
Determining the Optimal Conditions for the Production by Supercritical CO2 of Biodegradable PLGA Foams for the Controlled Release of Rutin as a Medical Treatment

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
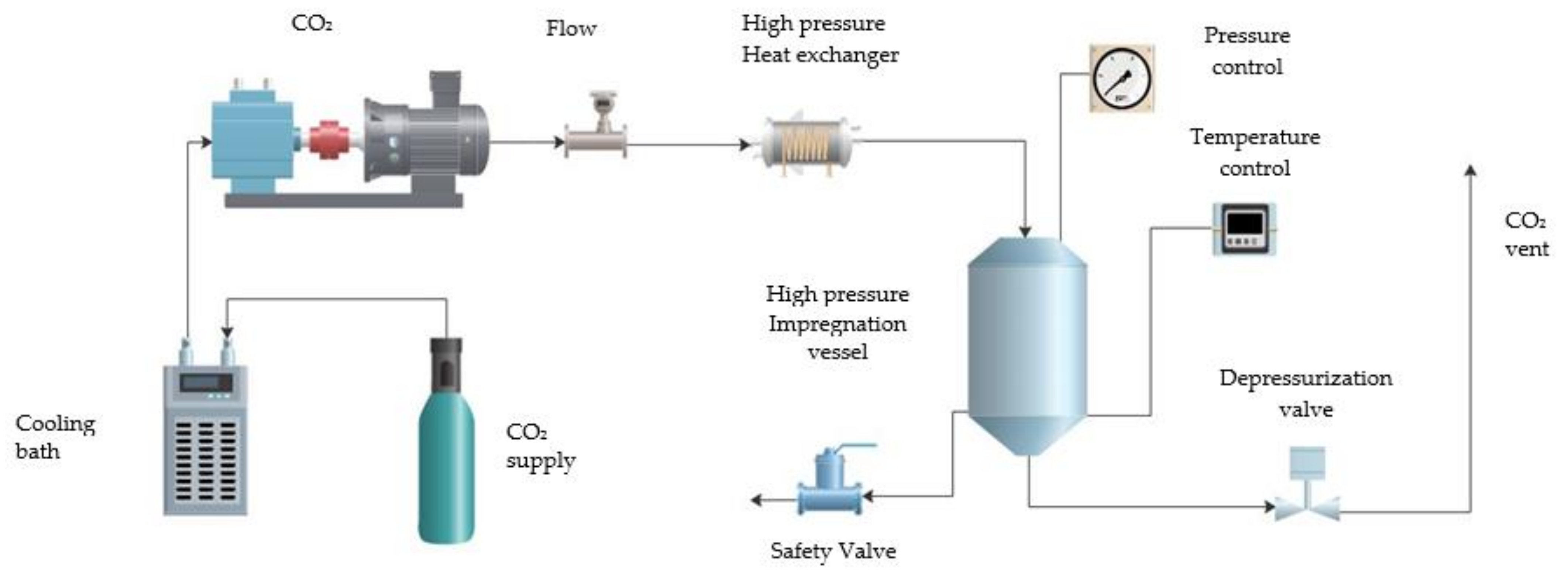
2.2. Foaming and Impregnation Process
2.3. Thermal Analysis
2.4. Mercury Intrusion Porosimetry
2.5. Scanning Electron Microscopy
2.6. In Vitro Release Test
3. Results and Discussion
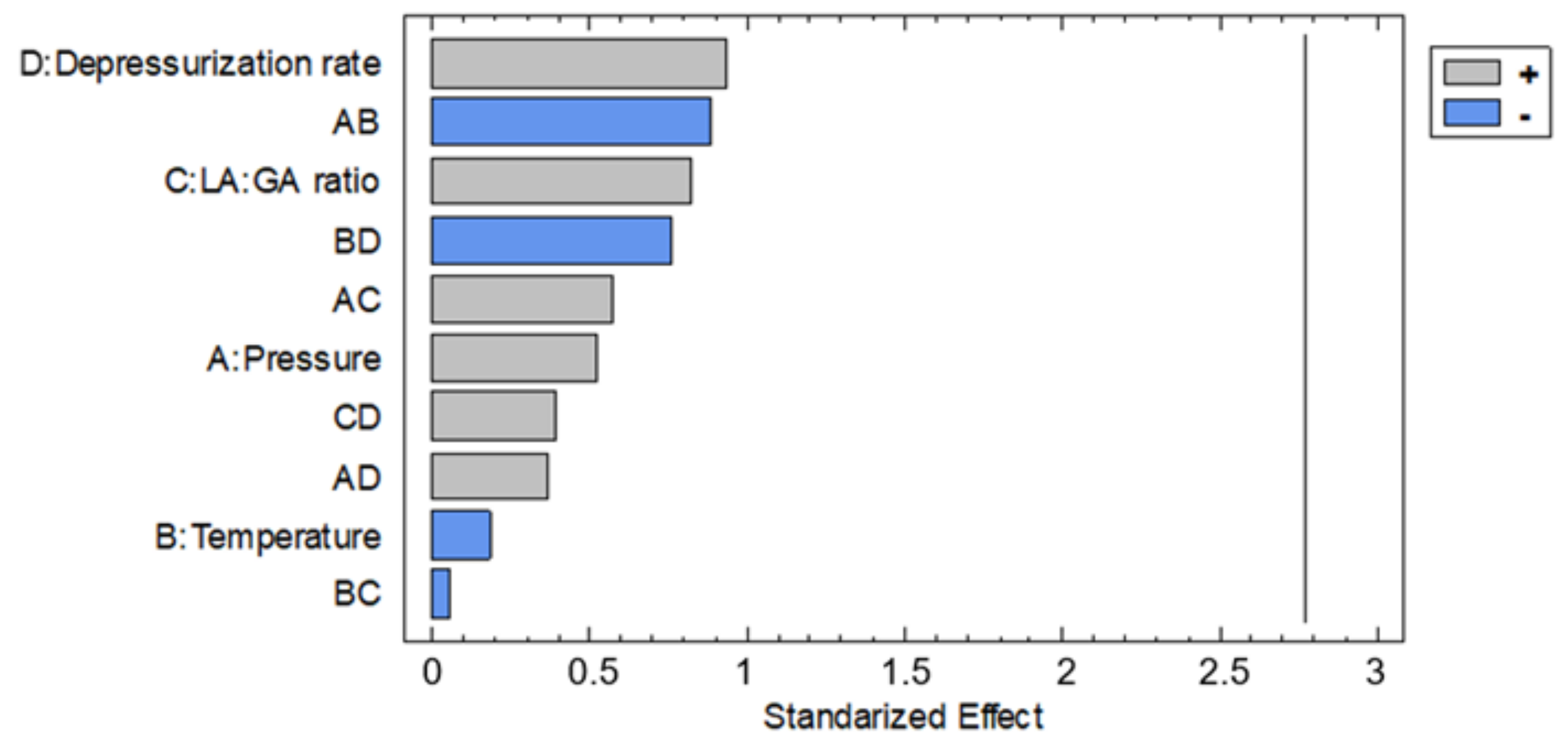
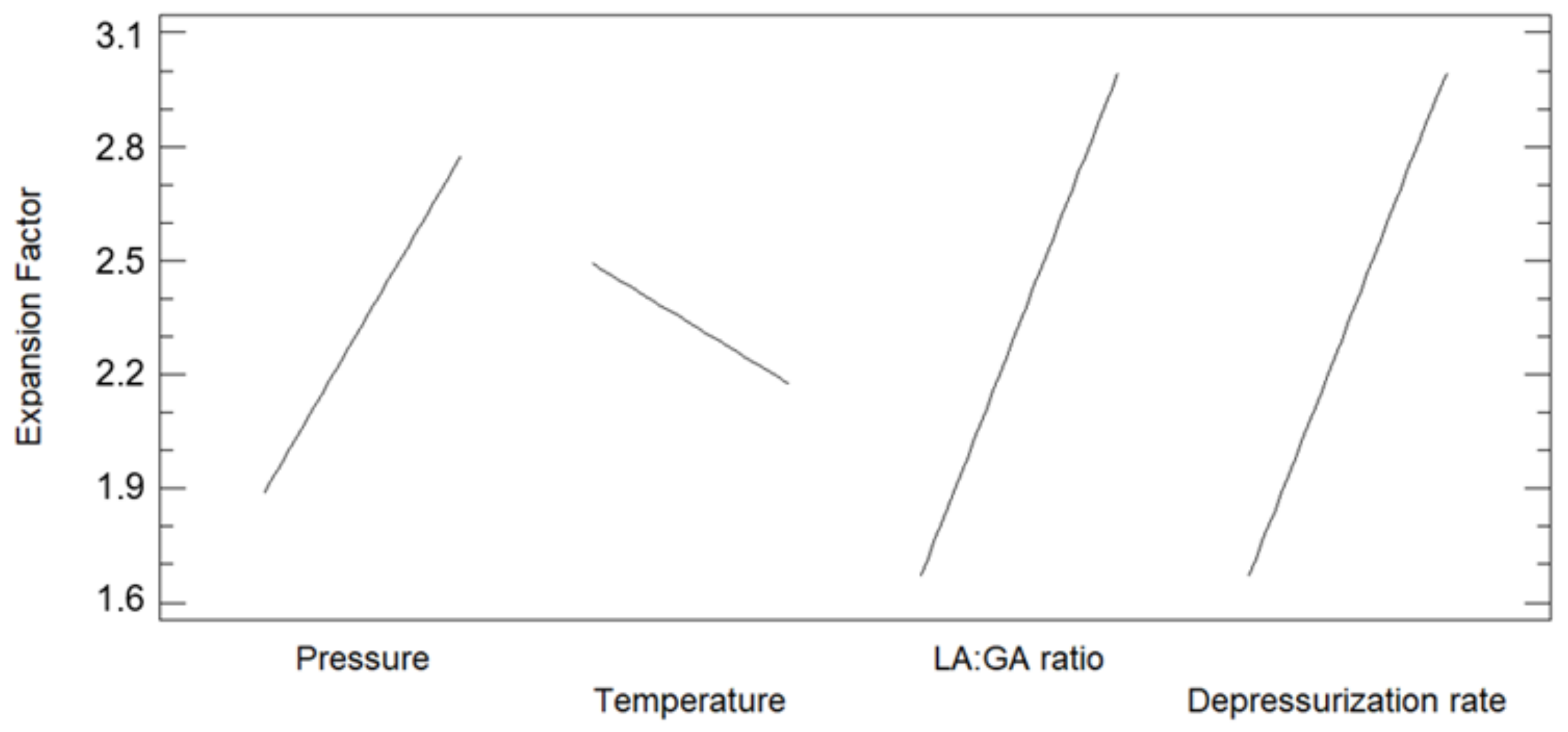
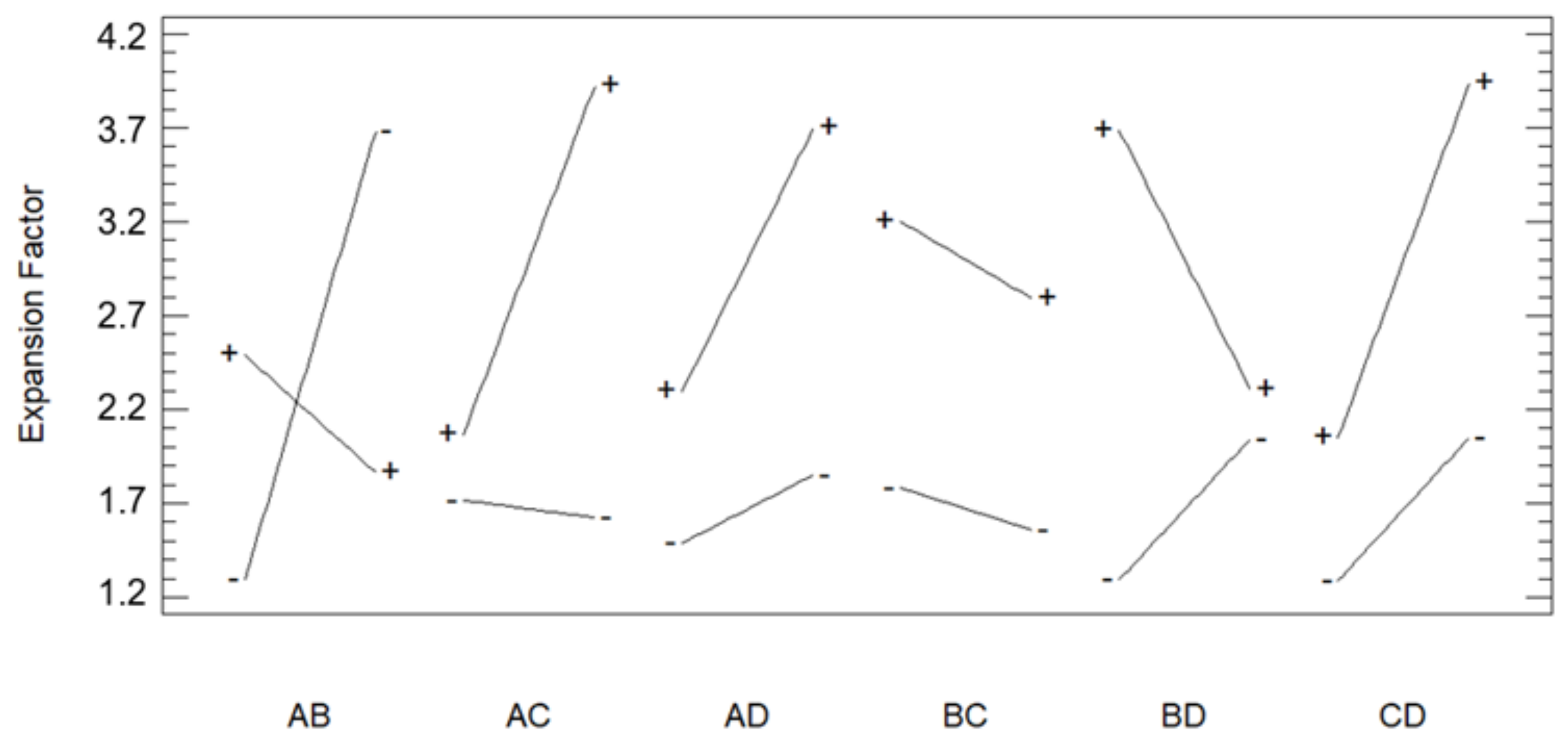
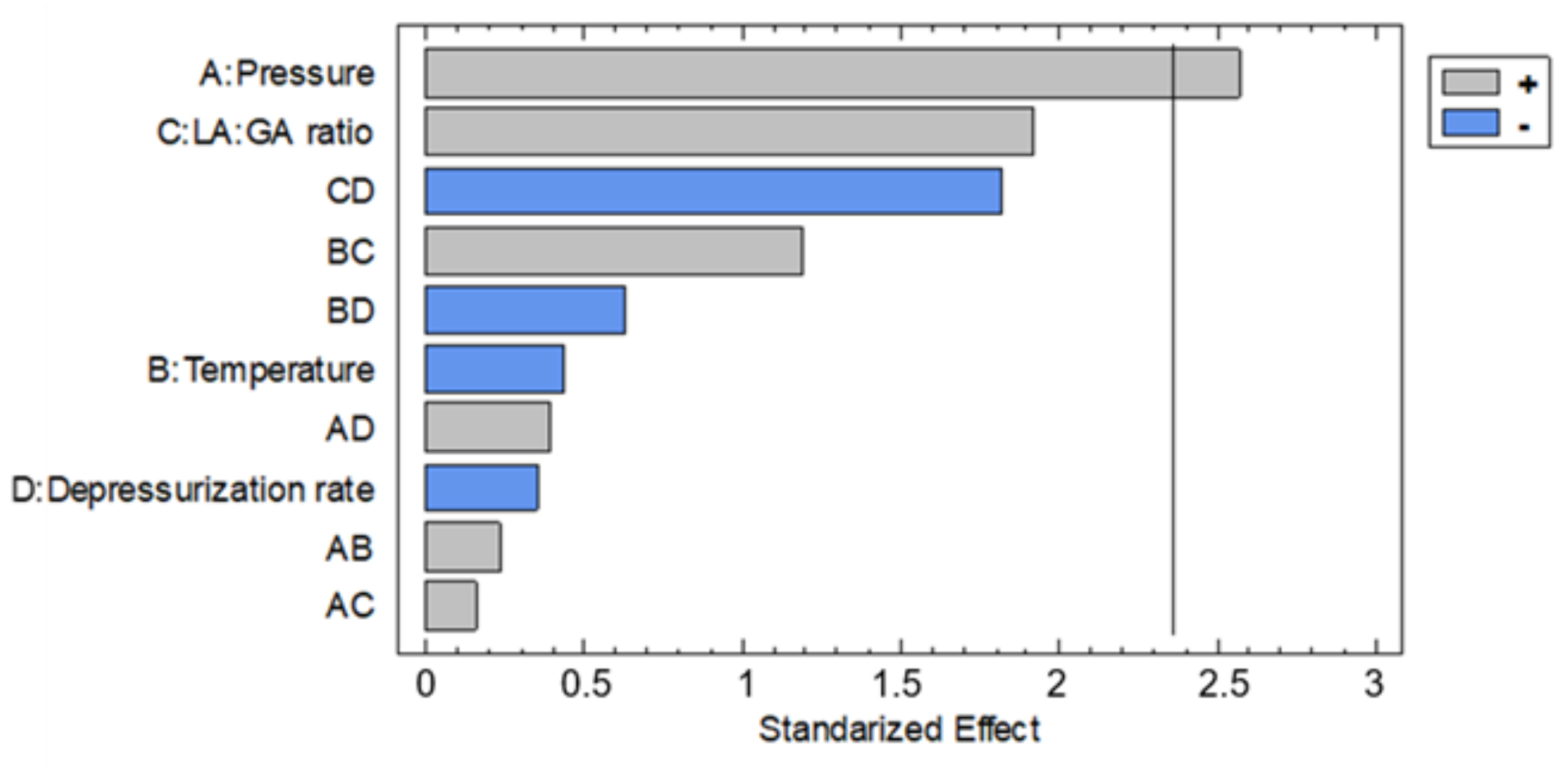
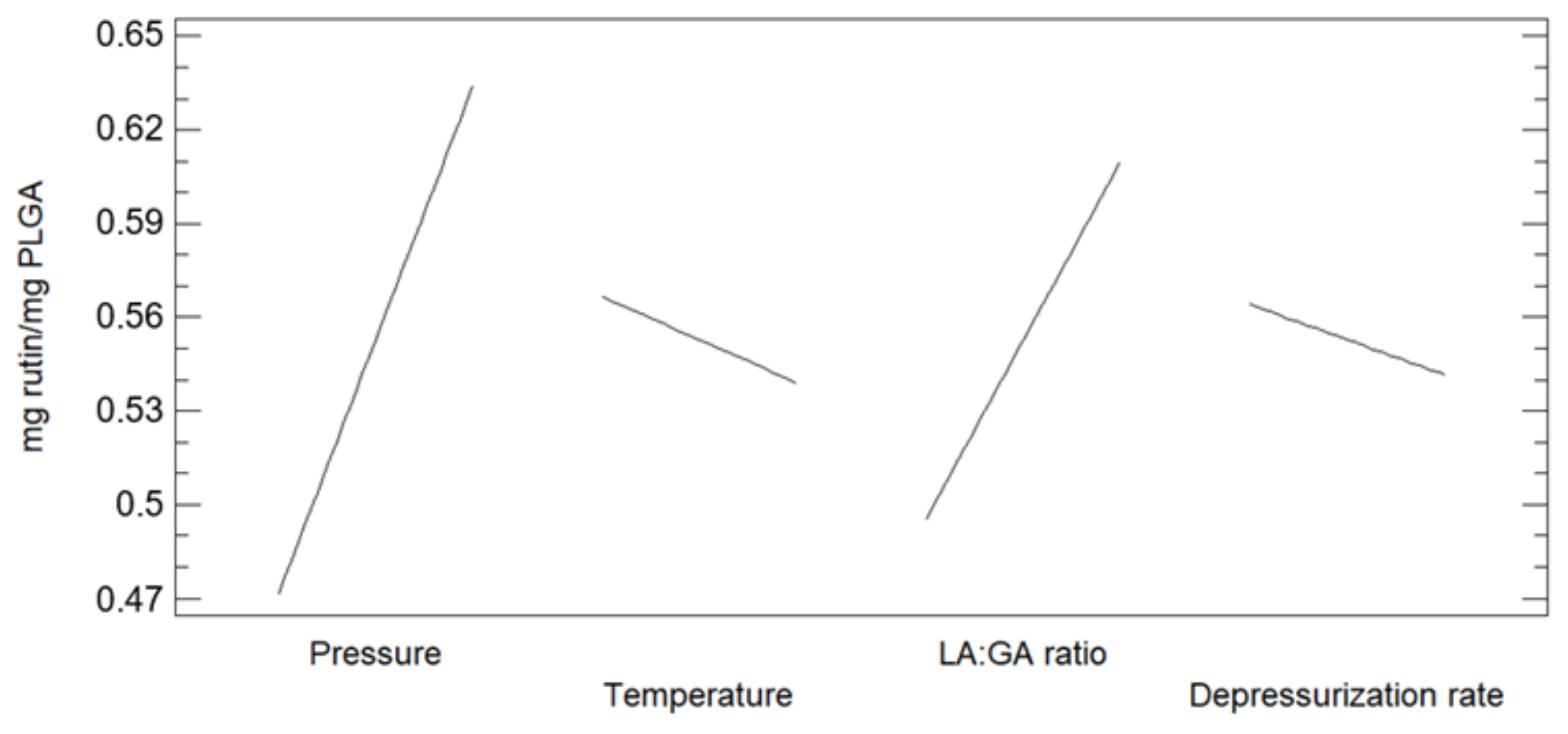
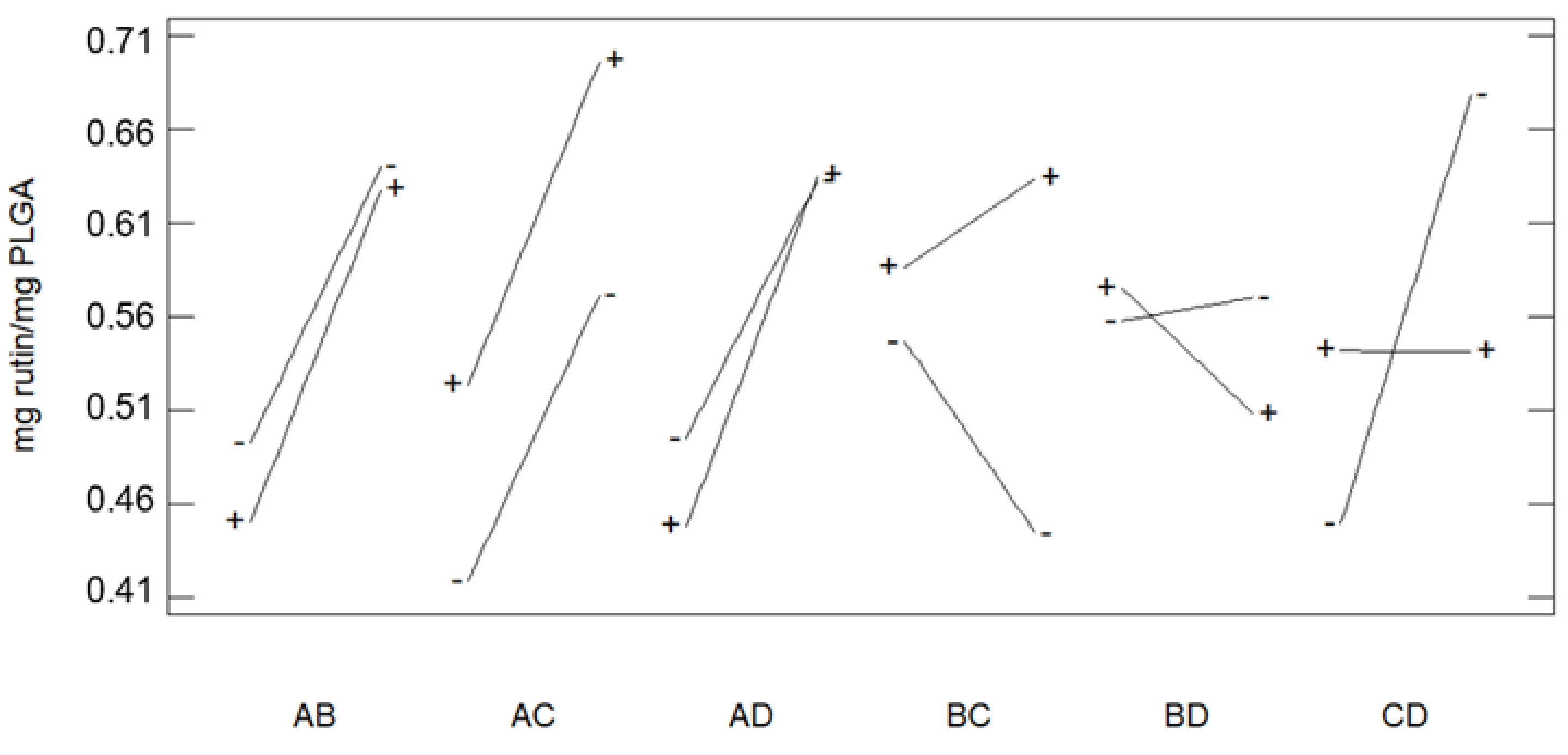
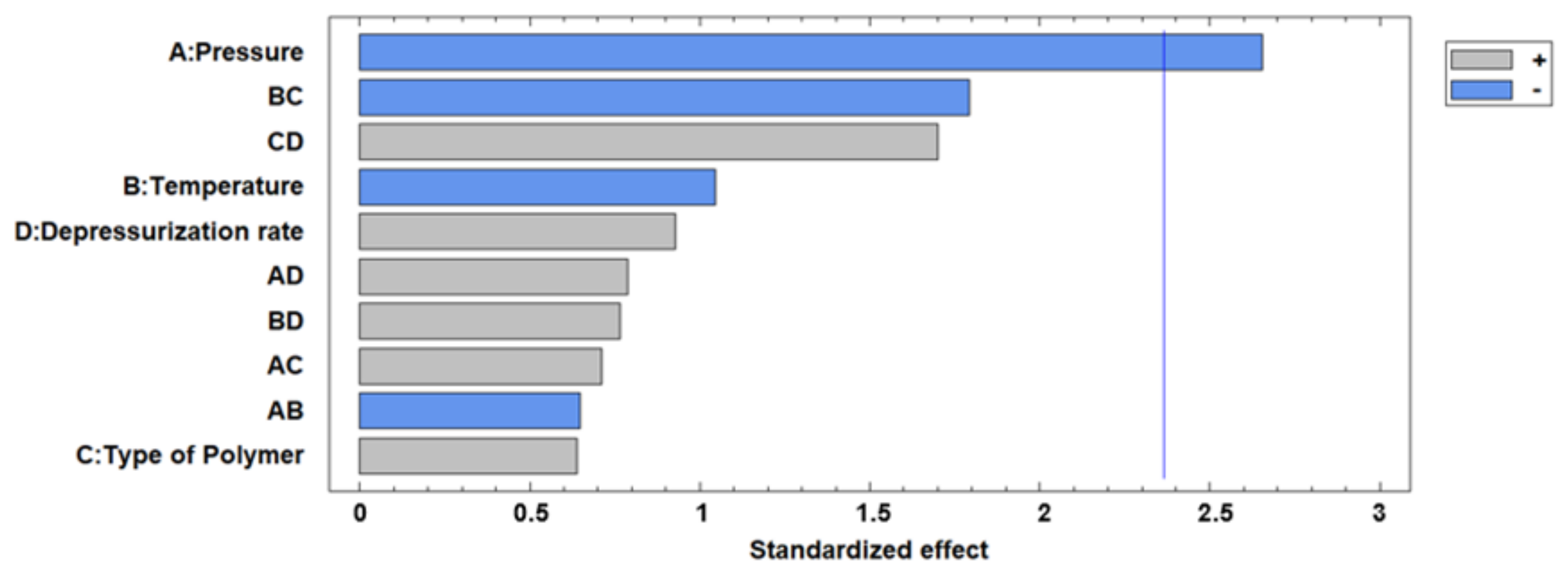
3.1. Foaming and Impregnation Runs
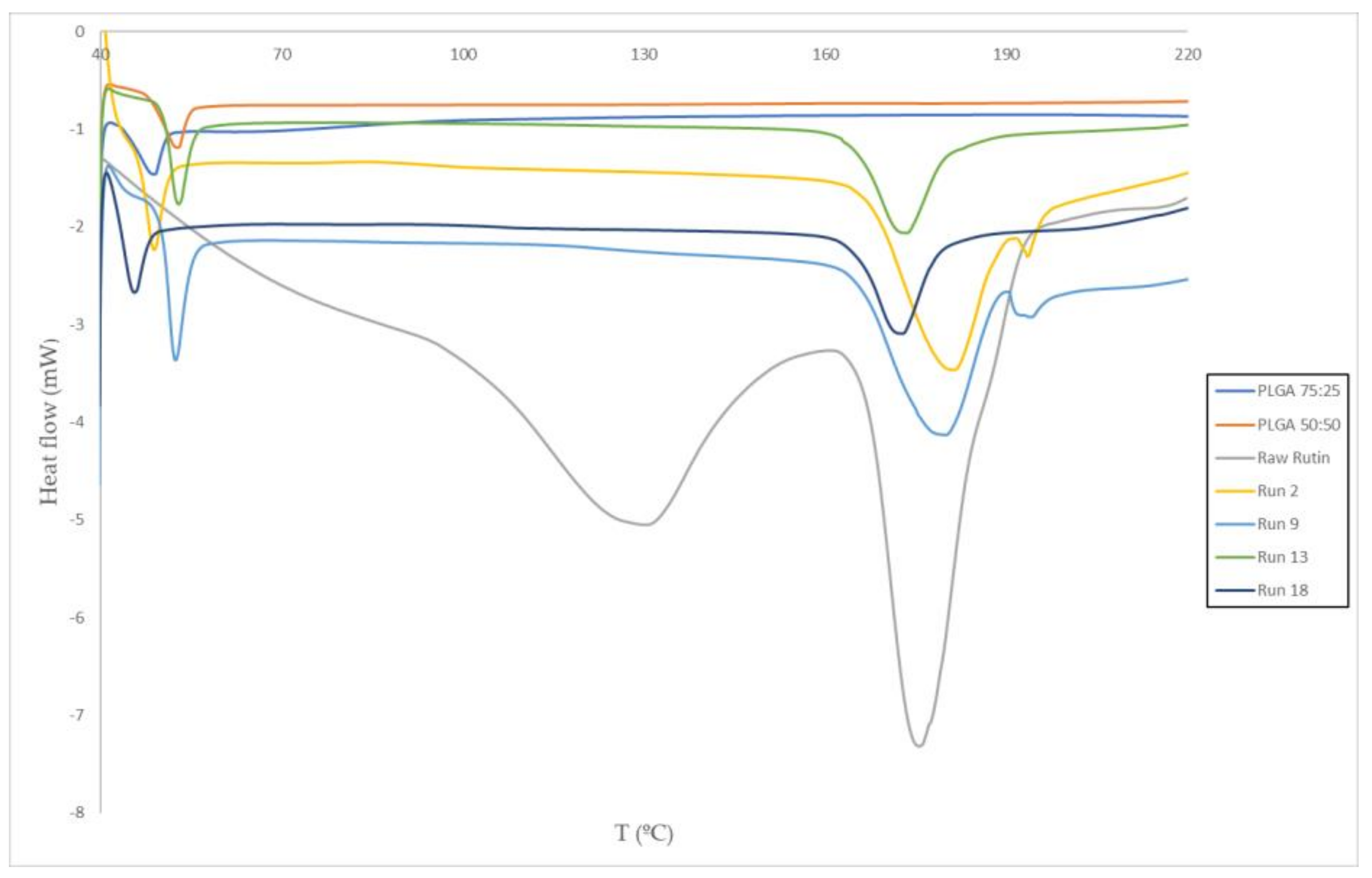
3.2. Thermal Analysis
3.3. Scanning Electron Microscopy (SEM)
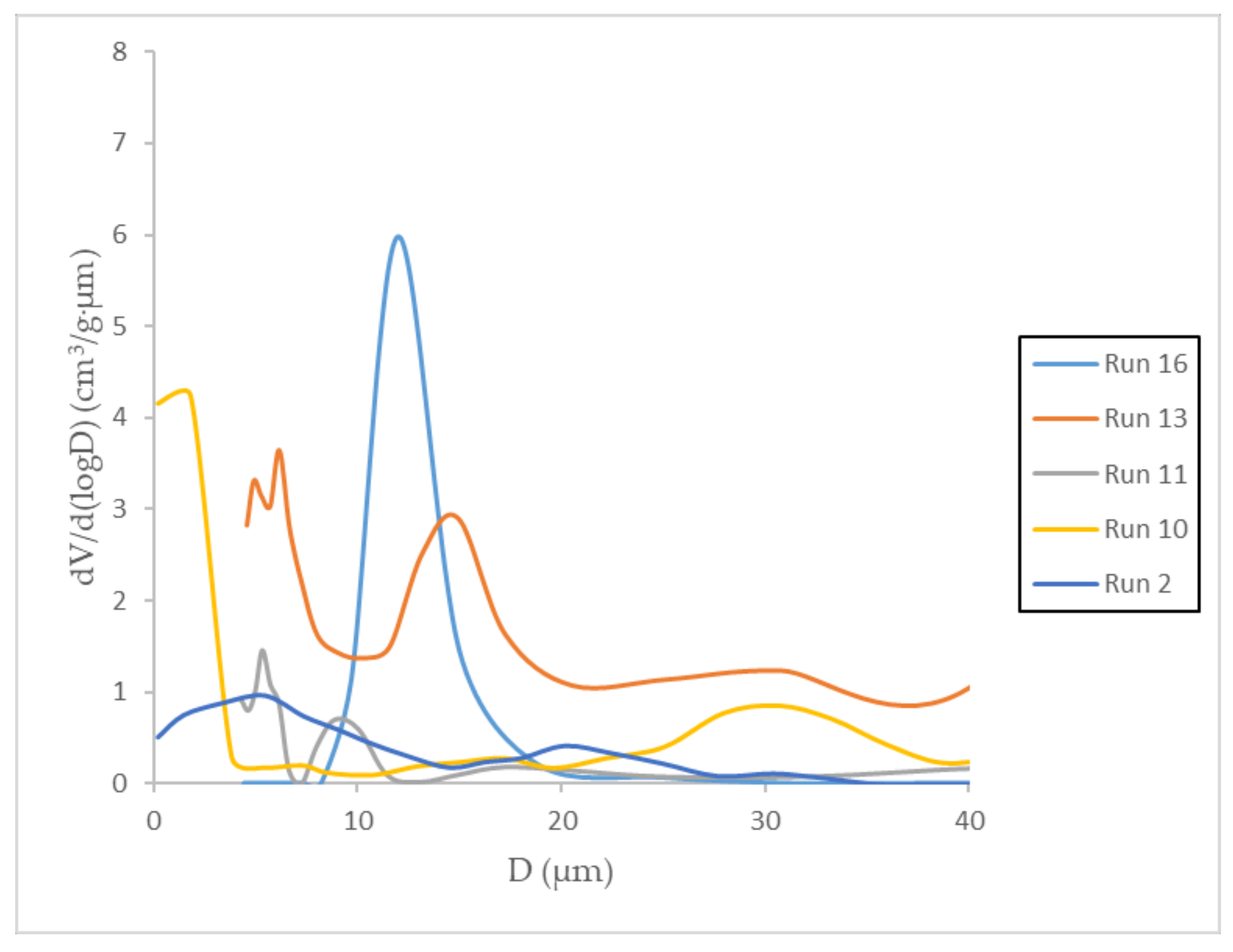
3.4. Mercury Intrusion Porosimetry
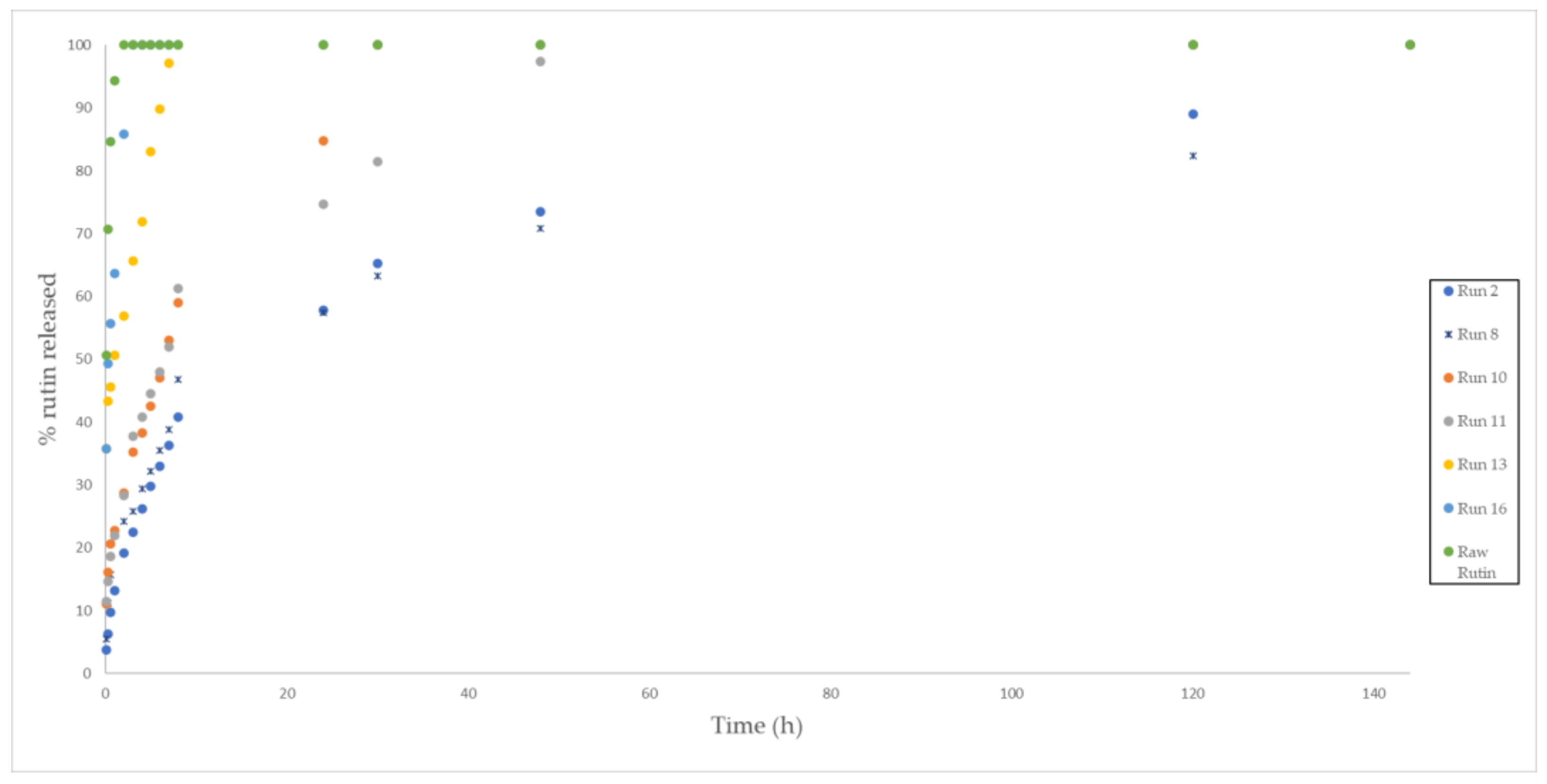
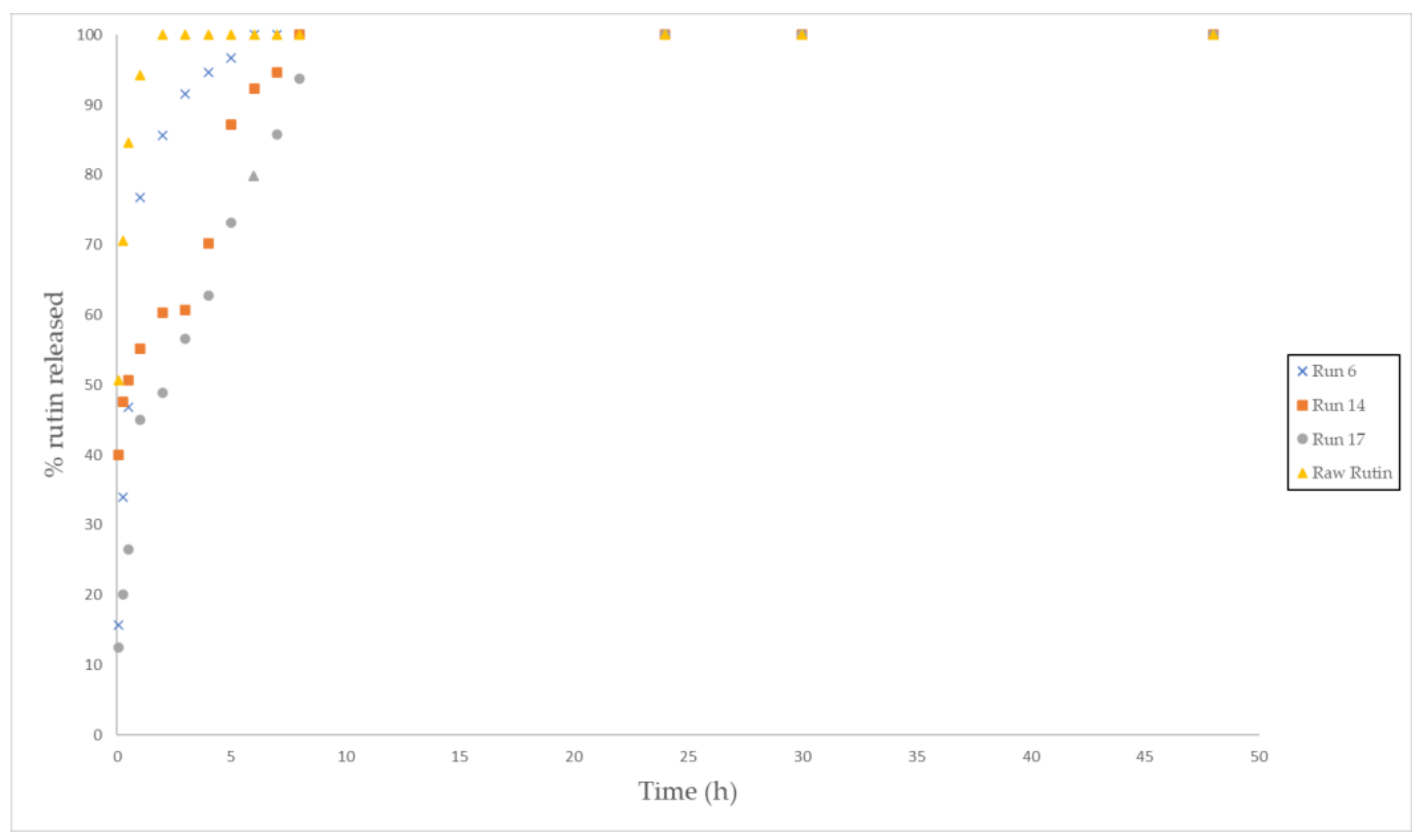
3.5. In Vitro Release Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sauceau, M.; Fages, J.; Common, A.; Nikitine, C.; Rodier, E. New challenges in polymer foaming: A review of extrusion processes assisted by supercritical carbon dioxide. Prog. Polym. Sci. 2011, 36, 749–766. [Google Scholar] [CrossRef] [Green Version]
- Kuang, T.; Chen, F.; Chang, L.; Zhao, Y.; Fu, D.; Gong, X.; Peng, X. Facile preparation of open-cellular porous poly (l-lactic acid) scaffold by supercritical carbon dioxide foaming for potential tissue engineering applications. Chem. Eng. J. 2017, 307, 1017–1025. [Google Scholar] [CrossRef]
