
Solid-Phase Extraction of Active Compounds from Natural Products by Molecularly Imprinted Polymers: Synthesis and Extraction Parameters

 ,
,  ,
,
Abstract
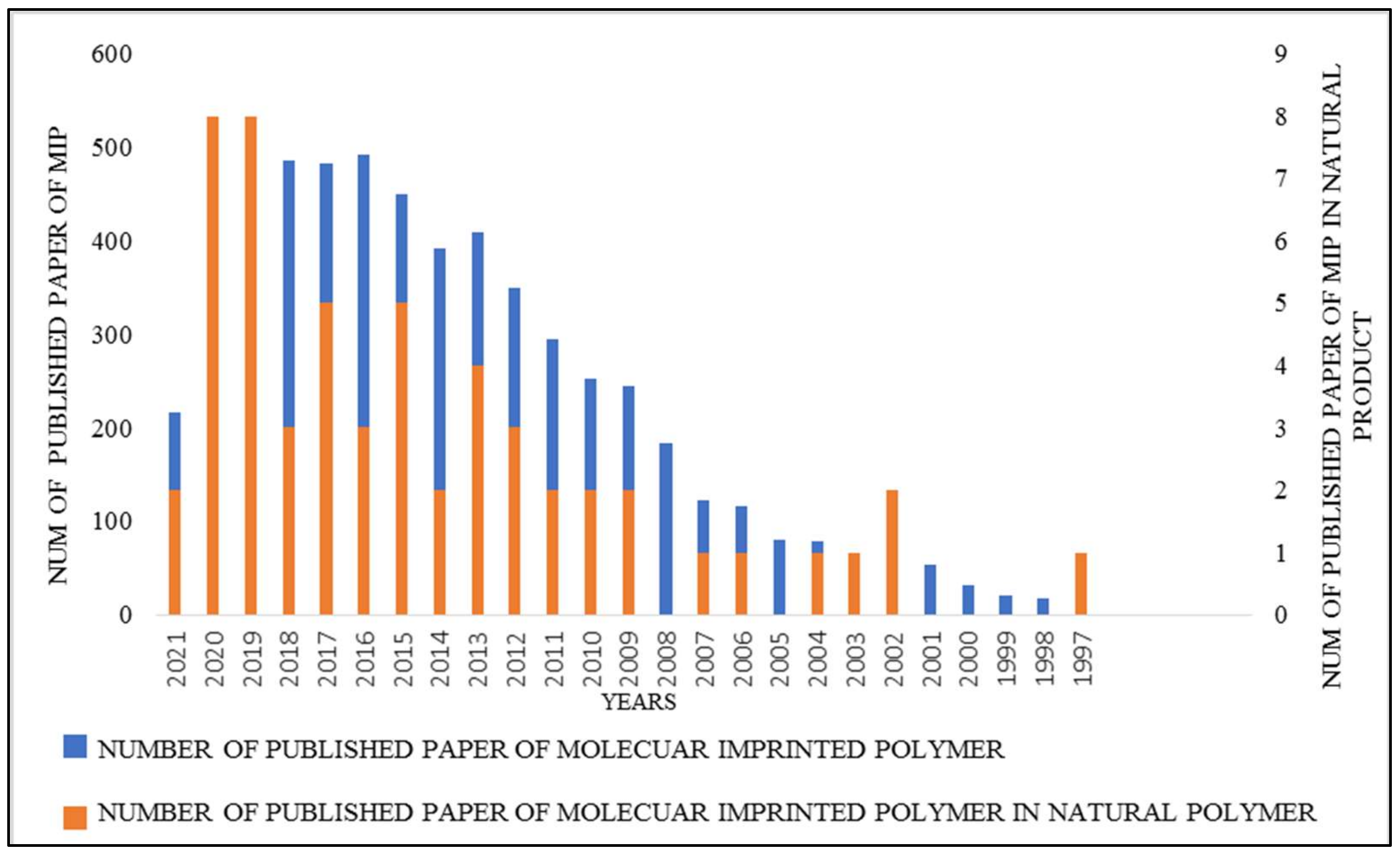
:1. Introduction
- “B”: Biological; usually a large (>50 residues) peptide or protein either isolated from an organism/cell line or produced by biotechnological means in a surrogate host
- “N”: Natural product
