In Vitro and In Vivo Biosafety Analysis of Resorbable Polyglycolic Acid-Polylactic Acid Block Copolymer Composites for Spinal Fixation


 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Experimental
2.1. Materials
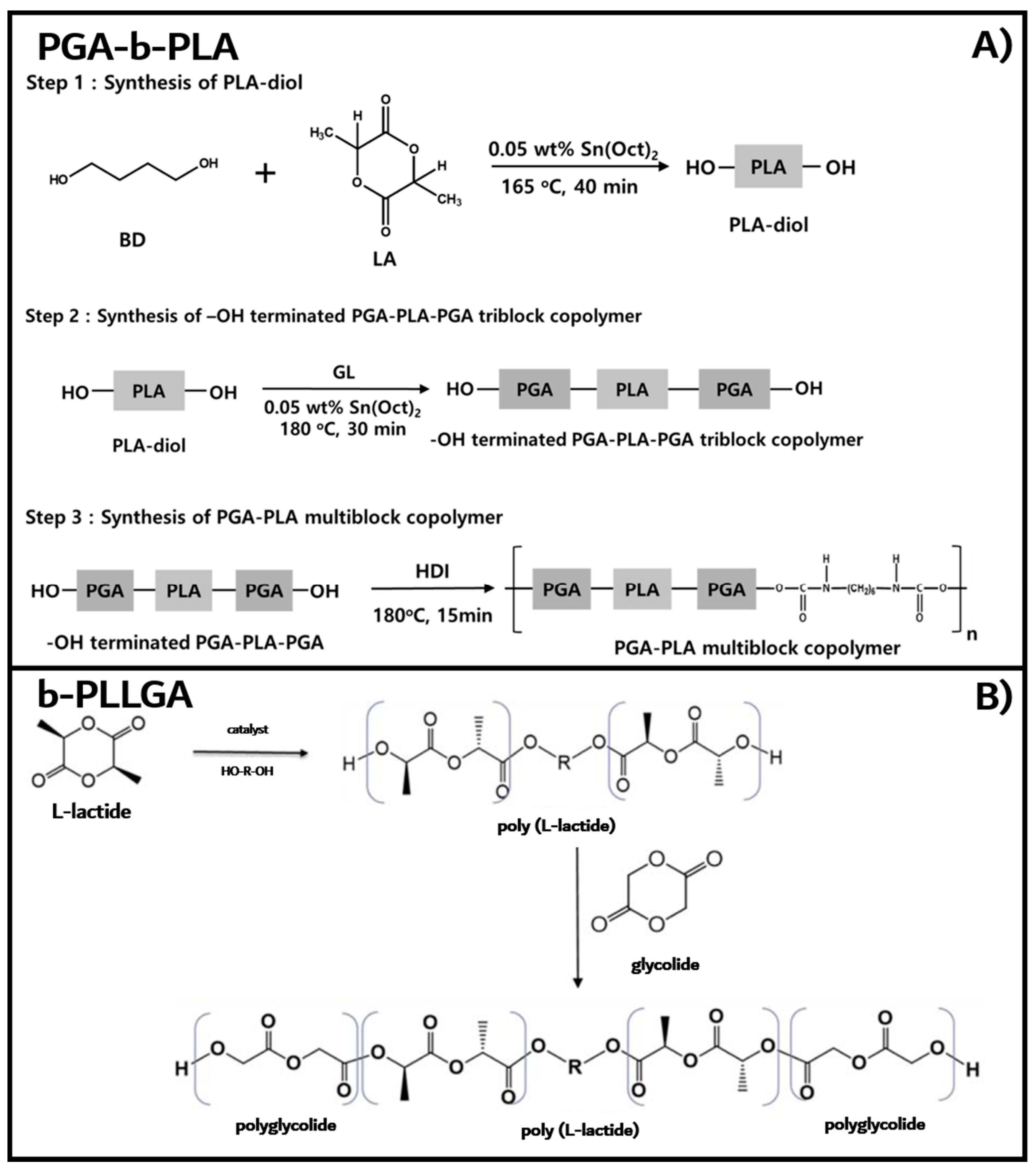
2.1.1. PGA–PLGA Multiblock Copolymer (PGA–b-PLA)
2.1.2. PGA–PLA Triblock Copolymer (b-PLLGA)
2.1.3. PGA–PLA Block Copolymer Composites
2.1.4. In Vitro Cytotoxicity Test
2.2. Experimental
2.2.1. Synthesis of Poly(L-lactic acid)-b-poly(glycolic acid) Multiblock Copolymer (PGA–b-PLA)
2.2.2. Synthesis of Poly(L-lactic acid)–b-poly(glycolic acid) Triblock Copolymers (b-PLLGA)
2.2.3. PGA–PLA Block Copolymer Composites
2.2.4. Biological Responses by In Vitro Cytotoxicity Tests
2.2.5. Biological Responses by In Vivo Animal Biosafety Tests
2.2.6. In Vivo Degradation Test and Histochemical Analysis
3. Results and Discussion
3.1. Synthesis of PGA–b-PLA, b-PLLGA, and Their Composites
3.2. In Vitro Cytotoxicity Test
3.3. In Vivo Animal Biosafety Tests
3.4. In Vivo Degradation Test and Histochemical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hughes, T.B. Bioabsorbable Implants in the Treatment of Hand Fractures. Clin. Orthop. Relat. Res. 2006, 445, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Waris, E.; Konttinen, Y.T.; Ashammakhi, N.; Suuronen, R.; Santavirta, S. Bioabsorbable fixation devices in trauma and bone surgery: Current clinical standing. Expert Rev. Med. Devices 2004, 1, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Viljanen, J.; Kinnunen, J.; Bondestam, S.; Majola, A.; Rokkanen, P.; Tormala, P. Bone changes after experimental osteotomies fixed with absorbable self-reinforced poly-L-lactide screws or metallic screws studied by plain radiographs, quantitative computed tomography and magnetic resonance imaging. Biomaterials 1995, 16, 1353–1358. [Google Scholar] [CrossRef]
- Litsky, A.S. Clinical Reviews: Bioabsorbable Implants for Orthopaedic Fracture Fixation. J. Appl. Biomater. 1993, 4, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, W.S. Principles of Development and SUse of Absorbable Internal Fixation. Tissue Eng. 2000, 6, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, P.; Bohm, H.; Driemel, O.; Muhling, J.; Pistner, H. Resorbable versus titanium osteosynthesis devices in bilateral sagittal split ramus osteotomy of the mandible—The results of a two centre randomized clinical study with an eight-year follow-up. J. Craniomaxillofac. Surg. 2010, 38, 522–528. [Google Scholar] [CrossRef]
- Mittal, R.; Morley, J.; Dinopoulos, H.; Drakoulakis, E.G.; Vermani, E.; Giannoudis, P.V. Use of bio-resorbable implants for stabilization of distal radius fractures: The United Kingdom patients’ perspective. Int. J. Care Inj. 2005, 36, 333–338. [Google Scholar] [CrossRef]
