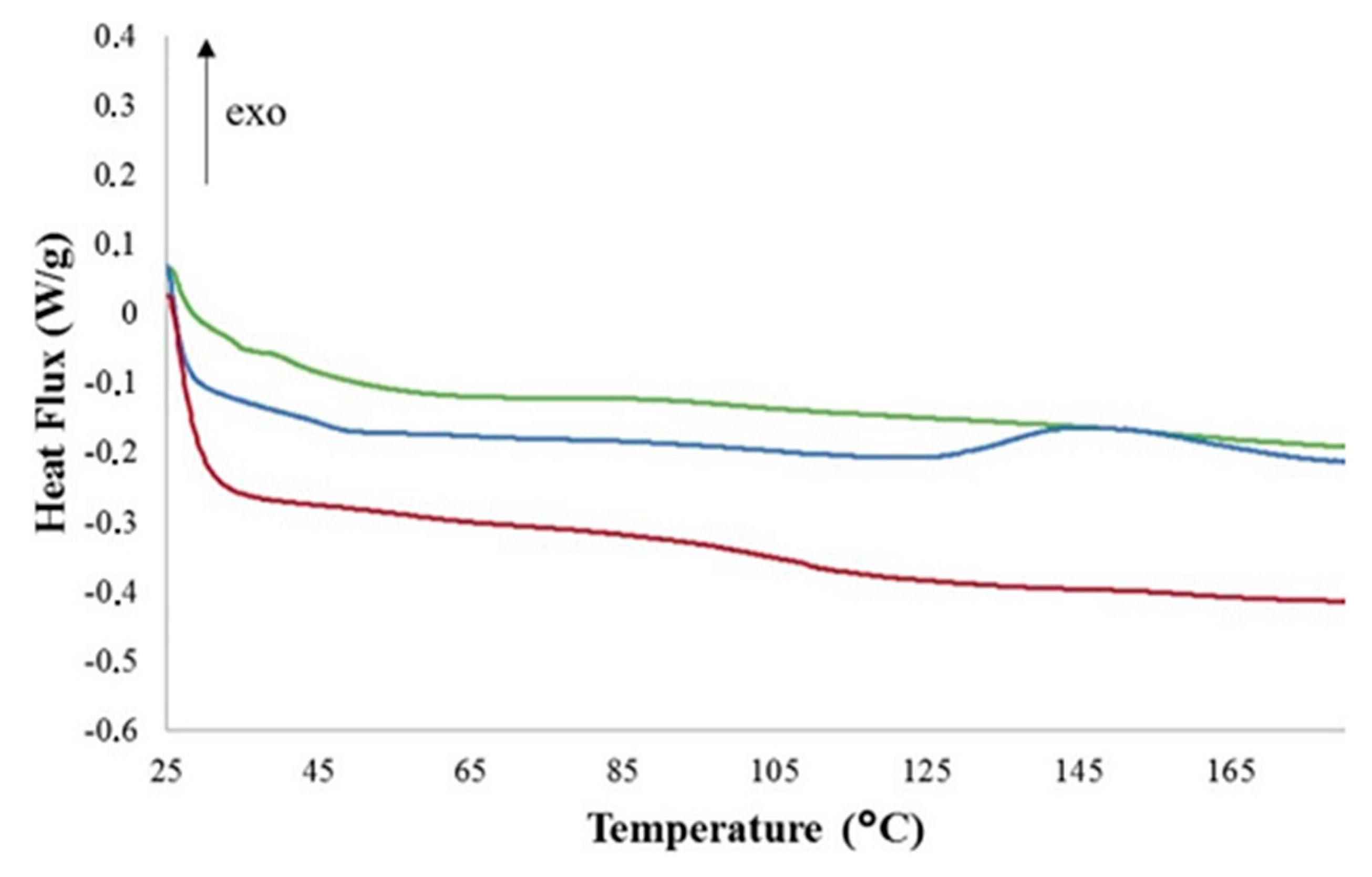
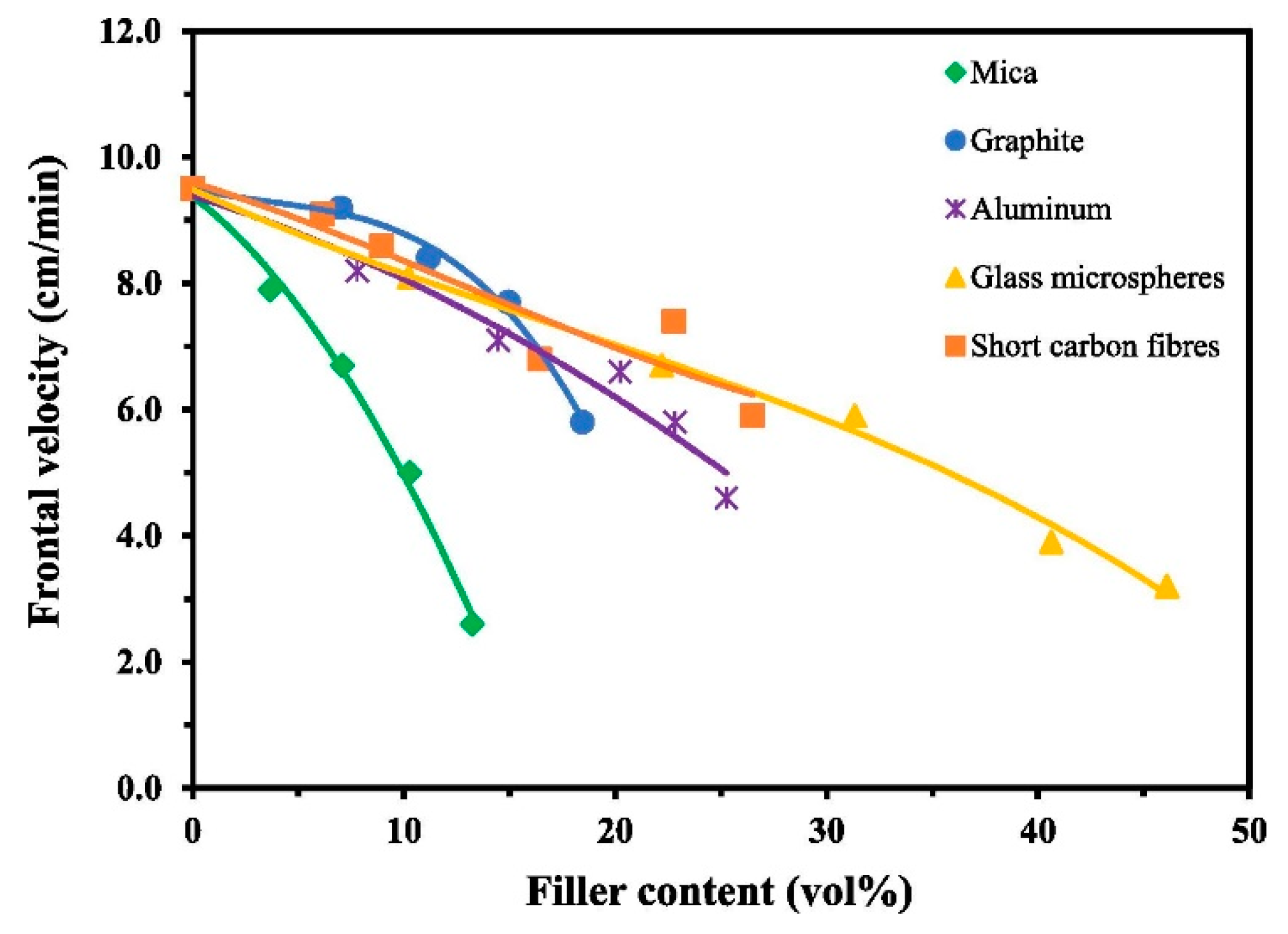
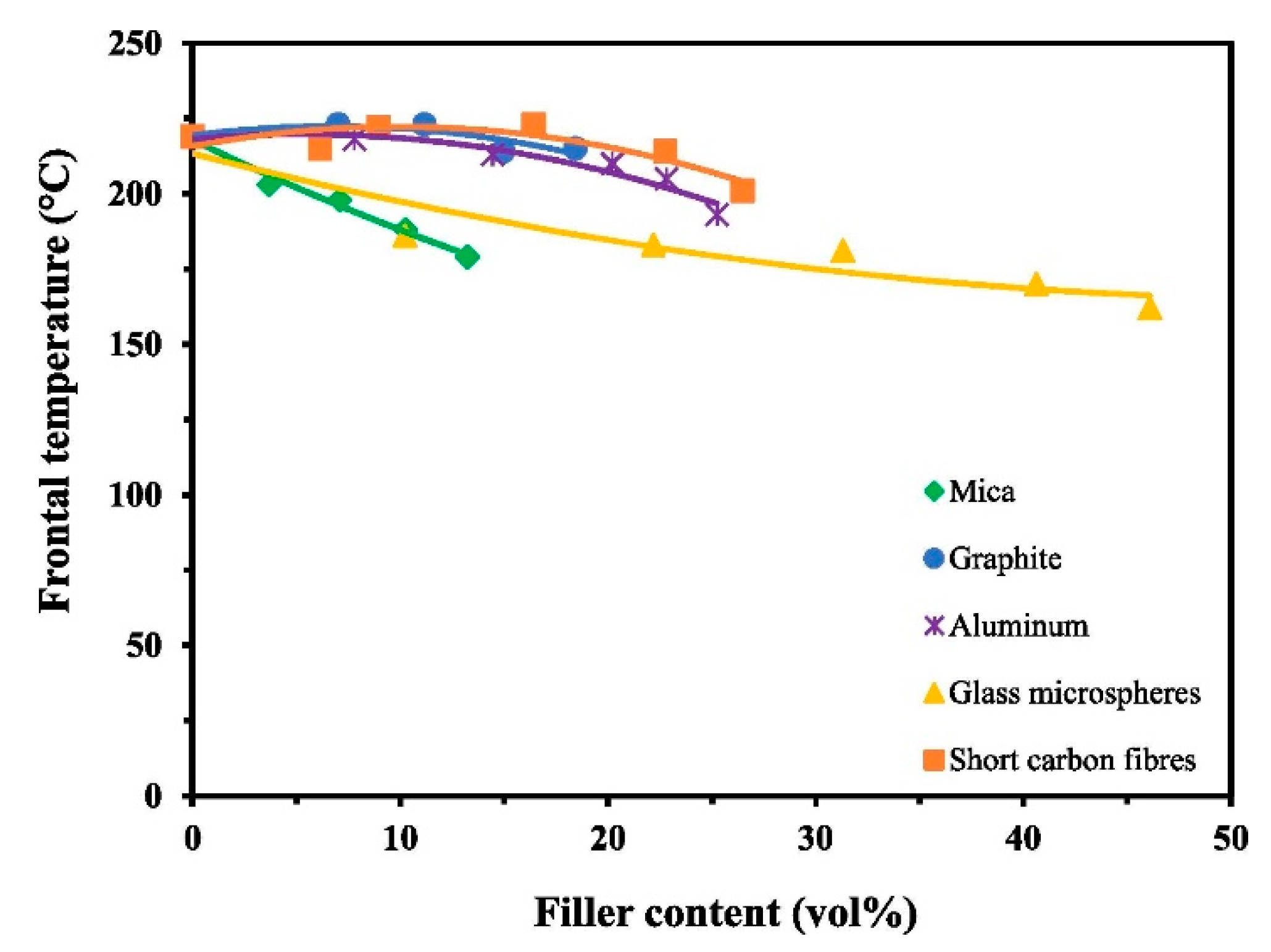
6. Effect of Fillers in Epoxy Composites
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, S.; Sui, J.; Chen, L.; Pojman, J.A. Polyurethane-nanosilica hybrid nanocomposites synthesized by frontal polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1670–1680. [Google Scholar] [CrossRef]
- Chekanov, Y.A.; Pojman, J.A. Preparation of functionally gradient materials via frontal polymerization. J. Appl. Polym. Sci. 2000, 78, 2398–2404. [Google Scholar] [CrossRef]
- Guo, X.; Wang, C.-F.; Fang, Y.; Chen, L.; Chen, S. Fast synthesis of versatile nanocrystal-embedded hydrogels toward the sensing of heavy metal ions and organoamines. J. Mater. Chem. 2011, 21, 1124–1129. [Google Scholar] [CrossRef]
- Yan, Q.-Z.; Zhang, W.; Lu, G.-D.; Su, X.-T.; Ge, C.-C. Frontal Polymerization Synthesis of Starch-Grafted Hydrogels: Effect of Temperature and Tube Size on Propagating Front and Properties of Hydrogels. Chem. A Eur. J. 2006, 12, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Sanna, R.; Alzari, V.; Nuvoli, D.; Scognamillo, S.; Marceddu, S.; Mariani, A. Polymer hydrogels of 2-hydroxyethyl acrylate and acrylic acid obtained by frontal polymerization. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1515–1520. [Google Scholar] [CrossRef]
- Nagy, I.P.; Sike, L.; Pojman, J.A. Thermochromic Composite Prepared via a Propagating Polymerization Front. J. Am. Chem. Soc. 1995, 117, 3611–3612. [Google Scholar] [CrossRef]
- White, S.R.; Kim, C. A Simultaneous Lay-Up and in situ Cure Process for Thick Composites. J. Reinf. Plast. Compos. 1993, 12, 520–535. [Google Scholar] [CrossRef]
- Pojman, J.A. Frontal Polymerization. Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; pp. 957–980. ISBN 9780080878621. [Google Scholar]
- Chechilo, N.M.; Khvilivitskii, R.J.; Enikolopyan, N.S. On the Phenomenon of Polymerization Reaction Spreading. Dokl. Akad. Nauk SSSR 1972, 204, 1180–1181. [Google Scholar]
- Davtyan, D.S.; Tonoyan, A.O.; Davtyan, S.P.; Savchenko, V.I. Geometric shape and stability of frontal regimes during radical polymerization of MMA in cylindrical flow reactor. Polym. Sci. Ser. A 1999, 41, 153–158. [Google Scholar]
- Davtyan, D.S.; Tonoyan, A.O.; Radugina, A.A.; Davtyan, S.P.; Savchenko, V.I.; Abrosimov, A.F. Control of conversion and molecular masses during the frontal polymerization of MMA in a cylindrical flow reactor. Polym. Sci. Ser. A 1999, 41, 147–152. [Google Scholar]
- Davtyan, D.S.; Tonoyan, A.O.; Radugina, A.A.; Davtyan, S.P.; Savchenko, V.I.; Abrosimov, A.F. Frontal radical polymerization of methyl methacrylate in a cylindrical flow reactor. Polym. Sci. Ser. A 1999, 41, 138–146. [Google Scholar]
- Decker, C. Photoinitiated crosslinking polymerisation. Prog. Polym. Sci. 1996, 21, 593–650. [Google Scholar] [CrossRef]
- Peiffer, R.W. Applications of photopolymer technology. In Photopolymerization: Fundamentals and Applications; Scranton, A.B., Bowman, C.N., Peiffer, R.W., Eds.; American Chemical Society: Washington, DC, USA, 1997; pp. 1–14. ISBN 0-8412-3520-1. [Google Scholar]
- Fouassier, J.P.; Rabek, J.F. Radiation Curing in Polymer Science and Technology; Elsevier Applied Science: London, UK, 1993; ISBN 1851669388. [Google Scholar]
- Pappas, S.P. Radiation Curing. Science and Technology; Springer US: Boston, MA, USA, 1992; ISBN 9781489907141. [Google Scholar]
