
Rubber Degrading Strains: Microtetraspora and Dactylosporangium


Abstract
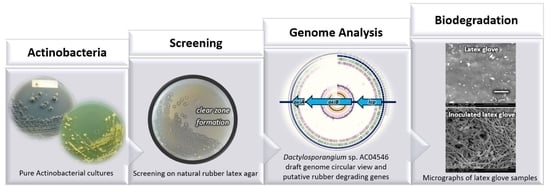
:
1. Introduction
2. Materials and Methods
2.1. Screening for Clear Zone Forming Strains on Natural Rubber (NR) Latex Agar
2.2. Identification of Clear Zone Forming Strains on Natural Rubber (NR) Latex Agar
2.3. Profiling of Latex Clearing Protein (Lcp) Genes
2.4. Genomic Deoxyribonucleic Acid (DNA) Isolation
2.5. Genome Attributes and Annotation
2.6. Taxonomic Delineation
2.7. Utilization of Rubber Materials
- Morphological Observation using Scanning Electron Microscope (SEM)
- Attenuated Total Reflection-Fourier Transform Infrared (ATR-FTIR)
3. Results
3.1. Screening for Clear Zone Forming Strains on Natural Rubber (NR) Latex Agar
3.2. Taxonomic Identification of Clear Zone Forming Strains
3.3. Profiling of Latex Clearing Protein (Lcp) Genes in Clear Zone Forming Strains
3.4. Strain of Interest: Microtetraspora sp. AC03309 (JCM 34240)
3.4.1. Morphological Characterization of Microtetraspora sp. AC03309
3.4.2. Microtetraspora sp. AC03309 Taxonomic Delineation
3.4.3. Microtetraspora sp. AC03309 Genomic Attributes
- Genomic & Protein Features
- Specialty Genes:
- Secondary Metabolite Clusters:
- Sulfur Metabolism:
3.4.4. Identification of Latex Clearing Protein (Lcp) Homolog for Microtetraspora sp. AC03309
3.4.5. Predicted Genes Involved in Rubber Degradation for Microtetraspora sp. AC03309
3.5. Strain of Interest: Dactylosporangium sp. AC04546 (JCM 34239)
3.5.1. Morphological Characterization of Dactylosporangium sp. AC04546
3.5.2. Dactylosporangium sp. AC04546 Taxonomic Delineation
3.5.3. Dactylosporangium sp. AC04546 Genomic Attributes
- Genomic & Protein Features
- Specialty Gene:
- Secondary metabolite clusters:
- Urea Metabolism:
- Polyhydroxybutyrate Metabolism
3.5.4. Identification of Latex Clearing Protein (Lcp) Homolog for Dactylosporangium sp. AC04546
3.5.5. Predicted Genes Involved in Rubber Degradation for Dactylosporangium sp. AC04546
3.6. Utilization of Rubber Materials by Microtetraspora sp. AC03309 and Dactylosporangium sp. AC04546
3.7. ATR-FTIR Analysis of Degraded Rubber Materials
3.7.1. Fresh Latex ATR-FTIR Profile
3.7.2. Latex Glove ATR-FTIR Profile
3.7.3. Tyre ATR-FTIR Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig | Start (bp) | Stop (bp) | |
|---|---|---|---|
| 1 | contig000014 | 94,175 | 94,462 |
| 2 | contig000145 | 766 | 977 |
| Contig | Start (bp) | Stop (bp) | Classification | Identity (%) | |
|---|---|---|---|---|---|
| 1 | contig000052 | 61,507 | 62,700 | elfamycin (ARO: 3003361) | 77 |
| 2 | contig 000084 | 35,081 | 36,280 | rapamycin antibiotic (ARO:3002883) | 93 |
| Contig | Product | Start (bp) | Stop (bp) | Length (bp) | Query Coverage | Identity (%) | |
|---|---|---|---|---|---|---|---|
| 1 | Contig000007 | RecA protein | 27,312 | 25,081 | 1104 | 88 | 85 |
| 2 | Contig000641 | Serine/threonine-protein kina, pknB (EC 2.7.11.1) | 35 | 322 | 288 | 90 | 88 |
| 3 | Contig000054 | Cell division protein, ftsZ | 15,100 | 13,667 | 1434 | 95 | 92 |
| Roles | ORF | |
|---|---|---|
| Inorganic sulfur: Sulfate/thiosulfate metabolism | ||
| 1 | 3’(2’),5’-bisphosphate nucleotidase (EC 3.1.3.7) | 1 |
| 2 | Adenylylsulfate kinase (EC 2.7.1.25) | 2 |
| 3 | Adenylyl-sulfate reductase [thioredoxin] (EC 1.8.4.10) | 3 |
| 4 | Ferredoxin | 12 |
| 5 | Ferredoxin-like protein involved in electron transfer | 1 |
| 6 | Ferredoxin--NADP(+) reductase, actinobacterial (eukaryote-like) type (EC 1.18.1.2) | 1 |
| 7 | Ferredoxin--sulfite reductase, actinobacterial type (EC 1.8.7.1) | 1 |
| 8 | Phosphoadenylyl-sulfate reductase [thioredoxin] (EC 1.8.4.8) | 1 |
| 9 | Putative sulfate permease | 1 |
| 10 | Sulfate adenylyltransferase subunit 1 (EC 2.7.7.4) | 1 |
| 11 | Sulfate adenylyltransferase subunit 2 (EC 2.7.7.4) | 1 |
| 12 | Sulfate adenylyltransferase, dissimilatory-type (EC 2.7.7.4) | 1 |
| 13 | Sulfate and thiosulfate binding protein CysP | 1 |
| 14 | Sulfate and thiosulfate import ATP-binding protein CysA (EC 3.6.3.25) | 2 |
| 15 | Sulfate transport system permease protein CysW | 1 |
| Organic sulfur: Alkanesulfonate assimilation | ||
| 1 | ABC-type nitrate/sulfonate/bicarbonate transport system, ATPase component | 1 |
| 2 | ABC-type nitrate/sulfonate/bicarbonate transport system, permease component | 2 |
