
A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Synthesis
2.2. Cyclic Voltammetry and EQCM Studies
2.3. In Situ Conductance Measurements
2.4. Scanning Electron Microscopy Measurements
2.5. UV-vis-NIR Spectroscopy and In Situ Spectroelectrochemical Studies
3. Results
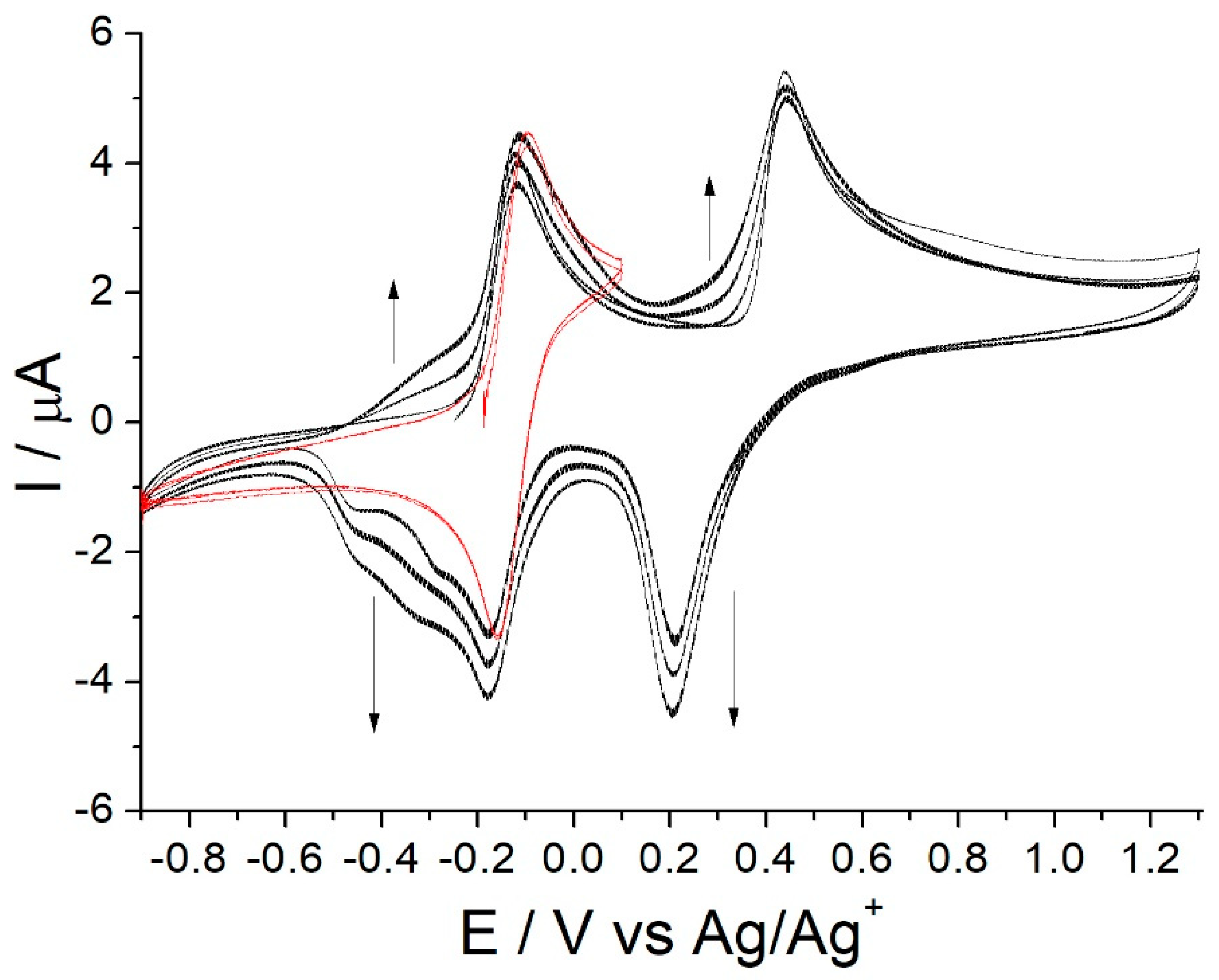
3.1. Oxidative Electrochemistry of [Co(Amben)]
3.2. EQCM Study of the Electro-Oxidative Polymerization of [Co(Amben)]
3.3. CV and EQCM Studies of Thin Poly-[Co(Amben)] Films
3.4. In Situ UV-vis-NIR Spectroelectrochemical Study of Poly-[Co(Amben)] Oxidation
3.5. In Situ Conductance and SEM Studies of Thick Poly-[Co(Amben)] Films
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heinze, J.; Frontana-Uribe, B.A.; Ludwigs, S. Electrochemistry of Conducting Polymers—Persistent Models and New Concepts. Chem. Rev. 2010, 110, 4724–4771. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Recent advances in the functional applications of conducting metallopolymers. Coord. Chem. Rev. 2018, 377, 237–258. [Google Scholar] [CrossRef]
- Clarke, R.M.; Herasymchuk, K.; Storr, T. Electronic structure elucidation in oxidized metal–salen complexes. Coord. Chem. Rev. 2017, 352, 67–82. [Google Scholar] [CrossRef]
- Chepurnaya, I.A.; Karushev, M.P.; Alekseeva, E.V.; Lukyanov, D.A.; Levin, O.V. Redox-conducting polymers based on metal-salen complexes for energy storage applications. Pure Appl. Chem. 2020, 92, 1239–1258. [Google Scholar] [CrossRef]
- Nunes, M.; Araújo, M.; Fonseca, J.; Moura, C.; Hillman, R.; Freire, C. High-Performance Electrochromic Devices Based on Poly[Ni(salen)]-Type Polymer Films. ACS Appl. Mater. Interfaces 2016, 8, 14231–14243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahm, C.E.; Peters, D.G. Catalytic reduction of Iodoethane and 2-Iodopropane at Carbon Electrodes Coated with Anodically Polymerized Films of Nickel(II) Salen. Anal. Chem. 1994, 66, 3117–3123. [Google Scholar] [CrossRef]
- Konev, A.S.; Kayumov, M.Y.; Karushev, M.P.; Novoselova, Y.V.; Lukyanov, D.A.; Alekseeva, E.V.; Levin, O.V. Polymeric Metal Salen-Type Complexes as Catalysts for Photoelectrocatalytic Hydrogen Peroxide Production. ChemElectroChem 2018, 5, 3138–3142. [Google Scholar] [CrossRef]
- Novozhilova, M.; Anischenko, D.; Chepurnaya, I.; Dmitrieva, E.; Malev, V.; Timonov, A.; Karushev, M. Metal-centered redox activity in a polymeric Cobalt(II) complex of a sterically hindered salen type ligand. Electrochim. Acta 2020, 353, 136496. [Google Scholar] [CrossRef]