- Green, W.A. Industrial Photoinitiators. A Technical Guide; CRC Press: Boca Raton, FL, USA, 2010; ISBN 1439827451. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W. New photoinitiators for cationic polymerization. J. Polym. Sci. Symp. 1977, 56, 383–395. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W. Diaryliodonium Salts. A New Class of Photoinitiators for Cationic Polymerization. Macromolecules 1977, 10, 1307–1315. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Photoinitiated cationic polymerization with triarylsulfonium salts. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 977–999. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Dye-sensitized photoinitiated cationic polymerization. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 2441–2451. [Google Scholar] [CrossRef]
- Crivello, J.V. Photopolymerization. In Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; pp. 919–955. ISBN 9780080878621. [Google Scholar]
- Crivello, J.V. The discovery and development of onium salt cationic photoinitiators. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4241–4254. [Google Scholar] [CrossRef]
- Sangermano, M. Advances in cationic photopolymerization. Pure Appl. Chem. 2012, 84, 2089–2101. [Google Scholar] [CrossRef]
- Sangermano, M.; Razza, N.; Crivello, J.V. Cationic UV-Curing: Technology and Applications. Macromol. Mater. Eng. 2014, 299, 775–793. [Google Scholar] [CrossRef]
- Kaur, M.; Srivastava, A.K. Photopolymerization: A Review. J. Macromol. Sci. Part C Polym. Rev. 2002, 42, 481–512. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. The photoinitiated cationic polymerization of epoxy resins. In Photopolymerization: Fundamentals and Applications; Scranton, A.B., Bowman, C.N., Peiffer, R.W., Eds.; American Chemical Society: Washington, DC, USA, 1997; pp. 1–16. ISBN 0-8412-3520-1. [Google Scholar]
- Zopf, R.F. UV Cationic Epoxy Systems for Dielectric Applications. Radiat. Curing 1982, 9, 10–26. [Google Scholar]
- Smith, G.H. Photocopolymerizable Compositions Based on Epoxy and Hydroxyl-Containing Organic Materials. US Patent US4256828A, 17 March 1981. [Google Scholar]
- Wang, A.A.; Knopf, R.J.; Osborn, C.L. Cationically Polymerizable Radiation Curable Compositions. Patent GB8110050A, 31 March 1981. [Google Scholar]
- Fouassier, J.-P.; Lalevée, J. Photoinitiators for Polymer Synthesis. Scope, Reactivity and Efficiency; Wiley-VCH: Weinheim, Germany, 2012; ISBN 9783527648245. [Google Scholar]
- Penczek, S.; Kubisa, P.; Matyjaszewski, K. Cationic Ring-Opening Polymerization of Heterocyclic Monomers; Springer: Berlin/Heidelberg, Germany, 1980; ISBN 9783540102090. [Google Scholar]
- Crivello, J.V.; Falk, B.; Zonca, M.R. Photoinduced cationic ring-opening frontal polymerizations of oxetanes and oxiranes. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1630–1646. [Google Scholar] [CrossRef]
- Crivello, J. Investigation of the photoactivated frontal polymerization of oxetanes using optical pyrometry. Polymer 2005, 46, 12109–12117. [Google Scholar] [CrossRef]
- Bulut, U.; Crivello, J.V. Investigation of the Reactivity of Epoxide Monomers in Photoinitiated Cationic Polymerization. Macromolecules 2005, 38, 3584–3595. [Google Scholar] [CrossRef]
- Mariani, A.; Bidali, S.; Fiori, S.; Sangermano, M.; Malucelli, G.; Bongiovanni, R.; Priola, A. UV-ignited frontal polymerization of an epoxy resin. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 2066–2072. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H. Polymer Handbook; Wiley: New York, NY, USA; London, UK, 1966. [Google Scholar]
- Bomze, D.; Knaack, P.; Liska, R. Successful radical induced cationic frontal polymerization of epoxy-based monomers by C–C labile compounds. Polym. Chem. 2015, 6, 8161–8167. [Google Scholar] [CrossRef]
- Bomze, D.; Knaack, P.; Koch, T.; Jin, H.; Liska, R. Radical induced cationic frontal polymerization as a versatile tool for epoxy curing and composite production. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3751–3759. [Google Scholar] [CrossRef]
- Knaack, P.; Klikovits, N.; Tran, A.D.; Bomze, D.; Liska, R. Radical induced cationic frontal polymerization in thin layers. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1155–1159. [Google Scholar] [CrossRef]