- Singh, A.; Banerjee, S.L.; Dhiman, V.; Bhadada, S.K.; Sarkar, P.; Khamrai, M.; Kumari, K.; Kundu, P.P. Fabrication of calcium hydroxyapatite incorporated polyurethane-graphene oxide nanocomposite porous scaffolds from poly (ethylene terephthalate) waste: A green route toward bone tissue engineering. Polymers 2020, 195, 122436. [Google Scholar] [CrossRef]
- Sola, A.; Bertacchini, J.; D’Avella, D.; Anselmi, L.; Maraldi, T.; Marmiroli, S.; Messori, M. Development of solvent-casting particulate leaching (SCPL) polymer scaffolds as improved three-dimensional supports to mimic the bone marrow niche. Mater. Sci. Eng. C 2019, 96, 153–165. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, M.C.; Dul, S.; Barra, G.M.O.; Pegoretti, A. Poly(vinylidene fluoride)/thermoplastic polyurethane flexible and 3D printable conductive composites. J. Appl. Polym. Sci. 2021, 138, 1–15. [Google Scholar] [CrossRef]
- Loh, G.H.; Sotayo, A.; Pei, E. Development and testing of material extrusion additive manufactured polymer–textile composites. Fash. Text. 2021, 8, 1–21. [Google Scholar] [CrossRef]
- Hernandez, J.H.M. Effect of the Incorporation of Polycaprolactone (PCL) on the Retrogradation of Binary Blends with Cassava Thermoplastic Starch (TPS). Polymers 2020, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Obaidat, R.; Alnaief, M.; Jaeger, P. Significant solubility of carbon dioxide in Soluplus® facilitates impregnation of ibuprofen using supercritical fluid technology. Pharm. Dev. Technol. 2017, 23, 697–705. [Google Scholar] [CrossRef]