- Novozhilova, M.V.; Smirnova, E.A.; Polozhentseva, J.A.; Danilova, J.A.; Chepurnaya, I.A.; Karushev, M.P.; Malev, V.V.; Timonov, A.M. Multielectron redox processes in polymeric cobalt complexes with N2O2 Schiff base ligands. Electrochim. Acta 2018, 282, 105–115. [Google Scholar] [CrossRef]
- Kingsborough, R.P.; Swager, T.M. Electroactivity Enhancement by Redox Matching in Cobalt Salen-Based Conducting Polymers. Adv. Mater. 1998, 10, 1100–1104. [Google Scholar] [CrossRef]
- Kingsborough, R.P.; Swager, T.M. Electrocatalytic Conducting Polymers: Oxygen Reduction by a Polythiophene−Cobalt Salen Hybrid. Chem. Mater. 2000, 12, 872–874. [Google Scholar] [CrossRef]
- Dmitrieva, E.; Rosenkranz, M.; Danilova, J.S.; Smirnova, E.A.; Karushev, M.P.; Chepurnaya, I.A.; Timonov, A.M. Radical formation in polymeric nickel complexes with N2O2 Schiff base ligands: An in situ ESR and UV–vis–NIR spectroelectrochemical study. Electrochim. Acta 2018, 283, 1742–1752. [Google Scholar] [CrossRef]
- Łępicka, K.; Pieta, P.; Shkurenko, A.; Borowicz, P.; Majewska, M.; Rosenkranz, M.; Avdoshenko, S.; Popov, A.A.; Kutner, W. Spectroelectrochemical Approaches to Mechanistic Aspects of Charge Transport in meso-Nickel(II) Schiff Base Electrochromic Polymer. J. Phys. Chem. C 2017, 121, 16710–16720. [Google Scholar] [CrossRef]
- Martins, M.; Boas, M.V.; de Castro, B.; Hillman, A.R.; Freire, C. Spectroelectrochemical characterisation of copper salen-based polymer-modified electrodes. Electrochimica Acta 2005, 51, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Polozhentseva, Y.A.; Novozhilova, M.V.; Bykov, V.A.; Karushev, M.P. Modification of Porous Carbon Material with Polymeric Cobalt Complex with a Schiff Base of Salen-Type for Electrodes of Electrochemical Supercapacitors. Tech. Phys. Lett. 2020, 46, 913–915. [Google Scholar] [CrossRef]
- Shioya, T.; Swager, T.M. A reversible resistivity-based nitric oxide sensorThis work was made possible by a Postdoctoral Fellowship from the Japan Society for the Promotion of Science to T. Shioya and the generous financial support of the Office of Naval Research. Chem. Commun. 2002, 1364–1365. [Google Scholar] [CrossRef]
- Swager, T.M. 50th Anniversary Perspective: Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future. Macromolecules 2017, 50, 4867–4886. [Google Scholar] [CrossRef]