- Tran, A.D.; Koch, T.; Knaack, P.; Liska, R. Radical induced cationic frontal polymerization for preparation of epoxy composites. Compos. Part A Appl. Sci. Manuf. 2020, 132, 105855. [Google Scholar] [CrossRef]
- Sangermano, M.; D’Anna, A.; Marro, C.; Klikovits, N.; Liska, R. UV-activated frontal polymerization of glass fibre reinforced epoxy composites. Compos. Part B Eng. 2018, 143, 168–171. [Google Scholar] [CrossRef]
- Sangermano, M.; Roppolo, I.; Chiappone, A. New Horizons in Cationic Photopolymerization. Polymer 2018, 10, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atif, M.; Bongiovanni, R.; Yang, J. Cationically UV-Cured Epoxy Composites. Polym. Rev. 2015, 55, 90–106. [Google Scholar] [CrossRef]
- Strohmeier, V.W.; Barbeau, C. Polymerisation von Propylenoxid mit Mangandecacarbonyl nach UV-Bestrahlung. Die Makromol. Chem. 1965, 81, 86–91. [Google Scholar] [CrossRef]
- Kaeriyama, K. Photocatalyzed polymerization of epichlorohydrin. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 1547–1548. [Google Scholar] [CrossRef]
- Licari, J.J.; Crepeau, P.C. Electromagnetic Radiation Polymerization. US Patent US3205157A, 7 September 1965. [Google Scholar]
- Schlesinger, S.I. Photopolymerization of Epoxides. Photogr. Sci. Eng. 1974, 18, 387–393. [Google Scholar]
- Crivello, J.V.; Lee, J.L. Alkoxy-substituted diaryliodonium salt cationic photoinitiators. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 3951–3968. [Google Scholar] [CrossRef]
- Crivello, J.V. Design and Synthesis of Photoacid Generating Systems. J. Photopolym. Sci. Technol. 2008, 21, 493–497. [Google Scholar] [CrossRef] [Green Version]
- Crivello, J.V. Cationic polymerization–Iodonium and sulfonium salt photoinitiators. In Initiators—Poly-Reactions—Optical Activity; Advances in Polymer Science; Springer: Berlin, Germany, 1984; pp. 1–48. [Google Scholar]
- Crivello, J.V.; Lam, J.H.W.; Volante, C.N. Photoinitiated Cationic Polymerization using Diaryliodonium Salts. J. Radiat. Curing 1977, 4, 2–16. [Google Scholar]
- Crivello, J.V.; Lam, J.H.N.; Moore, J.E.; Schroeter, S.H. Triarylsulfonium Salts: A New Class of Photoinitiators for Cationic Polymerization. J. Radiat. Curing 1978, 5, 2–17. [Google Scholar]
- Crivello, J.V.; Lee, J.L. The synthesis, characterization, and photoinitiated cationic polymerization of silicon-containing epoxy resins. J. Polym. Sci. Part A Polym. Chem. 1990, 28, 479–503. [Google Scholar] [CrossRef]
- Castellanos, F.; Fouassier, J.P.; Priou, C.; Cavezzan, J. Synthesis, reactivity, and properties of new diaryliodonium salts as photoinitiators for the cationic polymerization of epoxy silicones. J. Appl. Polym. Sci. 1996, 60, 705–713. [Google Scholar] [CrossRef]
- Klikovits, N.; Knaack, P.; Bomze, D.; Krossing, I.; Liska, R. Novel photoacid generators for cationic photopolymerization. Polym. Chem. 2017, 8, 4414–4421. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Szillat, F.; Fouassier, J.P.; Lalevée, J. Remarkable Versatility of Silane/Iodonium Salt as Redox Free Radical, Cationic, and Photopolymerization Initiators. Macromolecules 2019, 52, 5638–5645. [Google Scholar] [CrossRef]
- Yagci, Y.; Hepuzer, Y. A Novel Visible Light Initiatiating System for Cationic Polymerization. Macromolecules 1999, 32, 6367–6370. [Google Scholar] [CrossRef]
- Fouassier, J.P. Photoinitiator, Photopolymerization and Photocuring: Fundamentals and Applications; Hanser Publishers: New York, NY, USA, 1995. [Google Scholar]
- Pappas, S.P.; Gatechair, L.R.; Jilek, J.H. Photoinitiation of cationic polymerization. III. Photosensitization of diphenyliodonium and triphenylsulfonium salts. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 77–84. [Google Scholar] [CrossRef]