- Duarte, A.R.C.; Mano, J.F.; Reis, R.L. Dexamethasone-loaded scaffolds prepared by supercritical-assisted phase inversion. Acta Biomater. 2009, 5, 2054–2062. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.K.; Beckman, E.J. Generation of microcellular polymeric foams using supercritical carbon dioxide. I: Effect of pressure and temperature on nucleation. Polym. Eng. Sci. 1994, 34, 1137–1147. [Google Scholar] [CrossRef]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of factors influencing the hydrolytic degradation of single PLGA microparticles. Polym. Degrad. Stab. 2015, 119, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Jamshidi, K.; Hyon, S.-H.; Ikada, Y. Thermal characterization of polylactides. Polymers 1988, 29, 2229–2234. [Google Scholar] [CrossRef]
- Blasi, P. Poly(lactic acid)/poly(lactic-co-glycolic acid)-based microparticles: An overview. J. Pharm. Investig. 2019, 49, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Singh, G.; Kaur, T.; Kaur, R.; Kaur, A. Recent biomedical applications and patents on biodegradable polymer-PLGA. Int. J. Pharmacol. Pharm. Sci. 2014, 1, 30–42. [Google Scholar]
- Chang, N.-J.; Lam, C.-F.; Lin, C.-C.; Chen, W.-L.; Li, C.-F.; Lin, Y.-T.; Yeh, M.-L. Transplantation of autologous endothelial progenitor cells in porous PLGA scaffolds create a microenvironment for the regeneration of hyaline cartilage in rabbits. Osteoarthr. Cartil. 2013, 21, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, I.; Gutiérrez, C.; Rodríguez, J.; De Lucas, A.; García, M. Production of drug-releasing biodegradable microporous scaffold impregnated with gemcitabine using a CO2 foaming process. J. CO2 Util. 2020, 41, 101227. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J.; Li, S.; Yang, S.; Lu, E.; Xi, Z.; Cen, L.; Zhao, L.; Yuan, W. Highly interconnected macroporous MBG/PLGA scaffolds with enhanced mechanical and biological properties via green foaming strategy. Chin. J. Chem. Eng. 2021, 29, 426–436. [Google Scholar] [CrossRef]
- Mohammadi, M.S.; Rezabeigi, E.; Bertram, J.; Marelli, B.; Gendron, R.; Nazhat, S.N.; Bureau, M.N. Poly(d,l-Lactic acid) Composite Foams Containing Phosphate Glass Particles Produced via Solid-State Foaming Using CO2 for Bone Tissue Engineering Applications. Polymers 2020, 12, 231. [Google Scholar] [CrossRef] [Green Version]
- Milovanovic, S.; Markovic, D.; Mrakovic, A.; Kuska, R.; Zizovic, I.; Frerich, S.; Ivanovic, J. Supercritical CO2-assisted production of PLA and PLGA foams for controlled thymol release. Mater. Sci. Eng. C 2019, 99, 394–404. [Google Scholar] [CrossRef]
- Li, S.; Song, C.; Yang, S.; Yu, W.; Zhang, W.; Zhang, G.; Xi, Z.; Lu, E. Supercritical CO2 foamed composite scaffolds incorporating bioactive lipids promote vascularized bone regeneration via Hif-1α upregulation and enhanced type H vessel formation. Acta Biomater. 2019, 94, 253–267. [Google Scholar] [CrossRef]
- Yoon, S.J.; Park, K.S.; Kim, M.S.; Rhee, J.M.; Khang, G.; Lee, H.B. Repair of Diaphyseal Bone Defects with Calcitriol-Loaded PLGA Scaffolds and Marrow Stromal Cells. Tissue Eng. 2007, 13, 1125–1133. [Google Scholar] [CrossRef]
- Jeon, O.; Song, S.J.; Kang, S.-W.; Putnam, A.J.; Kim, B.-S. Enhancement of ectopic bone formation by bone morphogenetic protein-2 released from a heparin-conjugated poly(l-lactic-co-glycolic acid) scaffold. Biomaterials 2007, 28, 2763–2771. [Google Scholar] [CrossRef]
