
Synthesis, Structure, Crystallization and Mechanical Properties of Isodimorphic PBS-ran-PCL Copolyesters

 , , , and
, , , and
Abstract
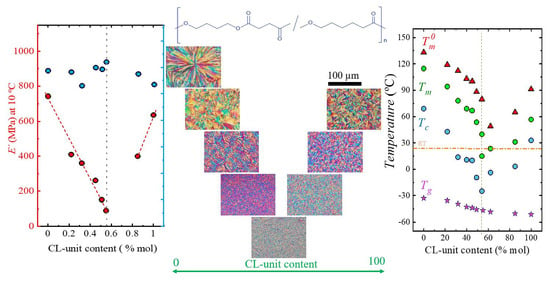
:
1. Introduction
- (1)
- Total inclusion behavior: It happens when the two comonomeric units contain similar repeating units. The chains can co-crystallize regardless of the composition, the so-called isomorphic behavior. In this case, the two comonomers are miscible in the crystalline state, and thermal and structural properties generally show a linear dependence on composition [18].
- (2)
- Inclusion-exclusion behavior: When the two homopolymers have similar repeating units, but they do not share any crystalline structure, an isodimorphic behavior in the random copolymers can result. This group of random copolymers shows crystallization over the whole composition range and a pseudo-eutectic behavior characterized by a dominant crystalline structure at each side of the pseudo-eutectic point or region. Moreover, the changes in the unit cell parameters of both crystal phases with composition evidence comonomer inclusion. Our group has recently studied isodimorphism in several random copolymer systems [19,20,21,22,23,24,25,26,27,28,29]. Isodimorphic crystallization has recently been found in different types of random copolymers such as poly (decamethylene succinate-ran-decamethylene fumarate), poly (butylene succinate-co-cis-butene succinate) and poly (hexamethylene carbonate)-co-poly (hexamethylene urethane) [30,31,32,33,34,35].
- (3)
- Total exclusion behavior: When the repeating units of two homopolymers are dissimilar, the minor comonomer would be completely kept out from the crystalline structure of the main component. Therefore, the transition temperatures and enthalpies decrease significantly with increasing comonomer content and the copolymer remains completely amorphous over a large range of copolymer compositions [19].
2. Materials and Methods
2.1. Materials
2.2. Synthesis Routes of BSxCLy Random Copolyesters
2.3. Nuclear Magnetic Resonance (NMR)
2.4. Gel Permeation Chromatography (GPC)
2.5. Differential Scanning Calorimetry (DSC)
2.6. Tensile Test
2.7. Dynamic Mechanical Thermal Analysis (DMTA)
3. Results
3.1. Synthesis Results
3.2. Non-Isothermal Melting-Crystallization Behavior
3.3. Controlling Single or Double Crystalline Phases at the Pseudo-Eutectic Point by Non-Isothermal and Isothermal Crystallization
3.4. Controlling the Crystalline Phase over Non-Isothermal and Isothermal Crystallization for Compositions at the Pseudo-Eutectic Point
3.5. Isothermal Crystallization
3.5.1. Nucleation Kinetics Studied by PLOM
3.5.2. Studying Overall Crystallization Kinetics by DSC
3.6. Mechanical Properties
3.6.1. Tensile Tests
3.6.2. DMTA results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lendlein, A.; Sisson, A. Handbook of Biodegradable Polymers: Isolation, Synthesis, Characterization and Applications; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Yu, L.; Dean, K.; Li, L. Polymer blends and composites from renewable resources. Prog. Polym. Sci. 2006, 31, 576–602. [Google Scholar] [CrossRef]
- Gandini, M.N.B.; Belgacem, M.N. Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Leja, K.; Lewandowicz, G. Polymer biodegradation and biodegradable polymers-a review. Polym. J. Environ. Stud. 2010, 19, 255–266. [Google Scholar]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Prog. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, P. Crystallization of biodegradable and biobased polyesters: Polymorphic crystallization, cocrystallization, and structure-property relationship. Prog. Polym. Sci. 2020, 109, 101291. [Google Scholar] [CrossRef]