- Yaǧci, Y.; Schnabel, W.; Yağcı, Y. Direct and sensitized photoinitiated cationic polymerization using pyridinium salts. Macromol. Symp. 1994, 85, 115–127. [Google Scholar] [CrossRef]
- Yagci, Y.; Endo, T. N-benzyl and N-alkoxy pyridinium salts as thermal and photochemical initiators for cationic polymerization. In Polymer Synthesis: Polymer Catalysis; Améduri, B., Ed.; Springer: Berlin, Germany; London, UK, 1997; pp. 59–86. ISBN 978-3-540-61288-9. [Google Scholar]
- Bi, Y.; Neckers, D.C. A Visible Light Initiating System for Free Radical Promoted Cationic Polymerization. Macromolecules 1994, 27, 3683–3693. [Google Scholar] [CrossRef]
- Chen, Y.; Yamamura, T.; Igarashi, K. Photosensitization of carbazole derivatives in cationic polymerization with a novel sensitivity to near-UV light. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 90–100. [Google Scholar] [CrossRef]
- Hua, Y.; Crivello, J.V. Synergistic interaction of epoxides andN-vinylcarbazole during photoinitiated cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3697–3709. [Google Scholar] [CrossRef]
- Hua, Y.; Crivello, J.V. Development of Polymeric Photosensitizers for Photoinitiated Cationic Polymerization. Macromolecules 2001, 34, 2488–2494. [Google Scholar] [CrossRef]
- Crivello, J.V.; Sangermano, M. Visible and long-wavelength photoinitiated cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 343–356. [Google Scholar] [CrossRef]
- Hua, Y.; Jiang, F.; Crivello, J.V. Photosensitized Onium-Salt-Induced Cationic Polymerization with Hydroxymethylated Polynuclear Aromatic Hydrocarbons. Chem. Mater. 2002, 14, 2369–2377. [Google Scholar] [CrossRef]
- Gomurashvili, Z.; Crivello, J.V. Phenothiazine photosensitizers for onium salt photoinitiated cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 1187–1197. [Google Scholar] [CrossRef]
- Gomurashvili, Z.; Crivello, J.V. Monomeric and Polymeric Phenothiazine Photosensitizers for Photoinitiated Cationic Polymerization. Macromolecules 2002, 35, 2962–2969. [Google Scholar] [CrossRef]
- Crivello, J.V.; Jiang, F. Development of Pyrene Photosensitizers for Cationic Photopolymerizations. Chem. Mater. 2002, 14, 4858–4866. [Google Scholar] [CrossRef]
- Crivello, J.V.; Jang, M. Anthracene electron-transfer photosensitizers for onium salt induced cationic photopolymerizations. J. Photochem. Photobiol. A Chem. 2003, 159, 173–188. [Google Scholar] [CrossRef]
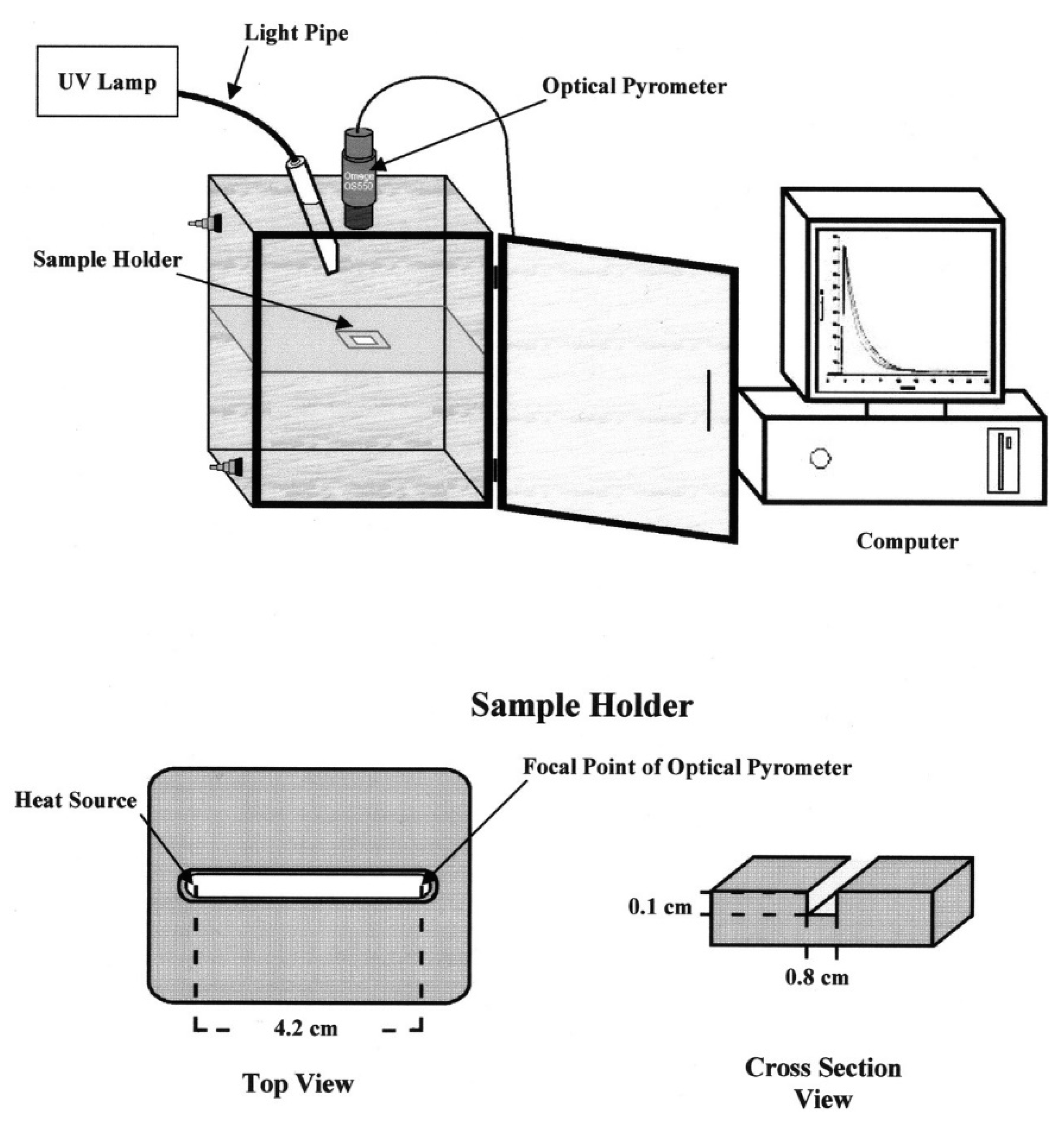
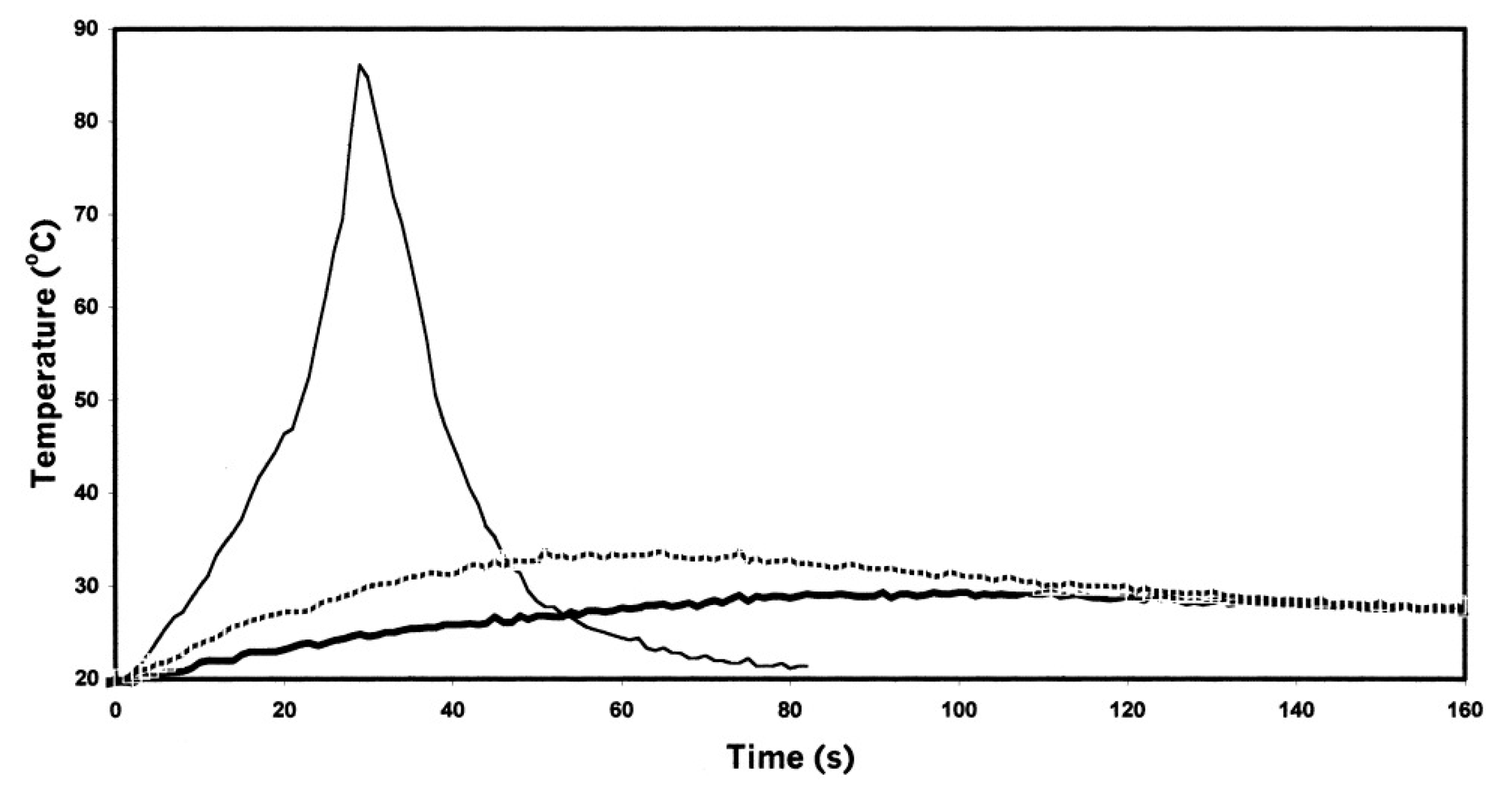
- Falk, B.; Vallinas, S.M.; Crivello, J.V. Monitoring photopolymerization reactions with optical pyrometry. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 579–596. [Google Scholar] [CrossRef]
- Crivello, J.V.; Jang, M. Synthesis of Monomer and Polymer-Bound Photosensitizers. J. Macromol. Sci. Part A 2005, 42, 1–19. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bulut, U. Curcumin: A naturally occurring long-wavelength photosensitizer for diaryliodonium salts. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5217–5231. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bulut, U. Indian Turmeric and its Use in Cationic Photopolymerizations. Macromol. Symp. 2006, 240, 1–11. [Google Scholar] [CrossRef]
- Crivello, J.V. Cationic photopolymerization of alkyl glycidyl ethers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 3036–3052. [Google Scholar] [CrossRef]
- Crivello, J.V. A new visible light sensitive photoinitiator system for the cationic polymerization of epoxides. J. Polym. Sci. Part A Polym. Chem. 2008, 47, 866–875. [Google Scholar] [CrossRef]
- Crivello, J.V. Radical-Promoted Visible Light Photoinitiated Cationic Polymerization of Epoxides. J. Macromol. Sci. Part A 2009, 46, 474–483. [Google Scholar] [CrossRef]