- Holliday, B.J.; Swager, T.M. Conducting metallopolymers: The roles of molecular architecture and redox matching. Chem. Commun. 2005, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Understanding the Effect of Metal Centers on Charge Transport and Delocalization in Conducting Metallopolymers. Macromolecules 2017, 50, 872–883. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Effect of conjugation length and metal-backbone interactions on charge transport properties of conducting metallopolymers. Polym. Chem. 2017, 8, 4359–4367. [Google Scholar] [CrossRef]
- Green, M.; Tasker, P.A. NN′-ethylenebis-(o-aminobenzylideneiminato)cobalt(II) and its derivatives. Part I. Physical properties and shape. J. Chem. Soc. A 1970, 3105–3108. [Google Scholar] [CrossRef]
- Higson, B.; McKenzie, E. The redox properties of some planar Schiff compounds of cobalt, nickel and copper. Inorg. Nucl. Chem. Lett. 1970, 6, 209–213. [Google Scholar] [CrossRef]
- Higson, B.M.; McKenzie, E.D. The structure, redox properties, and reactions of some planar [MIIN4] chelate compounds of cobalt, nickel, and copper, and their oxidised products, including paramagnetic cobalt(III) species. J. Chem. Soc. Dalton Trans. 1972, 269–280. [Google Scholar] [CrossRef]
- Karlsson, R.; Engelhardt, L.M.; Green, M. Crystal structure of [NN′-ethylenebis-(2-amino-5-chlorobenzylideneiminato)]cobalt(II). J. Chem. Soc. Dalton Trans. 1972, 2463–2465. [Google Scholar] [CrossRef]
- Kochem, A.; Gellon, G.; Jarjayes, O.; Philouze, C.; Leconte, N.; Van Gastel, M.; Bill, E.; Thomas, F. A singlet ground state for a cobalt(ii)–anilinosalen radical complex. Chem. Commun. 2014, 50, 4924–4926. [Google Scholar] [CrossRef] [PubMed]
- Kochem, A.; Thomas, F.; Jarjayes, O.; Gellon, G.; Philouze, C.; Weyhermüller, T.; Neese, F.; Van Gastel, M. Structural and Spectroscopic Investigation of an Anilinosalen Cobalt Complex with Relevance to Hydrogen Production. Inorg. Chem. 2013, 52, 14428–14438. [Google Scholar] [CrossRef] [PubMed]
- Shagisultanova, G.A.; Ardasheva, L.P. Electrochemical Polymerization of Ni(II) and Pd(II) Complexes with 1,2-Bis(o-aminobenzylidene)ethylenediamine. Russ. J. Coord. Chem. 2004, 30, 94–99. [Google Scholar] [CrossRef]
- Karushev, M.; Smirnova, E.; Chepurnaya, I. Nickel(II) Complex of N4 Schiff Base Ligand as a Building Block for a Conducting Metallopolymer with Multiple Redox States. Molecules 2021, 26, 2646. [Google Scholar] [CrossRef]
- Inzelt, G. Methods of Investigation. Underpotential Depos. 2012, 83–147. [Google Scholar] [CrossRef]
- Casado, N.; Mecerreyes, D. Chapter 1. Introduction to Redox Polymers: Classification, Characterization Methods and Main Applications. Polymer Chem. Series 2020, 1–26. [Google Scholar] [CrossRef]
- Salinas, G.; Frontana-Uribe, B.A. Analysis of Conjugated Polymers Conductivity by in situ Electrochemical-Conductance Method. ChemElectroChem 2019, 6, 4105–4117. [Google Scholar] [CrossRef]