| 3 | ABC-type nitrate/sulfonate/bicarbonate transport systems, periplasmic components | 1 |
| 4 | Alkanesulfonate monooxygenase (EC 1.14.14.5) | 2 |
| 5 | Alkanesulfonates ABC transporter ATP-binding protein | 1 |
| 6 | Alkanesulfonates transport system permease protein | 3 |
| 7 | Alkanesulfonates-binding protein | 1 |
| 8 | Alpha-ketoglutarate-dependent taurine dioxygenase (EC 1.14.11.17) | 1 |
| 9 | Arylsulfatase (EC 3.1.6.1) | 1 |
| 10 | FMN reductase (EC 1.5.1.29) | 1 |
| 11 | Putative arylsulfatase regulatory protein | 1 |
| Region | Type | From (bp) | To (bp) | Most Similar Known Cluster | Similarity | |
|---|---|---|---|---|---|---|
| Region 5.1 | other | 406 | 41,026 | |||
| Region 7.1 | NRPS | 58,409 | 104,012 | tetronasin | Polyketide | 3% |
| Region 8.1 | redox-cofactor | 1 | 16,071 | |||
| Region 8.2 | lanthipeptide-class-iv | 119,417 | 142,221 | |||
| Region 11.1 | lanthipeptide-class-iv | 4363 | 27,032 | blasticidin S | Other | 7% |
| Region 11.2 | NRPS,T1PKS | 87,888 | 145,746 | nostopeptolide A2 | Polyketide + NRP:Cyclic depsipeptide | 25% |
| Region 14.1 | T3PKS | 105,469 | 141,262 | alkylresorcinol | Polyketide | 66% |
| Region 16.1 | RiPP-like | 121,803 | 132,618 | |||
| Region 17.1 | terpene | 20,289 | 46,240 | hopene | Terpene | 46% |
| Region 26.1 | terpene | 88,473 | 108,659 | frankiamicin | Polyketide | 14% |
| Region 28.1 | NRPS | 1 | 28,384 | 5-acetyl-5,10-dihydrophenazine-1-carboxylic acid/5-(2-hydroxyacetyl)-5,10-dihydrophenazine-1-carboxylic acid/endophenazine A1/endophenazine F/endophenazine G | Other:Phenazine | 8% |
| Region 34.1 | betalactone,terpene | 62,313 | 94,083 | geosmin | Terpene | 100% |
| Region 39.1 | lanthipeptide-class-iii | 66,288 | 83,988 | |||
| Region 44.1 | NRPS | 1 | 42,513 | acarviostatin I03/acarviostatin II03/acarviostatin III03/acarviostatin IV03 | Saccharide | 11% |
| Region 44.2 | T1PKS,lanthipeptide-class-iv | 48,145 | 75,803 | labyrinthopeptin A2/labyrinthopeptin A1/labyrinthopeptin A3 | RiPP:Lanthipeptide | 40% |
| Region 45.1 | terpene | 24,504 | 45,502 | |||
| Region 49.1 | NRPS | 1 | 32,408 | herboxidiene | Polyketide | 3% |
| Region 57.1 | thiopeptide,LAP | 33,174 | 64,696 | muraymycin C1 | NRP + Polyketide | 7% |
| Region 61.1 | siderophore | 17,145 | 30,431 | |||
| Region 67.1 | NRPS | 1 | 50,370 | triacsins | Other | 25% |
| Region 71.1 | siderophore | 1 | 8702 | desferrioxamine E | Other | 75% |
| Region 91.1 | lanthipeptide-class-iv | 3240 | 26,017 | labyrinthopeptin A2/labyrinthopeptin A1/labyrinthopeptin A3 | RiPP:Lanthipeptide | 40% |
| Region 93.1 | NRPS | 1 | 28,814 | catenulisporolides | NRP + Polyketide | 5% |
| Region 135.1 | lanthipeptide-class-iii | 1 | 11,872 | catenulipeptin | RiPP:Lanthipeptide | 60% |
| Transcriptional Regulator, TetR Family on the Same Contig as lcp Gene | ||||
|---|---|---|---|---|
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000003 | 19,522 | 18,260 | 1263 |
| 2 | contig000003 | 28,599 | 29,279 | 681 |
| Twin-Arginine Translocation Protein, TAT Protein (TatA, TatB, TatC) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000002 | 137,788 | 137,363 | 426 |
| 2 | contig000113 | 18,270 | 18,629 | 360 |
| 3 | contig000133 | 9169 | 9507 | 339 |
| 4 | contig000133 | 9535 | 10,389 | 855 |
| Mammalian Cell Entry, Mce (Mce1D, Mce1D) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000186 | 1797 | 1339 | 459 |
| 2 | contig000629 | 96 | 284 | 189 |
| Acyl-CoA synthase | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000004 | 157,269 | 155,719 | 1551 |
| 2 | contig000004 | 152,549 | 150,963 | 1587 |
| 3 | contig000033 | 8187 | 6544 | 1644 |
| Acyl-CoA Dehydrogenase (EC 1.3.8.1) (EC 1.3.8.7) (Putative) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000016 | 19,917 | 20,870 | 954 |
| 2 | contig000016 | 24,758 | 23,676 | 1083 |
| 3 | contig000038 | 54,006 | 55,709 | 1704 |
| 4 | contig000038 | 55,738 | 57,438 | 1701 |
| 5 | contig000063 | 22,059 | 23,198 | 1140 |
| 6 | contig000070 | 24,723 | 23,602 | 1122 |
| 2,4-dienoyl-CoA Reductase, NADH: Flavin Oxidoreductases, Old Yellow Enzyme Family | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000070 | 29,836 | 28,598 | 1239 |
| Enoyl-CoA Hydratases (EC 4.2.1.17) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000004 | 157,510 | 158,325 | 816 |