- Schroeder, W.F.; Asmussen, S.V.; Sangermano, M.; Vallo, C.I. Visible light polymerization of epoxy monomers using an iodonium salt with camphorquinone/ethyl-4-dimethyl aminobenzoate. Polym. Int. 2013, 62, 1368–1376. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, X.; Xu, B.; Qian, X.; Song, G.-Q.; Nie, J. Cationic photopolymerization of bisphenol A diglycidyl ether epoxy under 385 nm. J. Appl. Polym. Sci. 2013, 130, 3698–3703. [Google Scholar] [CrossRef]
- Telitel, S.; Blanchard, N.; Schweizer, S.; Morlet-Savary, F.; Graff, B.; Fouassier, J.-P.; Lalevée, J. BODIPY derivatives and boranil as new photoinitiating systems of cationic polymerization exhibiting a tunable absorption in the 400–600 nm spectral range. Polymer 2013, 54, 2071–2076. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Garra, P.; Dumur, F.; Bui, T.-T.; Goubard, F.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; et al. Novel Carbazole Skeleton-Based Photoinitiators for LED Polymerization and LED Projector 3D Printing. Molecules 2017, 22, 2143. [Google Scholar] [CrossRef] [Green Version]
- Al Mousawi, A.; Arar, A.; Ibrahim-Ouali, M.; Duval, S.; Dumur, F.; Garra, P.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; et al. Carbazole-based compounds as photoinitiators for free radical and cationic polymerization upon near visible light illumination. Photochem. Photobiol. Sci. 2018, 17, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Al Mousawi, A.; Dumur, F.; Garra, P.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. Carbazole Scaffold Based Photoinitiator/Photoredox Catalysts: Toward New High Performance Photoinitiating Systems and Application in LED Projector 3D Printing Resins. Macromolecules 2017, 50, 2747–2758. [Google Scholar] [CrossRef]
- Xiao, P.; Lalevée, J.; Zhao, J.; Stenzel, M.H. N -Vinylcarbazole as Versatile Photoinaddimer of Photopolymerization under Household UV LED Bulb (392 nm). Macromol. Rapid Commun. 2015, 36, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
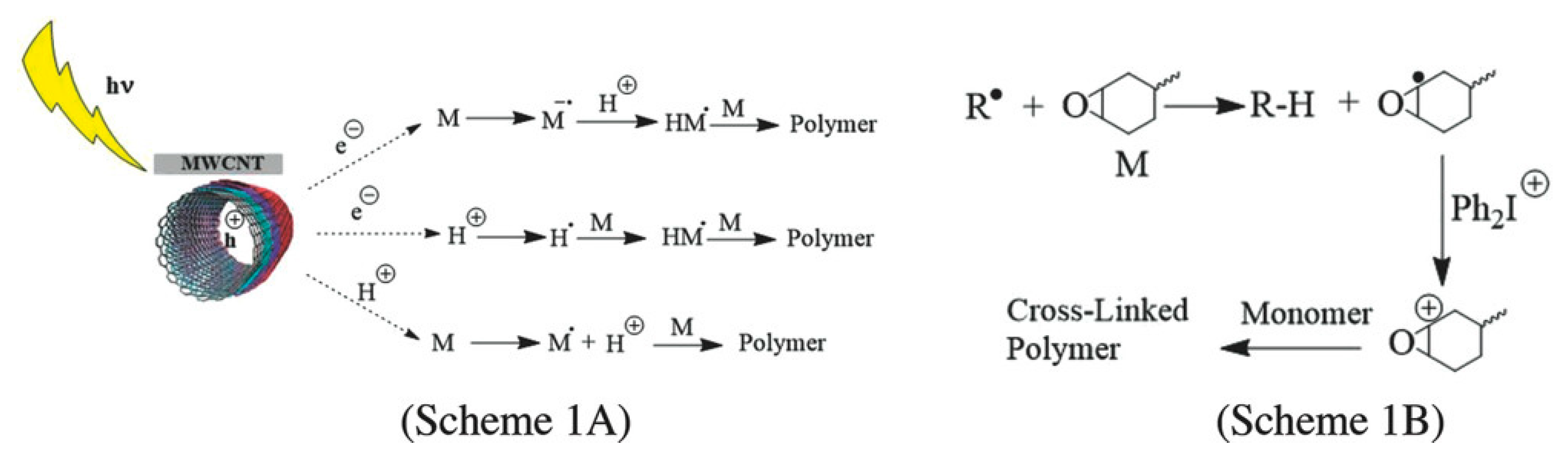
- Sangermano, M.; Rodríguez, D.; Gonzalez, M.C.; Laurenti, E.; Yagci, Y. Visible Light Induced Cationic Polymerization of Epoxides by Using Multiwalled Carbon Nanotubes. Macromol. Rapid Commun. 2018, 39, e1800250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dumur, F.; Horcajada, P.; Livage, C.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Iron-Based Metal-Organic Frameworks (MOF) as Photocatalysts for Radical and Cationic Polymerizations under Near UV and Visible LEDs (385–405 nm). Macromol. Chem. Phys. 2016, 217, 2534–2540. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Lara, D.M.; Noirbent, G.; Dumur, F.; Toufaily, J.; Hamieh, T.; Bui, T.-T.; Goubard, F.; Graff, B.; Gigmes, D.; et al. Carbazole Derivatives with Thermally Activated Delayed Fluorescence Property as Photoinitiators/Photoredox Catalysts for LED 3D Printing Technology. Macromolecules 2017, 50, 4913–4926. [Google Scholar] [CrossRef]
- Mokbel, H.; Anderson, D.; Plenderleith, R.; Dietlin, C.; Morlet-Savary, F.; Dumur, F.; Gigmes, D.; Fouassier, J.-P.; Lalevée, J. Copper photoredox catalyst “G1”: A new high performance photoinitiator for near-UV and visible LEDs. Polym. Chem. 2017, 8, 5580–5592. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Garra, P.; Sallenave, X.; Dumur, F.; Toufaily, J.; Hamieh, T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; Lalevée, J. π-Conjugated Dithienophosphole Derivatives as High Performance Photoinitiators for 3D Printing Resins. Macromolecules 2018, 51, 1811–1821. [Google Scholar] [CrossRef]
- Zhang, J.; Campolo, D.; Dumur, F.; Xiao, P.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. Iron Complexes in Visible-Light-Sensitive Photoredox Catalysis: Effect of Ligands on Their Photoinitiation Efficiencies. ChemCatChem 2016, 8, 2227–2233. [Google Scholar] [CrossRef]