- Sun, X.; Wang, J.; Wang, Y.; Zhang, Q. Collagen-based porous scaffolds containing PLGA microspheres for controlled kartogenin release in cartilage tissue engineering. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Guo, J.; Yuan, J. In vitro antioxidant properties of rutin. LWT 2008, 41, 1060–1066. [Google Scholar] [CrossRef]
- Montes, A.; Wehner, L.; Pereyra, C.; de la Ossa, E.M. Precipitation of submicron particles of rutin using supercritical antisolvent process. J. Supercrit. Fluids 2016, 118, 1–10. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. Formation of Rutin–β-Cyclodextrin Inclusion Complexes by Supercritical Antisolvent Precipitation. Polymers 2021, 13, 246. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Shi, J.L.; Li, Y.S.; Chen, H.R.; Shen, W.H.; Dong, X.P. Storage and release of ibuprofen drug molecules in hol-low mesoporous silica spheres with modified pore surface. Microporous Mesoporous Mater. 2005, 85, 75–81. [Google Scholar] [CrossRef]
- Chebil, L.; Humeau, C.; Anthoni, J.; Dehez, F.; Engasser, J.-M.; Ghoul, M. Solubility of Flavonoids in Organic Solvents. J. Chem. Eng. Data 2007, 52, 1552–1556. [Google Scholar] [CrossRef]
- Sang, J.; Wang, H.; Jin, J.; Meng, H. Comparison and modelling of rutin solubility in supercritical carbon dioxide and subcritical 1,1,1,2-tetrafluoroethane. J. CO2 Util. 2017, 21, 1–8. [Google Scholar] [CrossRef]
- Singh, I.; Gandhi, A.; Mohanty, S.; Nayak, S.K. Depressurization induced morphology control in solid-state microcellular batch foaming process. J. Macromol. Sci. Part. A 2019, 57, 409–420. [Google Scholar] [CrossRef]