| 2 | contig000008 | 7568 | 6855 | 711 |
| 3 | contig000008 | 11,332 | 12,129 | 798 |
| 4 | contig000013 | 81,498 | 80,584 | 915 |
| 5 | contig000014 | 109,166 | 109,984 | 819 |
| 6 | contig000016 | 7177 | 8541 | 1365 |
| 7 | contig000016 | 14,015 | 13,206 | 810 |
| 8 | contig000029 | 64,705 | 66,807 | 2103 |
| 9 | contig000030 | 66,724 | 65,951 | 774 |
| 10 | contig000034 | 65,180 | 64,380 | 801 |
| 11 | contig000049 | 15,615 | 14,776 | 840 |
| 12 | contig000066 | 20,230 | 20,994 | 765 |
| 13 | contig000084 | 24,228 | 25,004 | 777 |
| 14 | contig000113 | 12,043 | 11,240 | 804 |
| 3-hydroxyacyl-CoA Dehydrogenases (EC 1.1.1.35) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 58,250 | 56,769 | 1482 |
| 2 | contig000008 | 27,541 | 26,771 | 771 |
| 3 | contig000008 | 35,830 | 36,684 | 855 |
| 4 | contig000016 | 5912 | 5157 | 756 |
| 5 | contig000029 | 64,705 | 66,807 | 2103 |
| 6 | contig000628 | 289 | 86 | 204 |
| 3-ketoacyl-CoA Thiolase (EC 2.3.1.16) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 49,954 | 48,761 | 1194 |
| 2 | contig000002 | 54,042 | 52,825 | 1218 |
| 3 | contig000005 | 108,324 | 109,448 | 1125 |
| 4 | contig000008 | 19,025 | 17,337 | 1689 |
| 5 | contig000008 | 34,593 | 35,762 | 1170 |
| 6 | contig000008 | 70,364 | 69,177 | 1188 |
| 7 | contig000009 | 12,987 | 11,806 | 1182 |
| 8 | contig000025 | 61,696 | 62,844 | 1149 |
| 9 | contig000029 | 63,524 | 64,708 | 1185 |
| 10 | contig000033 | 3101 | 1878 | 1224 |
| 11 | contig000033 | 6551 | 5379 | 1173 |
| 12 | contig000034 | 68,356 | 67,148 | 1209 |
| 13 | contig000049 | 17,108 | 18,265 | 1158 |
| 14 | contig000066 | 45,751 | 46,896 | 1146 |
| 15 | contig000090 | 24,745 | 23,573 | 1173 |
| α-Methylacyl-CoA Racemase (Mcr) (EC 5.1.99.4) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000016 | 8557 | 9753 | 1194 |
| 2 | contig000016 | 110,010 | 111,122 | 1113 |
| 3 | contig000032 | 61,506 | 60,388 | 1119 |
| Superoxide Dismutase | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000020 | 73,292 | 72,687 | 606 |
| 2 | contig000056 | 23,468 | 23,869 | 402 |
Appendix B


| Contig | Start (bp) | Stop (bp) | |
|---|---|---|---|
| 1 | Contig000003 | 116,444 | 121,041 |
| 2 | Contig000003 | 136,865 | 140,549 |
| 3 | Contig000003 | 140,628 | 140,897 |
| 4 | Contig000003 | 140,978 | 141,433 |
| 5 | Contig000003 | 158,732 | 160,550 |
| 6 | Contig000003 | 167,167 | 169,203 |
| 7 | Contig 000002 | 176,434 | 176,695 |
| 8 | Contig000031 | 68,133 | 68,314 |
| 9 | Contig000051 | 75,685 | 76,012 |
| 10 | Contig000103 | 11,150 | 11,388 |
| 11 | Contig000106 | 41,651 | 41,808 |
| Contig | Start (bp) | Stop (bp) | Length (bp) | Classification | Identity (%) | |
|---|---|---|---|---|---|---|
| 1 | Contig000013 | 85,766 | 85,392 | 375 | aminoglycoside (ARO:3003395) | 83 |
| 2 | Contig000013 | 82,643 | 81,450 | 1194 | glycopeptide antibiotic (ARO:3005036) | 89 |
| Contig | Product | Start (bp) | Stop (bp) | Length (bp) | Query Coverage | Identity (%) | |
|---|---|---|---|---|---|---|---|
| 1 | contig00024 | Cell division protein FtsZ, ftsZ | 41,193 | 42,296 | 386 | 85 | 83 |
| 2 | contig00121 | RecA protein, recA | 1566 | 517 | 1050 | 93 | 88 |
| Contig | Start (bp) | Stop (bp) | Length (bp) | Function | |
|---|---|---|---|---|---|
| 1 | contig000144 | 11,078 | 11,380 | 303 | Urease gamma subunit (EC 3.5.1.5) UreA |
| 2 | contig000144 | 11,377 | 11,682 | 306 | Urease beta subunit (EC 3.5.1.5) UreB |
| 3 | contig000144 | 11,679 | 13,388 | 1710 | Urease alpha subunit (EC 3.5.1.5) UreC |
| 4 | contig000144 | 13,391 | 14,050 | 660 | Urease accessory protein UreF |
| 5 | contig000144 | 13,391 | 14,050 | 660 | Urease accessory protein UreF |
| 6 | contig000144 | 13,391 | 14,050 | 660 | Urease accessory protein UreF |
| 7 | contig000144 | 14,040 | 14,738 | 699 | Urease accessory protein UreG |
| 8 | contig000144 | 14,040 | 14,738 | 699 | Urease accessory protein UreG |
| 9 | contig000144 | 14,735 | 15,394 | 660 | Urease accessory protein UreD |
| 10 | contig000144 | 14,735 | 15,394 | 660 | Urease accessory protein UreD |
| 11 | contig000003 | 213,232 | 214,527 | 1296 | Urea ABC transporter, substrate-binding protein UrtA |
| 12 | contig000003 | 214,640 | 215,524 | 885 | Urea ABC transporter, permease protein UrtB |
| 13 | contig000003 | 215,521 | 216,615 | 1095 | Urea ABC transporter, permease protein UrtC |