- Sauerbrey, G. Verwendung von Schwingquarzen zur Wägung Dünner Schichten und zur Mikrowägung. Eur. Phys. J. A 1959, 155, 206–222. [Google Scholar] [CrossRef]
- Zotti, G.; Zecchin, S.; Schiavon, G.; Groenendaal, L. “Bert” Conductive and Magnetic Properties of 3,4-Dimethoxy- and 3,4-Ethylenedioxy-Capped Polypyrrole and Polythiophene. Chem. Mater. 2000, 12, 2996–3005. [Google Scholar] [CrossRef]
- Yurchenko, O.; Heinze, J.; Ludwigs, S. Electrochemically Induced Formation of Independent Conductivity Regimes in Polymeric Tetraphenylbenzidine Systems. ChemPhysChem 2010, 11, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Paynter, R.; Dao, L.H. Synthesis and characterization of poly(diarylamines): A new class of electrochromic conducting polymers. Macromolecules 1990, 23, 3598–3605. [Google Scholar] [CrossRef]
- Malacrida, C.; Lu, Y.; Dirnberger, K.; Gámez-Valenzuela, S.; Delgado, M.C.R.; Ludwigs, S. Towards highly conducting bicarbazole redox polymer films with plateau-like conductivities. J. Mater. Chem. C 2020, 8, 15393–15405. [Google Scholar] [CrossRef]
- Kochem, A.; Kanso, H.; Baptiste, B.; Arora, H.; Philouze, C.; Jarjayes, O.; Vezin, H.; Luneau, D.; Orio, M.; Thomas, F. Ligand Contributions to the Electronic Structures of the Oxidized Cobalt(II) salen Complexes. Inorg. Chem. 2012, 51, 10557–10571. [Google Scholar] [CrossRef] [PubMed]
- Otteny, F.; Perner, V.; Wassy, D.; Kolek, M.; Bieker, P.; Winter, M.; Esser, B. Poly(vinylphenoxazine) as Fast-Charging Cathode Material for Organic Batteries. ACS Sustain. Chem. Eng. 2019, 8, 238–247. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Range | Charge Carrier Molar Mass, g mol−1 |
|---|---|
| −0.5 to −0.15 V | 25 |
| −0.15 to +0.2 V | 94 |
| +0.35 to +0.95 V | 185 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karushev, M. A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers 2021, 13, 1667. https://doi.org/10.3390/polym13101667
Karushev M. A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers. 2021; 13(10):1667. https://doi.org/10.3390/polym13101667
Chicago/Turabian StyleKarushev, Mikhail. 2021. "A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex" Polymers 13, no. 10: 1667. https://doi.org/10.3390/polym13101667
APA StyleKarushev, M. (2021). A Novel Cobalt Metallopolymer with Redox-Matched Conjugated Organic Backbone via Electropolymerization of a Readily Available N4 Cobalt Complex. Polymers, 13(10), 1667. https://doi.org/10.3390/polym13101667