- Tai, H.; Upton, C.E.; White, L.J.; Pini, R.; Storti, G.; Mazzotti, M.; Shakesheff, K.M.; Howdle, S.M. Studies on the interactions of CO2 with biodegradable poly(dl-lactic acid) and poly(lactic acid-co-glycolic acid) copolymers using high pressure ATR-IR and high pressure rheology. Polymers 2010, 51, 1425–1431. [Google Scholar] [CrossRef]
- Fraile, M.; Buratto, R.; Gómez, B.; Martin, A.; Cocero, M.J. Enhanced Delivery of Quercetin by Encapsulation in Poloxamers by Supercritical Antisolvent Process. Ind. Eng. Chem. Res. 2014, 53, 4318–4327. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Pavlidou, E.; Panayiotou, C. Biodegradable polymer foams prepared with supercritical CO2–ethanol mixtures as blowing agents. J. Supercrit. Fluids 2007, 42, 265–272. [Google Scholar] [CrossRef]
- Reverchon, E.; Cardea, S. Production of controlled polymeric foams by supercritical CO2. J. Supercrit. Fluids 2007, 40, 144–152. [Google Scholar] [CrossRef]
- Dohrn, R.; Bertakis, E.; Behrend, O.; Voutsas, E.; Tassios, D. Melting point depression by using supercritical CO2 for a novel melt dispersion micronization process. J. Mol. Liq. 2007, 131–132, 53–59. [Google Scholar] [CrossRef]
- Jadhav, N.; Gaikwad, V.; Nair, K.; Kadam, H. Glass transition temperature: Basics and application in pharmaceutical sector. Asian J. Pharm. 2009, 3, 82. [Google Scholar] [CrossRef]