| 14 | contig000003 | 216,612 | 217,448 | 837 | Urea ABC transporter, ATPase protein UrtD |
| 15 | contig000003 | 217,450 | 218,133 | 684 | Urea ABC transporter, ATPase protein UrtE |
| 16 | contig000174 | 8890 | 9240 | 351 | Urea ABC transporter, ATPase protein UrtE |
| Region | Type | From | To | Most Similar Known Cluster | Similarity | |
|---|---|---|---|---|---|---|
| Region 7.1 | other | 1 | 32,122 | |||
| Region 7.2 | butyrolactone | 181,275 | 189,513 | colabomycin E | Polyketide:Type II | 4% |
| Region 9.1 | betalactone | 127,964 | 156,777 | |||
| Region 23.1 | LAP | 63,471 | 86,862 | |||
| Region 28.1 | terpene | 63,486 | 89,541 | hopene | Terpene | 46% |
| Region 29.1 | NRPS | 1 | 53,614 | gobichelin A/gobichelin B | NRP | 27% |
| Region 33.1 | redox-cofactor | 47,552 | 69,629 | lankacidin C | NRP + Polyketide | 20% |
| Region 38.1 | lanthipeptide-class-iv | 1 | 17,570 | salinilactam | Polyketide | 8% |
| Region 47.1 | RiPP-like | 1 | 5832 | |||
| Region 52.1 | thioamide-NRP | 23,205 | 83,117 | netropsin | NRP | 27% |
| Region 58.1 | redox-cofactor | 54,961 | 76,748 | |||
| Region 62.1 | NAPAA | 27,298 | 61,263 | |||
| Region 79.1 | terpene | 39,001 | 58,687 | isorenieratene | Terpene | 25% |
| Region 81.1 | lassopeptide | 19,124 | 41,635 | siamycin I | RiPP | 28% |
| Region 87.1 | T2PKS | 1 | 51,425 | arenimycin A | Polyketide:Type II + Saccharide:Hybrid/tailoring | 68% |
| Region 90.1 | terpene | 35,800 | 49,030 | calicheamicin | Polyketide | 5% |
| Region 99.1 | T3PKS | 20,966 | 45,958 | alkyl-O-dihydrogeranyl-methoxyhydroquinones | Terpene + Polyketide | 57% |
| Region 118.1 | NRPS | 8823 | 36,925 | A54145 | NRP | 3% |
| Region 229.1 | siderophore | 253 | 6171 | |||
| Twin-Arginine Translocation Protein, TAT Protein (TatA, TatB, TatC) | ||||
|---|---|---|---|---|
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000009 | 102,753 | 102,487 | 267 |
| 2 | contig000071 | 21,417 | 20,446 | 972 |
| 3 | contig000071 | 21,698 | 21,432 | 267 |
| 4 | contig000128 | 29,800 | 29,369 | 432 |
| Acyl-CoA Synthase (EC 6.2.1.1) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000098 | 31,823 | 33,694 | 1872 |
| 2 | contig000151 | 21,809 | 19,845 | 1965 |
| Transcriptional Regulator, TetR Family on the Same Contig as lcp Gene | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 362,871 | 363,434 | 564 |
| 2 | contig000082 | 7433 | 8032 | 600 |
| 3 | contig000082 | 41,434 | 40,763 | 672 |
| 4 | contig000082 | 43,443 | 42,790 | 654 |
| Acyl-CoA Dehydrogenase (EC 1.3.8.1) (EC 1.3.8.8) (Putative) | |||
| Contig | Start (bp) | Stop (bp) | Length (bp) |
| contig000001 | 308,373 | 309,527 | 1155 |
| contig000002 | 210,799 | 211,995 | 1197 |
| contig000002 | 226,160 | 224,997 | 1164 |
| contig000006 | 79,214 | 80,359 | 1146 |
| contig000006 | 91,911 | 93,056 | 1146 |
| contig000006 | 109,177 | 110,382 | 1206 |
| contig000006 | 123,342 | 124,502 | 1161 |
| contig000007 | 32,267 | 31,008 | 1260 |
| contig000007 | 42,815 | 43,978 | 1164 |
| contig000008 | 111,357 | 112,454 | 1098 |
| contig000009 | 125,869 | 124,712 | 1158 |
| contig000010 | 136,785 | 135,604 | 1182 |
| contig000010 | 151,583 | 150,426 | 1158 |
| contig000015 | 106,328 | 105,189 | 1140 |
| contig000015 | 125,651 | 124,686 | 966 |
| contig000015 | 126,787 | 125,651 | 1137 |
| contig000022 | 41,492 | 42,652 | 1161 |
| contig000026 | 55,545 | 54,331 | 1215 |
| contig000031 | 60,451 | 59240 | 1212 |
| contig000032 | 93,065 | 91,926 | 1140 |
| contig000039 | 4910 | 3720 | 1191 |
| contig000039 | 26,773 | 27,924 | 1152 |
| contig000043 | 24,574 | 26,259 | 1686 |
| contig000058 | 40,472 | 41,617 | 1146 |
| contig000058 | 44,004 | 45,245 | 1242 |
| contig000070 | 60,103 | 61,281 | 1179 |
| contig000079 | 56,242 | 57,411 | 1170 |
| contig000079 | 57,408 | 58,457 | 1050 |
| contig000082 | 5300 | 6439 | 1140 |
| contig000082 | 39,644 | 40,777 | 1134 |
| contig000095 | 11,194 | 12,378 | 1185 |
| contig000095 | 12,375 | 13,523 | 1149 |
| contig000102 | 18,437 | 17,304 | 1134 |
| contig000108 | 19,517 | 20,767 | 1251 |
| contig000116 | 8097 | 6244 | 1854 |
| contig000121 | 6633 | 5491 | 1143 |
| contig000128 | 19,042 | 17,828 | 1215 |
| contig000154 | 11,134 | 9848 | 1287 |
| contig000154 | 12,217 | 11,150 | 1068 |
| contig000154 | 13,405 | 12,230 | 1176 |
| contig000157 | 3155 | 4306 | 1152 |