- Vert, M.; Li, S.M.; Spenlehauer, G.; Guérin, P. Bioresorbability and biocompatibility of aliphatic polyesters. J. Mater. Sci. Mater. Med. 1992, 3, 432–446. [Google Scholar] [CrossRef]
- Chiulan, I.; Frone, A.N.; Brandabur, C.; Panaitescu, D.M. Recent advances in 3D printing of aliphatic polyesters. Bioengineering 2018, 5, 2. [Google Scholar] [CrossRef]
- Siracusa, V.; Rocculi, P.; Romani, S.; Dalla Rosa, M. Biodegradable polymers for food packaging: A review. Trends Food Sci. Technol. 2008, 19, 634–643. [Google Scholar] [CrossRef]
- Pillai, O.; Panchagnula, R. Polymers in drug delivery. Curr. Opin. Chem. Biol. 2001, 5, 447–451. [Google Scholar] [CrossRef]
- Montaudo, G.; Rizzarelli, P. Synthesis and enzymatic degradation of aliphatic copolyesters. Polym. Degrad. Stab. 2000, 70, 305–314. [Google Scholar] [CrossRef]
- Fields, R.D.; Rodriguez, F.; Finn, R.K. Microbial degradation of polyesters: Polycaprolactone degraded by P. pullulans. J. Appl. Polym. Sci. 1974, 18, 3571–3579. [Google Scholar] [CrossRef]
- Zheng, Y.; Yanful, E.K.; Bassi, A.S. A review of plastic waste biodegradation. Crit. Rev. Biotechnol. 2005, 25, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Abedalwafa, M.; Wang, F.; Wang, L.; Li, C. Biodegradable poly-epsilon-caprolactone (PCL) for tissue engineering applications: A review. Rev. Adv. Mater. Sci. 2013, 34, 123–140. [Google Scholar]
- Mohamed, R.M.; Yusoh, K. A review on the recent research of polycaprolactone (PCL). Adv. Mat. Res. 2016, 1134, 249–255. [Google Scholar] [CrossRef]
- Sisti, L.; Totaro, G.; Marchese, P. PBS makes its entrance into the family of biobased plastics. In Biodegradable and Biobased Polymers for Environmental and Biomedical Applications; John Wiley & Sons: Hobocan, NJ, USA, 2016; pp. 225–273. [Google Scholar]
- Kanemura, C.; Nakashima, S.; Hotta, A. Mechanical properties and chemical structures of biodegradable poly (butylene-succinate) for material reprocessing. Polym. Degrad. Stab. 2012, 97, 972–980. [Google Scholar] [CrossRef]
- Natta, G.; Corradini, P.; Sianesi, D.; Morero, D. Isomorphism phenomena in macromolecules. J. Poly. Sci. 1961, 51, 527–539. [Google Scholar] [CrossRef]
- Pérez-Camargo, R.A.; Arandia, I.; Safari, M.; Cavallo, D.; Lotti, N.; Soccio, M.; Müller, A.J. Crystallization of isodimorphic aliphatic random copolyesters: Pseudo-eutectic behavior and double-crystalline materials. Eur. Polym. J. 2018, 101, 233–247. [Google Scholar] [CrossRef]
- Arandia, I.; Mugica, A.; Zubitur, M.; Arbe, A.; Liu, G.; Wang, D.; Mincheva, R.; Dubois, P.; Müller, A.J. How composition determines the properties of isodimorphic poly(butylene succinate-ran-butylene azelate) random biobased copolymers: From single to double crystalline random copolymers. Macromolecules 2015, 48, 43–57. [Google Scholar] [CrossRef]
- Arandia, I.; Mugica, A.; Zubitur, M.; Iturrospe, A.; Arbe, A.; Liu, G.; Wang, D.; Mincheva, R.; Dubois, P.; Müller, A.J. Application of SSA Thermal fractionation and X-Ray diffraction to elucidate comonomer inclusion or exclusion from the crystalline phases in poly(butylene succinate-ran-butylene azelate) random copolymers. J. Poly. Sci. B Poly. Phy. 2016, 54, 2346–2358. [Google Scholar] [CrossRef]
- Arandia, I.; Mugica, A.; Zubitur, M.; Mincheva, R.; Dubois, P.; Müller, A.J.; Alegria, A. The complex amorphous phase in poly(butylene succinate-ran-butylene azelate) isodimorphic copolyesters. Macromolecules 2017, 50, 1569–1578. [Google Scholar] [CrossRef]