- Al Mousawi, A.; Dumur, F.; Garra, P.; Toufaily, J.; Hamieh, T.; Goubard, F.; Bui, T.-T.; Graff, B.; Gigmes, D.; Fouassier, J.P.; et al. Azahelicenes as visible light photoinitiators for cationic and radical polymerization: Preparation of photoluminescent polymers and use in high performance LED projector 3D printing resins. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1189–1199. [Google Scholar] [CrossRef]
- Xiao, P.; Zhang, J.; Graff, B.; Fouassier, J.P.; Lalevée, J. Rubrene-Based Green-Light-Sensitive Photoinitiating Systems of Polymerization. Macromol. Chem. Phys. 2017, 218, 1700314. [Google Scholar] [CrossRef]
- Zhang, J.; Zivic, N.; Dumur, F.; Xiao, P.; Graff, B.; Fouassier, J.P.; Gigmes, D.; Lalevée, J. N-[2-(Dimethylamino)ethyl]-1,8-naphthalimide derivatives as photoinitiators under LEDs. Polym. Chem. 2018, 9, 994–1003. [Google Scholar] [CrossRef]
- Mokbel, H.; Poriel, C.; Rault-Berthelot, J.; Dumur, F.; Gigmes, D.; Toufaily, J.; Hamieh, T.; Cordella, D.; Detrembleur, C.; Fouassier, J.P.; et al. A glance at violet LED sensitive photoinitiators based on the spiroxanthene scaffold. J. Appl. Polym. Sci. 2016, 133, 43213. [Google Scholar] [CrossRef]
- Mokbel, H.; Xiao, P.; Simonnet-Jégat, C.; Dumur, F.; Gigmes, D.; Toufaily, J.; Hamieh, T.; Fouassier, J.P.; Lalevée, J. Iodonium-polyoxometalate and thianthrenium-polyoxometalate as new one-component UV photoinitiators for radical and cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 981–989. [Google Scholar] [CrossRef]
- Meng, X.; Lu, H.; Li, Z.; Wang, C.; Liu, R.; Guan, X.; Yagci, Y. Near-infrared light induced cationic polymerization based on upconversion and ferrocenium photochemistry. Polym. Chem. 2019, 10, 5574–5577. [Google Scholar] [CrossRef]
- Bouzrati-Zerelli, M.; Guillaume, N.; Goubard, F.; Bui, T.-T.; Villotte, S.; Dietlin, C.; Morlet-Savary, F.; Gigmes, D.; Fouassier, J.P.; Dumur, F.; et al. A novel class of photoinitiators with a thermally activated delayed fluorescence (TADF) property. New J. Chem. 2018, 42, 8261–8270. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.; Seba, M.; Fujita, T.; Yamago, S.; Yagci, Y. Visible light-induced free radical promoted cationic polymerization using organotellurium compounds. Polym. Chem. 2018, 9, 5639–5643. [Google Scholar] [CrossRef]
- Kaya, K.; Kreutzer, J.; Yagci, Y. A Charge-Transfer Complex of Thioxanthonephenacyl Sulfonium Salt as a Visible-Light Photoinitiator for Free Radical and Cationic Polymerizations. ChemPhotoChem 2019, 3, 1187–1192. [Google Scholar] [CrossRef]
- Wang, D.; Kaya, K.; Garra, P.; Fouassier, J.P.; Graff, B.; Yagci, Y.; Lalevée, J. Sulfonium salt based charge transfer complexes as dual thermal and photochemical polymerization initiators for composites and 3D printing. Polym. Chem. 2019, 10, 4690–4698. [Google Scholar] [CrossRef]
- Garra, P.; Caron, A.; Al Mousawi, A.; Graff, B.; Morlet-Savary, F.; Dietlin, C.; Yagci, Y.; Fouassier, J.-P.; Lalevée, J. Photochemical, Thermal Free Radical, and Cationic Polymerizations Promoted by Charge Transfer Complexes: Simple Strategy for the Fabrication of Thick Composites. Macromolecules 2018, 51, 7872–7880. [Google Scholar] [CrossRef]
- Zhou, J.; Jia, S.; Fu, W.; Liu, Z.; Tan, Z. Fast curing of thick components of epoxy via modified UV-triggered frontal polymerization propagating horizontally. Mater. Lett. 2016, 176, 228–231. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lee, J.L. The synthesis and characterization of polymer-bound diaryliodonium salts and their use in photo and thermally initiated cationic polymerization. Polym. Bull. 1986, 16, 243–248. [Google Scholar] [CrossRef]
- Crivello, J.V.; Narayan, R. Epoxidized triglycerides as renewable monomers in photoinitiated cationic polymerization. Chem. Mater. 1992, 4, 692–699. [Google Scholar] [CrossRef]