| contig000202 | 7053 | 8201 | 1149 |
| contig000218 | 2054 | 126 | 1929 |
| 2,4-dienoyl-CoA Reductase (EC 1.3.1.34) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000007 | 126,818 | 125,733 | 1086 |
| Enoyl-CoA Hydratases (EC 4.2.1.17) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 296,029 | 295,283 | 747 |
| 2 | contig000001 | 300,636 | 301,421 | 786 |
| 3 | contig000001 | 307,165 | 306,362 | 804 |
| 4 | contig000001 | 329,756 | 330,586 | 831 |
| 5 | contig000001 | 344,214 | 344,978 | 765 |
| 6 | contig000002 | 202,945 | 202,163 | 783 |
| 7 | contig000002 | 206,845 | 205,931 | 915 |
| 8 | contig000006 | 81,885 | 81,088 | 798 |
| 9 | contig000006 | 89,484 | 90,293 | 810 |
| 10 | contig000006 | 97,828 | 96,953 | 876 |
| 11 | contig000006 | 114,743 | 11,3946 | 798 |
| 12 | contig000006 | 118,886 | 118,125 | 762 |
| 13 | contig000006 | 120,324 | 122,495 | 2172 |
| 14 | contig000006 | 124,537 | 125,340 | 804 |
| 15 | contig000008 | 131,053 | 131,946 | 894 |
| 16 | contig000009 | 130,569 | 131,345 | 777 |
| 17 | contig000015 | 104,062 | 103,208 | 855 |
| 18 | contig000017 | 101,441 | 100,656 | 786 |
| 19 | contig000019 | 132,075 | 132,860 | 786 |
| 20 | contig000025 | 2819 | 2001 | 819 |
| 21 | contig000025 | 43,741 | 42,917 | 825 |
| 22 | contig000027 | 47,243 | 48,025 | 783 |
| 23 | contig000028 | 22,979 | 23,818 | 840 |
| 24 | contig000028 | 26,480 | 28,549 | 2070 |
| 25 | contig000031 | 14,833 | 12,764 | 2070 |
| 26 | contig000032 | 69,957 | 70,736 | 780 |
| 27 | contig000032 | 90,750 | 89,941 | 810 |
| 28 | contig000039 | 8258 | 8986 | 729 |
| 29 | contig000039 | 12,002 | 11,265 | 738 |
| 30 | contig000039 | 33,697 | 34,668 | 972 |
| 31 | contig000039 | 35,986 | 36,708 | 723 |
| 32 | contig000043 | 48,021 | 50,117 | 2097 |
| 33 | contig000045 | 37,575 | 36,832 | 744 |
| 34 | contig000047 | 5992 | 5237 | 756 |
| 35 | contig000048 | 27,562 | 26,795 | 768 |
| 36 | contig000070 | 58,232 | 59,101 | 870 |
| 37 | contig000070 | 61,278 | 62,096 | 819 |
| 38 | contig000082 | 28,779 | 30,890 | 2112 |
| 39 | contig000095 | 10,175 | 9405 | 771 |
| 40 | contig000095 | 20,285 | 21,049 | 765 |
| 41 | contig000095 | 30,027 | 29,176 | 852 |
| 42 | contig000114 | 7605 | 8369 | 765 |
| 43 | contig000174 | 16,732 | 17,184 | 453 |
| 44 | contig000227 | 3486 | 2695 | 792 |
| 3-Hydroxyacyl-CoA Dehydrogenases (EC 1.1.1.35) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000017 | 39,722 | 41,509 | 1788 |
| 2 | contig000018 | 104,278 | 105,594 | 1317 |
| 3 | contig000095 | 18,898 | 18,137 | 762 |
| 4 | contig000095 | 21,871 | 21,101 | 771 |
| Thiolase/3-Ketoacyl-CoA Thiolase (EC 2.3.1.16) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 189,682 | 190,872 | 1191 |
| 2 | contig000001 | 318,428 | 317,205 | 1224 |
| 3 | contig000001 | 337,505 | 336,360 | 1146 |
| 4 | contig000006 | 78,027 | 79,214 | 1188 |
| 5 | contig000006 | 119,071 | 120,294 | 1224 |
| 6 | contig000015 | 116,318 | 115,128 | 1191 |
| 7 | contig000015 | 118,590 | 119,738 | 1149 |
| 8 | contig000017 | 68,346 | 69,533 | 1188 |
| 9 | contig000018 | 104,185 | 102,911 | 1275 |
| 10 | contig000027 | 53,803 | 54,987 | 1185 |
| 11 | contig000028 | 25,272 | 26,474 | 1203 |
| 12 | contig000031 | 16,023 | 14,830 | 1194 |
| 13 | contig000032 | 86,429 | 85,236 | 1194 |
| 14 | contig000032 | 97,872 | 95,914 | 1959 |
| 15 | contig000043 | 46,807 | 48,018 | 1212 |
| 16 | contig000058 | 45,434 | 46,648 | 1215 |
| 17 | contig000082 | 27,565 | 28,776 | 1212 |
| 18 | contig000095 | 20,128 | 18,941 | 1188 |
| 19 | contig000105 | 40,143 | 38,899 | 1245 |
| 20 | contig000007 | 41,311 | 42,423 | 1113 |
| 21 | contig000039 | 18,566 | 19,750 | 1185 |
| α-Methylacyl-CoA Racemase (Mcr) (EC 5.1.99.4) | ||||
| Contig | Start (bp) | Stop (bp) | Length (bp) | |
| 1 | contig000001 | 326,963 | 328,114 | 1152 |
| 2 | contig000027 | 43,150 | 41,963 | 1188 |
| 3 | contig000032 | 91,899 | 90,757 | 1143 |
| 4 | contig000082 | 3034 | 1871 | 1164 |
| 5 | contig000095 | 15,365 | 16,525 | 1161 |
References
- Cornish, K. Alternative natural rubber crops: Why should we care? Technol. Innov. 2017, 18, 244–255. [Google Scholar] [CrossRef]
- Blaettler, K.G. The Manufacturing Process of Rubber. 2018. Available online: https://sciencing.com/manufacturing-process-rubber-5206099.html (accessed on 23 August 2021).