- Pistner, H.; Bend, D.R.; Mühling, J.; Reuther, J.F. Poly(L-lactide): A long-term degradation study in vivo PartIII. Analytical characterization. Biomaterials 1993, 14, 291–298. [Google Scholar] [CrossRef]
- De Jong, W.H.; Bersgma, J.E.; Robinson, J.E.; Bos, R.R.M. Tissue response to partially in vitro predegraded poly-L-lactide implants. Biomaterials 2005, 26, 1781–1791. [Google Scholar] [CrossRef]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar] [CrossRef]
- Fargnoli, A.S.; Mu, A.; Katz, M.G.; Williams, R.D.; Margulies, K.B.; Weiner, D.B.; Yang, S.; Bridges, C.R. Anti-inflammatory loaded poly-lactic glycolic acid nanoparticle formulations to enhance myocardial gene transfer: An in-vitro assessment of a drug/gene combination therapeutic approach for direct injection. J. Transl. Med. 2014, 12, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.J.; Meredith, C.; Johnson, C.; Galis, Z.S. The effect of scaffold degradation rate on three-dimensional cell growth and angiogenesis. Biomaterials 2004, 25, 5735–5742. [Google Scholar] [CrossRef] [PubMed]
- Yetkin, H.; Senköylü, A.; Cila, E.; Öztürk, A.M.; Simsek, A. Biodegradable Implants in Orthopaedics and Traumatology. Turk. J. Med. Sci. 2000, 30, 297–301. [Google Scholar]
- Peponi, L.; Navarro-Baena, I.; Sonseca, A.; Gimenez, E.; Marcos-Fernandez, A.; Kenny, J.M. Synthesis and characterization of PCL–PLLA polyurethane with shape memory behavior. Eur. Polym. J. 2013, 49, 893–903. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.T.; Panthani, T.R.; Bates, F.S. Sustainable Poly(lactide-b-butadiene) Multiblock Copolymers with Enhanced Mechanical Properties. Macromolecules 2013, 46, 7387–7398. [Google Scholar] [CrossRef]
- Ayyoob, M.; Lee, S.M.; Kim, Y.J. Well-defined high molecular weight polyglycolide-b-poly(L-)lactide-b-polyglycolide triblock copolymers: Synthesis, characterization and microstructural analysis. J. Polym. Res. 2020, 27, 109–120. [Google Scholar] [CrossRef]
- Hong, Z.; Qiu, X.; Sun, J.; Deng, M.; Chen, X.; Jing, X. Grafting polymerization of L-lactide on the surface of hydroxyapatite nano-crystals. Polymer 2004, 45, 6699–6706. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer Molecular Weight Analysis by 1H NMR Spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Gao, Q.; Lan, P.; Shao, H.; Hu, X. Direct Synthesis with Melt Polycondensation and Microstructure Analysis of Poly(L-lactic acid-co-glycolic acid). Polym. J. 2002, 34, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Ayyoob, M.; Yang, X.; Park, H.J.; Park, S.Y.; Kim, J.H.; Nam, S.W.; Kim, Y.J. Synthesis of Bioresorbable Poly(Lactic-co-Glycolic Acid)s Through Direct Polycondensation: An Economical Substitute for the Synthesis of Polyglactin via ROP of Lactide and Glycolide. Fiber Polym. 2019, 20, 887–895. [Google Scholar] [CrossRef]
- Yoon, S.K.; Chung, D.J. Development of Blood Compatible Composite Using MPC Copolymer and Polyolefin for Non-PVC Blood Bag Application. Macromol. Res. 2020, 28, 319–326. [Google Scholar] [CrossRef]