- Crivello, J.V. The synthesis and cationic polymerization of novel epoxide monomers. Polym. Eng. Sci. 1992, 32, 1462–1465. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bi, D. Regioselective hydrosilations. IV. The synthesis and polymerization of monomers containing epoxy and alkoxysilane groups. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 3121–3132. [Google Scholar] [CrossRef]
- Crivello, J.V.; Carter, A.M. Photochemical synthesis and cationic photopolyermization of α,ω-diepoxyalkanes. J. Polym. Sci. Part A Polym. Chem. 1993, 31, 2663–2666. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bi, D. The synthesis and cationic polymerization of multifunctional silicon-containing epoxy monomers and oligomers. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 683–697. [Google Scholar] [CrossRef]
- Crivello, J.V.; Kim, W. Synthesis and photopolymerization of 1-propenyl glycidyl ether. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 1639–1648. [Google Scholar] [CrossRef]
- Crivello, J.; Bi, D.; Fan, M. Synthesis and Polymerization of Novel Cationically Polymerizable Monomers. J. Macromol. Sci. Part A 1994, 31, 1001–1029. [Google Scholar] [CrossRef]
- Crivello, J.V.; Malik, R. Synthesis and photoinitiated cationic polymerization of monomers with the silsesquioxane core. J. Polym. Sci. A Polym. Chem. 1997, 35, 407–425. [Google Scholar] [CrossRef]
- Crivello, J.V.; Liu, S.S. Synthesis and cationic photopolymerization of 1-butenyl glycidyl ether. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 1179–1187. [Google Scholar] [CrossRef]
- Crivello, J.V.; Varlemann, U. Structure and reactivity relationships in the photoinitiated cationic polymerization of 3,4-epoxycyclohexylmethyl-3′,4′-epoxycyclohexane carboxylate. In Photopolymerization: Fundamentals and Applications; Scranton, A.B., Bowman, C.N., Peiffer, R.W., Eds.; American Chemical Society: Washington, DC, USA, 1997; pp. 82–94. ISBN 0-8412-3520-1. [Google Scholar]
- Crivello, J.V.; Varlemann, U. The synthesis and study of the photoinitiated cationic polymerization of novel cycloaliphatic epoxides. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 2463–2471. [Google Scholar] [CrossRef]
- Crivello, J.V.; Varlemann, U. Mechanistic study of the reactivity of 3,4-epoxycyclohexylmethyl 3′,4′-epoxycyclohexancarboxylate in photoinitiated cationic polymerizations. J. Polym. Sci. Part A Polym. Chem. 1995, 33, 2473–2486. [Google Scholar] [CrossRef]
- Crivello, J.V.; Liu, S. Photoinitiated cationic polymerization of epoxy alcohol monomers. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 389–401. [Google Scholar] [CrossRef]
- Crivello, J.V.; Falk, B.; Zonca, M.R., Jr. Study of cationic ring-opening photopolymerizations using optical pyrometry. J. Appl. Polym. Sci. 2004, 92, 3303–3319. [Google Scholar] [CrossRef]
- Bulut, U.; Crivello, J.V. Reactivity of oxetane monomers in photoinitiated cationic polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 3205–3220. [Google Scholar] [CrossRef]
- Falk, B.; Zonca, M.R.; Crivello, J.V. Photoactivated Cationic Frontal Polymerization. Macromol. Symp. 2005, 226, 97–107. [Google Scholar] [CrossRef]
- Crivello, J.V. Design and synthesis of multifunctional glycidyl ethers that undergo frontal polymerization. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6435–6448. [Google Scholar] [CrossRef]
- Crivello, J.V.; Bulut, U. Dual photo- and thermally initiated cationic polymerization of epoxy monomers. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6750–6764. [Google Scholar] [CrossRef]
- Crivello, J.V. Effect of Temperature on the Cationic Photopolymerization of Epoxides. J. Macromol. Sci. Part A 2008, 45, 591–598. [Google Scholar] [CrossRef]
- Sangermano, M.; Antonazzo, I.; Sisca, L.; Carello, M. Photoinduced cationic frontal polymerization of epoxy–carbon fibre composites. Polym. Int. 2019, 68, 1662–1665. [Google Scholar] [CrossRef] [Green Version]