- Valdés, C.; Hernández, C.; Morales-Vera, R.; Andler, R. Desulfurization of vulcanized rubber particles using biological and couple microwave-chemical methods. Front. Environ. Sci. 2021, 9, 1–10. [Google Scholar] [CrossRef]
- Andler, R. Bacterial and enzymatic degradation of poly(cis-1,4-isoprene) rubber: Novel biotechnological applications. Biotechnol. Adv. 2020, 44, 107606. [Google Scholar] [CrossRef] [PubMed]
- Kasai, D. Poly(cis-1,4-isoprene)-cleavage enzymes from natural rubber-utilizing bacteria. Biosci. Biotechnol. Biochem. 2020, 84, 1089–1097. [Google Scholar] [CrossRef]
- Spence, D.; Van Niel, C.B. Bacterial decomposition of the rubber in Hevea latex. Ind. Eng. Chem. 1936, 28, 847–850. [Google Scholar] [CrossRef]
- De Vries, O. Decomposition of rubber hydrocarbons by fungi. Zentbl. Bakteriol. Parasitenkd. Infekt. 1928, 74, 22–24. [Google Scholar]
- Shaposhnikov, V.; Rabotnova, I.; Iarmola, G.; Kuznetsova, V.; Mozokhina-Porshniakova, N.N. Destructive effect of bacterial development on natural rubber. Mikrobiologiia 1952, 21, 47–54. [Google Scholar]
- Röther, W.; Birke, J.; Grond, S.; Beltran, J.M.; Jendrossek, D. Production of functionalized oligo-isoprenoids by enzymatic cleavage of rubber. Microb. Biotechnol. 2017, 10, 1426–1433. [Google Scholar] [CrossRef]
- Hiessl, S.; Schuldes, J.; Thürmer, A.; Halbsguth, T.; Bröker, D.; Angelov, A.; Liebl, W.; Daniel, R.; Steinbüchel, A. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl. Environ. Microbiol. 2012, 78, 2874–2887. [Google Scholar] [CrossRef] [Green Version]
- Andler, R.; Vivod, R.; Steinbüchel, A. Synthesis of polyhydroxyalkanoates through the biodegradation of poly(cis-1,4-isoprene) rubber. J. Biosci. Bioeng. 2019, 127, 360–365. [Google Scholar] [CrossRef]
- Tsuchii, A.; Suzuki, T.; Takeda, K. Microbial degradation of natural rubber vulcanizates. Appl. Environ. Microbiol. 1985, 50, 965–970. [Google Scholar] [CrossRef] [Green Version]
- Azadi, D.; Shojaei, H.; Mobasherizadeh, S.; Naser, A.D. Screening, isolation and molecular identification of biodegrading mycobacteria from Iranian ecosystems and analysis of their biodegradation activity. AMB Express 2017, 7, 180. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, M.; Williams, S.T. Ecology of actinomycetes. Annu. Rev. Microbiol. 1983, 37, 189–216. [Google Scholar] [CrossRef]
- Manurung, R.; Abdullah, Z.C.; Ahmad, F.B.; Kuek, C. Biodiversity-biotechnology: Gateway to discoveries. In Proceedings of the Sustainable Utilization and Wealth Creation, Kuching, Sarawak, Malaysia, 19–21 November 2008. [Google Scholar]
- Hassan, Q.P.; Bhat, A.M.; Shah, A.M. Chapter 6: Bioprospecting actinobacteria for bioactive secondary metabolites from untapped ecoregions of the Northwestern Himalayas. In New and Future Developments in Microbial Biotechnology and Bioengineering: Microbial Secondary Metabolites Biochemistry and Applications; Elsevier: Amsterdam, The Netherlands, 2019; pp. 77–85. Available online: https://doi.org/10.1016/B978-0-444-63504-4.00006-2 (accessed on 25 August 2021).
- Clark, T. Enhancing the biodegradation of waste rubber discarded rubber materials. In Proceedings of the International Latex Conference, Fairlawn, OH, USA; 2015. Available online: https://www.rubbernews.com/assets/PDF/RN10077285.PDF (accessed on 24 July 2020).
- Chengalroyen, M.D.; Dabbs, E.R. The biodegradation of latex rubber: A minireview. J. Polym. Environ. 2013, 21, 874–880. [Google Scholar] [CrossRef]
- Shivlata, L.; Satyanarayana, T. Thermophilic and alkaliphilic actinobacteria: Biology and potential applications. Front. Microbiol. 2015, 6, 1014. [Google Scholar] [CrossRef]
- Linh, D.V.; Huong, L.N.; Tabata, M.; Imai, S.; Iijima, S.; Kasai, D.; Anh, T.K.; Fukuda, M. Characterization and functional expression of a rubber degradation gene of a Nocardia degrader from a rubber-processing factory. J. Biosci. Bioeng. 2017, 123, 412–418. [Google Scholar] [CrossRef]
- Jendrossek, D.; Tomasi, G.; Kroppenstedt, R.M. Bacterial degradation of natural rubber: A privilege of actinomycetes? FEMS Microbiol. Lett. 1997, 150, 179–188. [Google Scholar] [CrossRef]
- Vivod, R.; Andler, R.; Oetermann, S.; Altenhoff, A.L.; Seipel, N.; Holtkamp, M.; Hogeback, J.; Karst, U.; Steinbüchel, A. Characterization of the latex clearing protein of the poly(cis-1,4-isoprene) and poly(trans-1,4-isoprene) degrading bacterium Nocardia nova SH22a. J. Gen. Appl. Microbiol. 2019, 65, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Hiessl, S.; Poehlein, A.; Daniel, R.; Steinbüchel, A. Insights into the microbial degradation of rubber and gutta-percha by analysis of the complete genome of Nocardia nova SH22a. Appl. Environ. Microbiol. 2014, 80, 3895–3907. [Google Scholar] [CrossRef] [Green Version]
- Watcharakul, S.; Röther, W.; Birke, J.; Umsakul, K.; Hodgson, B.; Jendrossek, D. Biochemical and spectroscopic characterization of purified latex clearing protein (Lcp) from newly isolated rubber degrading Rhodococcus rhodochrous strain RPK1 reveals novel properties of Lcp. BMC Microbiol. 2016, 16, 92. [Google Scholar] [CrossRef] [Green Version]
- Nawong, C.; Umsakul, K.; Sermwittayawong, N. Rubber gloves biodegradation by a consortium, mixed culture and pure culture isolated from soil samples. Braz. J. Microbiol. 2018, 49, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Akkharabunditsakul, V.; Tanrattanakul, V.; Umsakul, K. Biodegradation of synthetic rubbers by a mixed culture isolated from various rubber factory soils in Songkhla, Thailand. Int. J. Adv. Agric. Environ. Eng. 2017, 4, 33–37. [Google Scholar]
- Parte, A.C. LPSN-list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2018, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braaz, R.; Fischer, P.; Jendrossek, D. Novel type of Heme-dependent oxygenase catalyzes oxidative Cleavage of rubber (poly-cis-1,4-isoprene). Appl. Environ. Microbiol. 2004, 70, 7388–7395. [Google Scholar] [CrossRef] [Green Version]
- Chia, K.-H.; Nanthini, J.; Thottathil, G.P.; Najimudin, N.; Haris, M.R.H.M.; Sudesh, K. Identification of new rubber-degrading bacterial strains from aged latex. Polym. Degrad. Stab. 2014, 109, 354–361. [Google Scholar] [CrossRef]
- Heisey, R.M.; Papadatos, S. Isolation of microorganisms able to metabolize purified natural rubber. Appl. Environ. Microbiol. 1995, 61, 3092–3097. [Google Scholar] [CrossRef] [Green Version]
- Shirling, E.B.; Gottlieb, D. Methods for characterization of Streptomyces species. Int. J. Syst. Bacteriol. 1966, 16, 340. [Google Scholar] [CrossRef] [Green Version]
- Hamaki, T.; Suzuki, M.; Fudou, R.; Jojima, Y.; Kajiura, T.; Tabuchi, A.; Sen, K.; Shibai, H. Isolation of novel bacteria and actinomycetes using soil-extract agar medium. J. Biosci. Bioeng. 2005, 99, 485–492. [Google Scholar] [CrossRef]
- Applied Biosystems, Sequence Scanner; Thermo Fisher Scientific: Waltham, MA, USA; Available online: https://www.thermofisher.cn/cn/zh/home.html (accessed on 5 August 2021).