- Arandia, I.; Zaldua, N.; Maiz, J.; Pérez-Camargo, R.A.; Mugica, A.; Zubitur, M.; Mincheva, R.; Dubois, P.; Müller, A.J. Tailoring the isothermal crystallization kinetics of isodimorphic poly(butylene succinate-ran-butylene azelate) random copolymers by changing composition. Polymer 2019, 183, 121863. [Google Scholar] [CrossRef]
- Arandia, I.; Meabe, L.; Aranburu, N.; Sardon, H.; Mecerreyes, D.; Müller, A.J. Influence of chemical structures on isodimorphic behavior of three different copolycarbonate random copolymer series. Macromolecules 2020, 53, 669–681. [Google Scholar] [CrossRef]
- Pérez-Camargo, R.A.; Fernández-d’Arlas, B.; Cavallo, D.; Debuissy, T.; Pollet, E.; Avérous, L.; Müller, A.J. Tailoring the Structure, Morphology, and Crystallization of Isodimorphic Poly(butylene succinate-ran-butylene adipate) Random Copolymers by Changing Composition and Thermal History. Macromolecules 2017, 50, 597–608. [Google Scholar] [CrossRef]
- Basterretxea, A.; Gabirondo, E.; Flores, I.; Etxeberria, A.; Gonzalez, A.; Müller, A.J.; Mecerreyes, D.; Coulembier, O.; Sardon, H. Isomorphic polyoxyalkylene copolyethers obtained by copolymerization of aliphatic diols. Macromolecules 2019, 52, 3506–3515. [Google Scholar] [CrossRef]
- Safari, M.; Leon Boigues, L.; Shi, G.; Maiz, J.; Liu, G.; Wang, D.; Mijangos, C.; Müller, A.J. Effect of Nanoconfinement on the Isodimorphic Crystallization of Poly(butylene succinate-ran-caprolactone) Random Copolymers. Macromolecules 2020, 53, 6486–6497. [Google Scholar] [CrossRef]
- Pérez-Camargo, R.A.; Liu, G.; Cavallo, D.; Wang, D.; Müller, A.J. Effect of the crystallization conditions on the exclusion/inclusion balance in biodegradable poly(butylene succinate-ran-butylene adipate) copolymers. Biomacromolecules 2020, 21, 3420–3435. [Google Scholar] [CrossRef]
- Schäfer, M.; Yuan, S.; Petzold, A.; Pérez-Camargo, R.A.; Müller, A.J.; Thurn-Albrecht, T.; Saalwächter, K.; Schmidt-Rohr, K. Asymmetric co-unit inclusion in statistical copolyesters. Macromolecules 2021, 54, 835–845. [Google Scholar] [CrossRef]
- Wei, X.W.; Huang, G.; Wang, J.; Meng, X.; Zhou, Q.; Ye, H.M. Tailoring Crystallization of Random Terpolyester: Combination of Isodimorphism and Isomorphism. Macromolecules 2020, 53, 8918–8927. [Google Scholar] [CrossRef]
- Zhang, B.; Wei, Z.; Zhao, Y.; Che, R.; Wang, Y.; Leng, X.; Li, Y. Isodimorphic aliphatic copolyester as midblock of poly (l-lactide)-based triblock copolymers towards largely enhanced impact toughness. Eur. Polym. J. 2019, 111, 28–37. [Google Scholar] [CrossRef]
- Ye, H.M.; Wang, J.; Wang, C.S.; Li, H.F. Unique Isodimorphism of poly (decamethylene succinate-ran-decamethylene fumarate): Large pseudoeutectic region and fantastic crystallization/melting behavior. Macromolecules 2019, 52, 1447–1457. [Google Scholar] [CrossRef]
- Zhang, C.; Pérez-Camargo, R.A.; Zheng, L.; Zhao, Y.; Liu, G.; Wang, L.; Wang, D. Crystallization of poly (hexamethylene carbonate)-co-poly (hexamethylene urethane) segmental block copolymers: From single to double crystalline phases. Polymer 2021, 222, 123675. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, M.; Xu, Y.; Wang, W.; Wang, Z.; Zhang, L. Bio-based polyesters: Recent progress and future prospects. Prog. Polym. Sci. 2021, 120, 101430. [Google Scholar] [CrossRef]
- Yu, Y.; Wei, Z.; Zheng, L.; Jin, C.; Leng, X.; Li, Y. Competition and miscibility of isodimorphism and their effects on band spherulites and mechanical properties of poly (butylene succinate-co-cis-butene succinate) unsaturated aliphatic copolyesters. Polymer 2018, 150, 52–63. [Google Scholar] [CrossRef]