- Frutos, M.J.; Rincón-Frutos, L.; Valero-Cases, E. Rutin in Nonvitamin and Nonmineral Nutritional Supplements; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Mielczarek, C. Acid–base properties of selected flavonoid glycosides. Eur. J. Pharm. Sci. 2005, 25, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Bae, Y.H.; Okano, T. Hydrogels: Swelling, Drug Loading, and Release. Pharm. Res. 1992, 9, 283–290. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Level 1 | Level 2 |
|---|---|---|
| Ratio lactide:glycolide (LA:GA) | 75:25 | 50:50 |
| Pressure (bar) | 80 | 200 |
| Temperature (°C) | 35 | 55 |
| Depressurization rate (bar/min) | 5 | 100 |
| Runs | P (bar) | T (°C) | Dr (bar/min) | Ratio LA:GA | CO2 Density (kg/m3) | Expansion Factor (Vf/V0) 1 | mg Rutin Loaded/ mg PLGA |
|---|---|---|---|---|---|---|---|
| 1 | 80 | 55 | 5 | 50:50 | 103.12 | 1 | 0.63 |
| 2 | 200 | 55 | 5 | 50:50 | 753.71 | 2.60 | 0.75 |
| 3 | 80 | 35 | 100 | 75:25 | 490.62 | 1.63 | 0.69 |
| 4 | 200 | 35 | 5 | 75:25 | 865.65 | 1.52 | 0.57 |
| 5 | 200 | 55 | 100 | 75:25 | 753.71 | 1 | 0.59 |
| 6 | 80 | 35 | 100 | 50:50 | 490.62 | 1.11 | 0.37 |
| 7 | 140 | 45 | 50 | 75:25 | 709.66 | 1 | 0.39 |
| 8 | 140 | 45 | 50 | 50:50 | 709.66 | 5.60 | 0.48 |
| 9 | 200 | 55 | 5 | 75:25 | 753.71 | 1.42 | 0.52 |
| 10 | 200 | 55 | 100 | 50:50 | 753.71 | 2.03 | 0.71 |
| 11 | 200 | 35 | 100 | 75:25 | 865.65 | 2.81 | 0.66 |
| 12 | 200 | 35 | 5 | 50:50 | 865.65 | 1.22 | 0.75 |
| 13 | 80 | 55 | 100 | 75:25 | 103.12 | 3.02 | 0.28 |
| 14 | 80 | 35 | 5 | 50:50 | 490.62 | 1 | 0.65 |
| 15 | 80 | 35 | 5 | 75:25 | 490.62 | 1.17 | 0.32 |
| 16 | 200 | 35 | 100 | 50:50 | 865.65 | 8.55 | 0.64 |
| 17 | 80 | 55 | 5 | 75:25 | 103.12 | 1.05 | 0.44 |
| 18 | 80 | 55 | 100 | 50:50 | 103.12 | 1 | 0.51 |
| Runs | Vp | S | Mean Dp (μm) | ||
|---|---|---|---|---|---|