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acid Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bateman, A.; Bridge, A.; Wu, C. UniProt. UniProt Consortium (Online). Available online: https://www.uniprot.org/blast/ (accessed on 5 August 2021).
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Moore, E.; Arnscheidt, A.; Krüger, A.; Strömpl, C.; Mau, M. Section 1 update: Simplified protocols for the preparation of genomic DNA from bacterial cultures. Mol. Microb. Ecol. Man. 2008. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC bioinformatics resource center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; O′Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.K.; Durkin, A.S.; et al. RefSeq: Expanding the prokaryotic genome annotation pipeline reach with protein family model curation. Nucleic Acids Res. 2020, 49, 1020–1028. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Huerta-Cepas, J. EggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015, 44, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.; Cheng, A.A.; Liu, S.; et al. CARD: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acid Symp. Ser. 2013, 42, D206–D214. [Google Scholar] [CrossRef]
- UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef]
- Bendtsen, J.D.; Nielsen, H.; Widdick, D.; Palmer, T.; Brunak, S. Prediction of twin-arginine signal peptides. BMC Bioinform. 2005, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Auch, A.F.; Klenk, H.P.; Göker, M. Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2010, 2, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.M.; Tindall, B.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1000+ type-strain genomes substantially improves taxonomic classification of Alphaproteobacteria. Front. Bioeng. Biotechnol. 2020, 11, 468. [Google Scholar]
- Berekaa, M.M.; Linos, A.; Reichelt, R.; Keller, U.; Steinbüchel, A. Effect of pretreatment of rubber material on its biodegradability by various rubber degrading bacteria. FEMS Microbiol. Lett. 2000, 184, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Basik, A.A.; Juboi, H.; Shamsul, S.S.G.; Sanglier, J.J.; Yeo, T.C. Actinomycetes isolated from Wetland and Hill Paddy during the warm and cool Seasons in Sarawak, East Malaysia. J. Microbiol. Biotechnol. Food Sci. 2020, 9, 774–781. [Google Scholar]
- Rose, K.; Tenberge, K.B.; Steinbüchel, A. Identification and characterization of genes from Streptomyces sp. strain K30 responsible for clear zone formation on natural rubber latex and poly(cis-1,4-isoprene) rubber degradation. Biomacromolecules 2005, 6, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Röther, W.; Austen, S.; Birke, J.; Jendrossek, D. Molecular insights in the cleavage of rubber by the latex-clearing- protein (Lcp) of Streptomyces sp. strain K30. Appl. Environ. Microbiol. 2016, 82, 6593–6602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Oh, H.S.; Park, S.C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Volpiano, C.G.; Sant′Anna, F.H.; Ambrosini, A.; de São José, J.F.; Beneduzi, A.; Whitman, W.B.; de Souza, E.M.; Lisboa, B.B.; Vargas, L.K.; Passaglia, L.M. Genomic metrics applied to Rhizobiales (Hyphomicrobiales): Species reclassification, identification of unauthentic genomes and false type strains. Front. Microbiol. 2021, 12, 734850. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–667. [Google Scholar] [CrossRef]
- Wang, B.; Huang, C.; Zhao, H.; Benkovic, S.J. Unraveling the iterative type I polyketide synthases hidden in Streptomyces. Proc. Natl. Acad. Sci. USA 2020, 117, 8449–8454. [Google Scholar] [CrossRef]
- Tsuchii, A.; Takeda, K. Rubber-degrading enzyme from a bacterial culture. Appl. Environ. Microbiol. 1990, 56, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Rose, K.; Steinbüchel, A. Biodegradation of natural rubber and related compounds: Recent insights into a hardly understood catabolic capability of microorganisms. Appl. Environ. Microbiol. 2005, 71, 2803–2812. [Google Scholar] [CrossRef] [Green Version]
- Bode, H.B.; Zeeck, A.; Pluckhahn, K.; Jendrossek, D. Physiological and chemical investigations into microbial degradation of synthetic poly(cis-1,4-isoprene). Appl. Environ. Microbiol. 2000, 66, 3680–3685. [Google Scholar] [CrossRef] [Green Version]
- Hiessl, S.; Böse, D.; Oetermann, S.; Eggers, J.; Pietruszka, J.; Steinbüchel, A. Latex clearing protein-An oxygenase cleaving poly(cis-1,4-isoprene) rubber at the cis double bonds. Appl. Environ. Microbiol. 2014, 80, 5231–5240. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Siedenburg, G.; Birke, J.; Mobeen, F.; Jendrossek, D.; Prakash, T. Metabolic and taxonomic insights into the gram-negative natural rubber degrading bacterium Steroidobacter cummioxidans sp. nov., strain 35Y. PLoS ONE 2018, 13, e0200399. [Google Scholar]
- Oetermann, S.; Jongsma, R.; Coenen, A.; Keller, J.; Steinbüchel, A. LcpR VH2—Regulating the expression of latex-clearing proteins in Gordonia polyisoprenivorans VH2. Microbiology 2019, 165, 343–354. [Google Scholar] [CrossRef]