- Song, Y.J.; Chon, J.W.; Yoo, H.; Kim, J.H.; Chung, D.J. Cytotoxicity and In vivo Biosafety Studies of the Poly(alkylphenol) Derivatives as Vulcanizing Agents. Macromol. Res. 2019, 27, 1081–1088. [Google Scholar] [CrossRef]
- Chon, J.W.; Jang, I.K.; Bae, K.W.; Suh, S.W.; Chung, I.K.; Song, Y.J.; Kim, J.H.; Chung, D.J. Cytotoxicity and In vivo Biosafety of the Polylactide Based Bone Composite Materials. Macromol. Res. 2017, 25, 648–655. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Score for Skin Sensitization Test (Magnussin and Kilgman) | Grade for Skin Sensitization Test (Magnussin and Kilgman) | |||
|---|---|---|---|---|
| Standard | Score | Sensitization Rate (%) | Grade | Class |
| No reaction | 0 | 0~8 | I | Weak |
| Scattered mild redness | 1 | 9~28 | II | Mild |
| Moderate and diffuse redness | 2 | 29~64 | III | Moderate |
| Intense redness and swelling | 3 | 65~80 | IV | Strong |
| 81~100 | V | Extreme | ||
| Position | δ (ppm) | Multiplicity | Peak Area | |
|---|---|---|---|---|
| a | –CH2 of 1,4-butanediol | 1.82 | multiplet | 0.32 |
| b | –CH2 of 1,4-butanediol | 4.34 | multiplet | 0.32 |
| c | –CH of PLA | 5.35 | quartet | 0.96 |
| d | –CH3 of PLA | 1.66 | doublet | 3.0 |
| e | –CH2 of PGA | 5.02 | singlet | 0.59 |
| f | –NHCOO– of the urethane bond | 3.23 | quartet | 0.32 |
| Sample Name | Tensile Strength (MPa) |
|---|---|
| PGA–b-PLA | 82.605 |
| b-PLLGA | 41.365 |
| PGA–b-PLA composite | 113.28 |
| b-PLLGA composite | 104.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.K.; Yang, J.H.; Lim, H.T.; Chang, Y.-W.; Ayyoob, M.; Yang, X.; Kim, Y.J.; Ko, H.-S.; Jho, J.Y.; Chung, D.J. In Vitro and In Vivo Biosafety Analysis of Resorbable Polyglycolic Acid-Polylactic Acid Block Copolymer Composites for Spinal Fixation. Polymers 2021, 13, 29. https://doi.org/10.3390/polym13010029
Yoon SK, Yang JH, Lim HT, Chang Y-W, Ayyoob M, Yang X, Kim YJ, Ko H-S, Jho JY, Chung DJ. In Vitro and In Vivo Biosafety Analysis of Resorbable Polyglycolic Acid-Polylactic Acid Block Copolymer Composites for Spinal Fixation. Polymers. 2021; 13(1):29. https://doi.org/10.3390/polym13010029
Chicago/Turabian StyleYoon, Seung Kyun, Jin Ho Yang, Hyun Tae Lim, Young-Wook Chang, Muhammad Ayyoob, Xin Yang, Young Jun Kim, Han-Seung Ko, Jae Young Jho, and Dong June Chung. 2021. "In Vitro and In Vivo Biosafety Analysis of Resorbable Polyglycolic Acid-Polylactic Acid Block Copolymer Composites for Spinal Fixation" Polymers 13, no. 1: 29. https://doi.org/10.3390/polym13010029
APA StyleYoon, S. K., Yang, J. H., Lim, H. T., Chang, Y. -W., Ayyoob, M., Yang, X., Kim, Y. J., Ko, H. -S., Jho, J. Y., & Chung, D. J. (2021). In Vitro and In Vivo Biosafety Analysis of Resorbable Polyglycolic Acid-Polylactic Acid Block Copolymer Composites for Spinal Fixation. Polymers, 13(1), 29. https://doi.org/10.3390/polym13010029