- Ciulik, C.; Safari, M.; Martinez de Ilarduya, A.; Morales-Huerta, J.C.; Iturrospe, A.; Arbe, A.; Müller, A.J.; Muñoz-Guerra, S. Poly (butylene succinate-ran-ε-caprolactone) copolyesters: Enzymatic synthesis and crystalline isodimorphic character. Eur. Polym. J. 2017, 95, 795–808. [Google Scholar] [CrossRef]
- Safari, M.; Martínez de Ilarduya, A.; Mugica, A.; Zubitur, M.; Muñoz-Guerra, S.; Müller, A.J. Tuning the thermal properties and morphology of isodimorphic poly [(butylene succinate)-ran-(ε-caprolactone)] copolyesters by changing composition, molecular weight, and thermal history. Macromolecules 2018, 51, 9589–9601. [Google Scholar] [CrossRef]
- Safari, M.; Mugica, A.; Zubitur, M.; Martínez de Ilarduya, A.; Muñoz-Guerra, S.; Müller, A.J. Controlling the isothermal crystallization of isodimorphic PBS-ran-PCL random copolymers by varying composition and supercooling. Polymers 2020, 12, 17. [Google Scholar] [CrossRef]
- Cao, A.; Okamura, T.; Ishiguro, C.; Nakayama, K.; Inoue, Y.; Masuda, T. Studies on syntheses and physical characterization of biodegradable aliphatic poly (butylene succinate-co-ε-caprolactone) s. Polymer 2002, 43, 671–679. [Google Scholar] [CrossRef]
- Jin, C.; Wei, Z.; Yu, Y.; Sui, M.; Leng, X.; Li, Y. Copolymerization of ethylene brassylate with δ-valerolactone towards isodimorphic random copolyesters with continuously tunable mechanical properties. Eur. Polym. J. 2018, 102, 90–100. [Google Scholar] [CrossRef]
- Chartoff, R.P.; Turi, E.A. Thermal Characterization of Polymeric Materials; Academic Press: New York, NY, USA, 1997; Volume 1, p. 513. [Google Scholar]
- Hiemenz, P.C.; Lodge, T.P. Polymer Chemistry; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Fox, T.G. Influence of diluent and of copolymer composition on the glass temperature of a polymer system. Bull. Am. Phys. Soc. 1956, 1, 123. [Google Scholar]
- Tsagaropoulos, G.; Eisenberg, A. Dynamic mechanical study of the factors affecting the two glass transition behavior of filled polymers. Similarities and differences with random ionomers. Macromolecules 1995, 28, 6067–6077. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Tg (°C), tan δ | Tg (°C), E″ | Tg (°C), DSC |
|---|---|---|---|
| HMW PBS | −10.0 | −16.7 | −31.7 |
| HMW-BS78CL22 | −18.1 | −24.0 | −38.6 |
| HMW-BS68CL32 | −23.3 | −28.6 | −42.8 |
| HMW-BS55CL45 | −28.5 | −33.4 | −43.5 |
| HMW-BS51CL49 | −30.0 | −35.3 | −45.3 |
| HMW-BS46CL54 | −33.8 | −40.9 | −47.3 |
| HMW-BS15CL85 | −36.8 | −41.1 | −55.1 |
| HMW-PCL | −37.5 | −43.3 | −60.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safari, M.; Otaegi, I.; Aramburu, N.; Guerrica-Echevarria, G.; de Ilarduya, A.M.; Sardon, H.; Müller, A.J. Synthesis, Structure, Crystallization and Mechanical Properties of Isodimorphic PBS-ran-PCL Copolyesters. Polymers 2021, 13, 2263. https://doi.org/10.3390/polym13142263
Safari M, Otaegi I, Aramburu N, Guerrica-Echevarria G, de Ilarduya AM, Sardon H, Müller AJ. Synthesis, Structure, Crystallization and Mechanical Properties of Isodimorphic PBS-ran-PCL Copolyesters. Polymers. 2021; 13(14):2263. https://doi.org/10.3390/polym13142263
Chicago/Turabian StyleSafari, Maryam, Itziar Otaegi, Nora Aramburu, Gonzalo Guerrica-Echevarria, Antxon Martínez de Ilarduya, Haritz Sardon, and Alejandro J. Müller. 2021. "Synthesis, Structure, Crystallization and Mechanical Properties of Isodimorphic PBS-ran-PCL Copolyesters" Polymers 13, no. 14: 2263. https://doi.org/10.3390/polym13142263
APA StyleSafari, M., Otaegi, I., Aramburu, N., Guerrica-Echevarria, G., de Ilarduya, A. M., Sardon, H., & Müller, A. J. (2021). Synthesis, Structure, Crystallization and Mechanical Properties of Isodimorphic PBS-ran-PCL Copolyesters. Polymers, 13(14), 2263. https://doi.org/10.3390/polym13142263