- Yikmis, M.; Arenskötter, M.; Rose, K.; Lange, N.; Wernsmann, H.; Wiefel, L.; Steinbüchel, A. Secretion and transcriptional regulation of the latex-clearing protein, Lcp, by the rubber-degrading bacterium Streptomyces sp. strain K30. Appl. Environ. Microbiol. 2008, 74, 5373–5382. [Google Scholar] [CrossRef] [Green Version]
- Coenen, A.; Oetermann, S.; Steinbüchel, A. Identification of LcpRB A3 (2), a novel regulator of Lcp expression in Streptomyces coelicolor A3 (2). Appl. Microbiol. Biotechnol. 2019, 103, 5715–5726. [Google Scholar] [CrossRef]
- Schulte, C.; Arenskötter, M.; Berekaa, M.M.; Arenskötter, Q.; Priefert, H.; Steinbüchel, A. Possible involvement of an extracellular superoxide dismutase (SodA) as a radical scavenger in poly(cis-1,4-isoprene) degradation. Appl. Environ. Microbiol. 2008, 74, 7643–7653. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing cultivation to identify bacterial species. Microbe 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef] [Green Version]
- Behera, B.K.; Chakraborty, H.J.; Patra, B.; Rout, A.K.; Dehury, B.; Das, B.K.; Sarkar, D.J.; Parida, P.K.; Raman, R.K.; Rao, A.R.; et al. Metagenomic analysis reveals bacterial and fungal diversity and their bioremediation potential from sediments of River Ganga and Yamuna in India. Front. Microbiol. 2020, 11, 2531. [Google Scholar] [CrossRef]
- Elustondo, P.; Zakharian, E.; Pavlov, E. Identification of the polyhydroxybutyrate granules in mammalian cultured cells. Chem. Biodivers. 2012, 9, 2597–2604. [Google Scholar] [CrossRef] [Green Version]
- Nanthini, J.; Ong, S.Y.; Sudesh, K. Identification of three homologous latex-clearing protein (lcp) genes from the genome of Streptomyces sp. strain CFMR 7. Gene 2017, 628, 146–155. [Google Scholar] [CrossRef]
- Gibu, N.; Arata, T.; Kuboki, S.; Linh, D.V.; Fukuda, M.; Steinbüchel, A.; Kasai, D. Characterization of the genes responsible for rubber degradation in Actinoplanes sp. strain OR16. Appl. Microbiol. Biotechnol. 2020, 104, 7367–7376. [Google Scholar] [CrossRef]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to read and interpret FTIR spectroscope of organic material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Birke, J.; Jendrossek, D. Rubber oxygenase and latex clearing protein cleave rubber to different products and use different cleavage mechanisms. Appl. Environ. Microbiol. 2014, 80, 5012–5020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fainleib, A.; Pires, R.V.; Lucas, E.F.; Soares, B.G. Degradation of non-vulcanized natural rubber—Renewable resource for fine chemicals used in polymer synthesis. Polimeros 2013, 23, 441–450. [Google Scholar] [CrossRef]
- Craciun, G.; Manaila, E.; Stelescu, M.D. Materials new elastomeric materials based on natural rubber obtained by electron beam irradiation for food and pharmaceutical use. Materials 2018, 9, 999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, K.M. The Microbial Devulcanisation of Waste Ground Tyre Rubber Using Acidophilic Microorganisms. Ph.D. Thesis, Stellenbosch University, Stellenbosch, South Africa, March 2018. [Google Scholar]
- Kole, P.J.; Löhr, A.J.; Van Belleghem, F.G.A.J.; Ragas, A.M.J. Wear and tear of tyres: A stealthy source of microplastics in the environment. Int. J. Environ. Res. Public Health 2017, 14, 1265. [Google Scholar] [CrossRef] [PubMed]
- Sieber, R.; Kawecki, D.; Nowack, B. Dynamic probabilistic material flow analysis of rubber release from tires into the environment. Environ. Pollut. 2020, 258, 113573. [Google Scholar] [CrossRef]










| Family | Genera | No. of Strains Tested | Clear Zone Formation |
|---|---|---|---|
| Streptomycetaceae | Streptomyces | 526 | 2 |
| Kitasatospora | 12 | 0 | |
| Micromonosporaceae | Micromonospora | 86 | 6 |
| Dactylosporangium | 3 | 1 | |
| Streptosporangiaceae | Nonomuraea | 31 | 2 |
| Microtetraspora | 11 | 7 | |
| Microbispora | 4 | 0 | |
| Herbidospora | 2 | 0 | |
| Thermoactinomycetaceae | Shimazuella | 1 | 0 |
| Thermomonosporaceae | Actinomadura | 8 | 0 |
| Actinoallomurus | 1 | 0 | |
| Pseudonocardiaceae | Actinomycetospora | 1 | 0 |
| Thermosporotrichaceae | Thermosporothrix | 1 | 0 |
| Mycobacteriaceae | Mycobacterium | 2 | 0 |
| Nocardiaceae | Nocardia | 46 | 0 |
| Others | 205 | 0 | |
| 940 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basik, A.A.; Nanthini, J.; Yeo, T.C.; Sudesh, K. Rubber Degrading Strains: Microtetraspora and Dactylosporangium. Polymers 2021, 13, 3524. https://doi.org/10.3390/polym13203524
Basik AA, Nanthini J, Yeo TC, Sudesh K. Rubber Degrading Strains: Microtetraspora and Dactylosporangium. Polymers. 2021; 13(20):3524. https://doi.org/10.3390/polym13203524
Chicago/Turabian StyleBasik, Ann Anni, Jayaram Nanthini, Tiong Chia Yeo, and Kumar Sudesh. 2021. "Rubber Degrading Strains: Microtetraspora and Dactylosporangium" Polymers 13, no. 20: 3524. https://doi.org/10.3390/polym13203524
APA StyleBasik, A. A., Nanthini, J., Yeo, T. C., & Sudesh, K. (2021). Rubber Degrading Strains: Microtetraspora and Dactylosporangium. Polymers, 13(20), 3524. https://doi.org/10.3390/polym13203524