- Crivello, J.V. Hybrid free radical/cationic frontal photopolymerizations. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4331–4340. [Google Scholar] [CrossRef]
- Crivello, J.V. Synergistic effects in hybrid free radical/cationic photopolymerizations. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 3759–3769. [Google Scholar] [CrossRef]
- Crivello, J.V. Hybrid acrylate-oxetane photopolymerizable systems. J. Polym. Sci. Part A Polym. Chem. 2014, 53, 594–601. [Google Scholar] [CrossRef]
- Ryu, C.Y.; Spencer, M.J.; Crivello, J.V. Involvement of Supramolecular Complexes in the Capture and Release of Protonic Acids During the Cationic Ring-Opening Polymerization of Epoxides. Macromolecules 2012, 45, 2233–2241. [Google Scholar] [CrossRef]
- Crivello, J.V.; Narayan, R.; Sternstein, S. Fabrication and mechanical characterization of glass fiber reinforced UV-cured composites from epoxidized vegetable oils. J. Appl. Polym. Sci. 1997, 64, 2073–2087. [Google Scholar] [CrossRef]
- Klikovits, N.; Liska, R.; D’Anna, A.; Sangermano, M. Successful UV-Induced RICFP of Epoxy-Composites. Macromol. Chem. Phys. 2017, 218, 1700313. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.D.; Yourdkhani, M.; Centellas, P.J.; Aw, J.E.; Ivanoff, D.G.; Goli, E.; Lloyd, E.M.; Dean, L.M.; Sottos, N.R.; Geubelle, P.; et al. Rapid energy-efficient manufacturing of polymers and composites via frontal polymerization. Nature 2018, 557, 223–227. [Google Scholar] [CrossRef]
- Crivello, J.V.; Yang, B. Synthesis and Photoinitiated Cationic Polymerization of Epoxidized Elastomers. J. Macromol. Sci. Part A 1994, 31, 517–533. [Google Scholar] [CrossRef]
- Aerospace Sciences Meetings. 43rd AIAA Aerospace Sciences Meeting and Exhibit; Pojman, J.A., Ed.; American Institute of Aeronautics and Astronautics, Inc.: Reston, VA, USA, 2005. [Google Scholar]
- Briskman, V.A.; Kostarev, K.G.; Yudina, T.M.; Kondyurin, A.V.; Leontyev, V.B.; Levkovich, M.G.; Mashinsky, A.L.; Nechitailo, G.S. Polymerization in microgravity as a new process in space technology. Acta Astronaut. 2001, 48, 169–180. [Google Scholar] [CrossRef]
- Pojman, J.; Ainsworth, W.; Chekanov, Y.; Masere, J.; Volpert, V.; Dumont, T.; Wilke, H. Bubble behavior and convection in frontal polymerization on the KC-135 aircraft. In Proceedings of the 38th Aerospace Sciences Meeting and Exhibit, Reno, NV, USA, 10–13 January 2000; American Institute of Aeronautics and Astronautics: Reston, VA, USA, 2000; p. 01102000. [Google Scholar]
- Connell, J.W.; Crivello, J.V.; Bi, D. Effect of low earth orbit atomic oxygen exposure on epoxy functionalized siloxanes. J. Appl. Polym. Sci. 1995, 57, 1251–1259. [Google Scholar] [CrossRef]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | Irrad. Time (min) | Intensity (mJ/cm2min) | Tf (oC) | tf (s) | Vf (cm/min) |
|---|---|---|---|---|---|
 | 2.5 | 439 | 180 | 35 | 7.2 |
 | 6 | 413 | 232 | 22 | 11 |
 | 5 | 483 | 169 | 14 | 18 |
 | 8 | 575 | 203 | 6 | 20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, M.S.; Schlögl, S.; Wolfahrt, M.; Sangermano, M. Review on UV-Induced Cationic Frontal Polymerization of Epoxy Monomers. Polymers 2020, 12, 2146. https://doi.org/10.3390/polym12092146
Malik MS, Schlögl S, Wolfahrt M, Sangermano M. Review on UV-Induced Cationic Frontal Polymerization of Epoxy Monomers. Polymers. 2020; 12(9):2146. https://doi.org/10.3390/polym12092146
Chicago/Turabian StyleMalik, Muhammad Salman, Sandra Schlögl, Markus Wolfahrt, and Marco Sangermano. 2020. "Review on UV-Induced Cationic Frontal Polymerization of Epoxy Monomers" Polymers 12, no. 9: 2146. https://doi.org/10.3390/polym12092146
APA StyleMalik, M. S., Schlögl, S., Wolfahrt, M., & Sangermano, M. (2020). Review on UV-Induced Cationic Frontal Polymerization of Epoxy Monomers. Polymers, 12(9), 2146. https://doi.org/10.3390/polym12092146