| (cm3/g) | (m2/g) | Peak1 | Peak2 | Peak3 | |
| 2 | 1.37 | 0.11 | 5.48 | 20.17 | ---- |
| 10 | 6.64 | 0.25 | 1.80 | 30.66 | ---- |
| 11 | 1.95 | 0.22 | 5.32 | 9.04 | ---- |
| 13 | 1.25 | 1.17 | 5.26 | 6.15 | 14.93 |
| 16 | 0.84 | 0.27 | 11.97 | ---- | ---- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valor, D.; Montes, A.; Monteiro, M.; García-Casas, I.; Pereyra, C.; Martínez de la Ossa, E. Determining the Optimal Conditions for the Production by Supercritical CO2 of Biodegradable PLGA Foams for the Controlled Release of Rutin as a Medical Treatment. Polymers 2021, 13, 1645. https://doi.org/10.3390/polym13101645
Valor D, Montes A, Monteiro M, García-Casas I, Pereyra C, Martínez de la Ossa E. Determining the Optimal Conditions for the Production by Supercritical CO2 of Biodegradable PLGA Foams for the Controlled Release of Rutin as a Medical Treatment. Polymers. 2021; 13(10):1645. https://doi.org/10.3390/polym13101645
Chicago/Turabian StyleValor, Diego, Antonio Montes, Marilia Monteiro, Ignacio García-Casas, Clara Pereyra, and Enrique Martínez de la Ossa. 2021. "Determining the Optimal Conditions for the Production by Supercritical CO2 of Biodegradable PLGA Foams for the Controlled Release of Rutin as a Medical Treatment" Polymers 13, no. 10: 1645. https://doi.org/10.3390/polym13101645
APA StyleValor, D., Montes, A., Monteiro, M., García-Casas, I., Pereyra, C., & Martínez de la Ossa, E. (2021). Determining the Optimal Conditions for the Production by Supercritical CO2 of Biodegradable PLGA Foams for the Controlled Release of Rutin as a Medical Treatment. Polymers, 13(10), 1645. https://doi.org/10.3390/polym13101645