- “NB”: Natural product “Botanical” (in general these have been recognized as drug entities by the FDA)
- “ND”: Derived from a natural product and is usually a semisynthetic modification
- “S*”: Made by total synthesis, but the pharmacophore is/was from a natural product
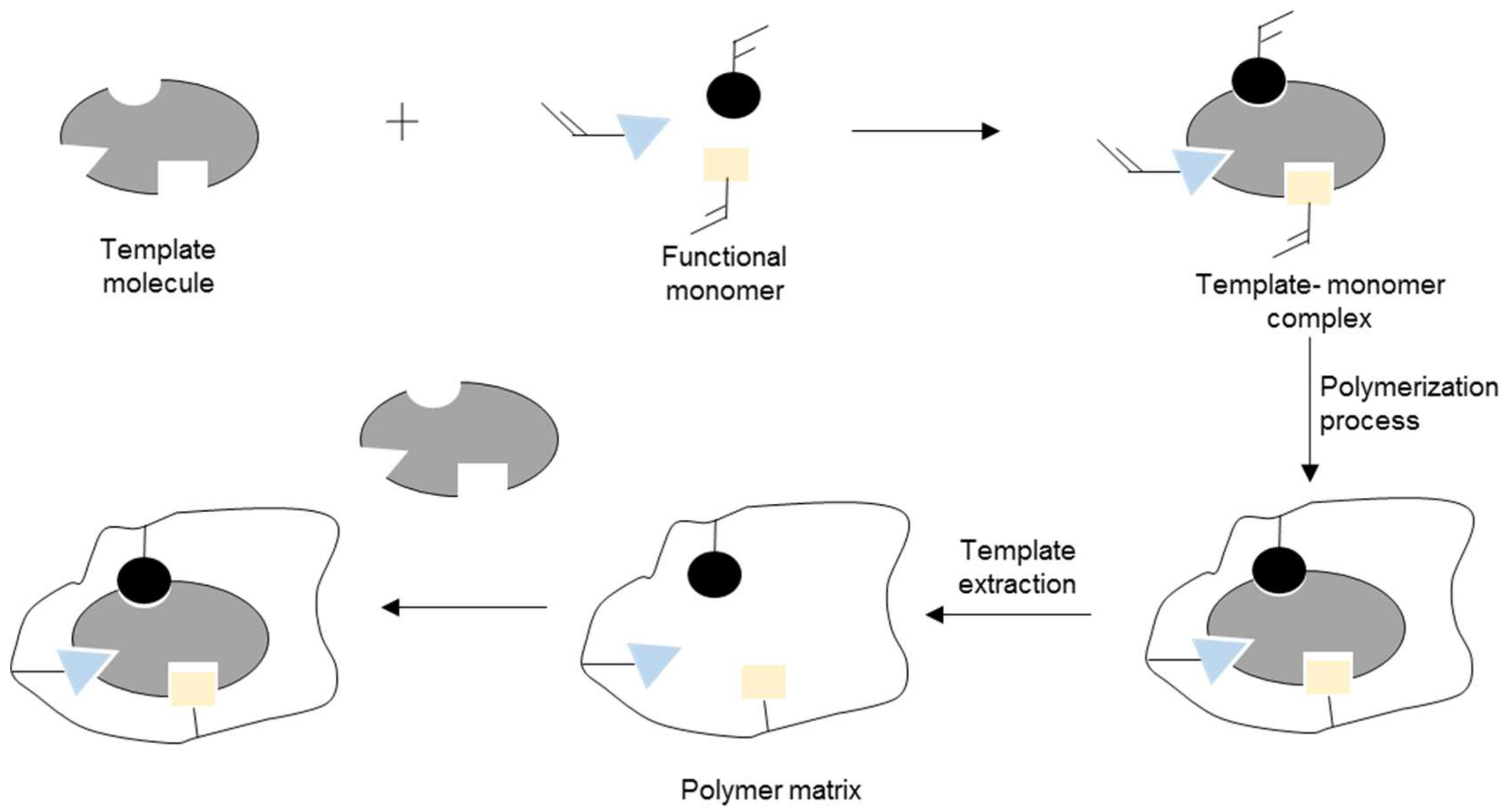
2. Synthesis and Characterization of MIP
2.1. Polymerization Parameter in MIP Synthesis
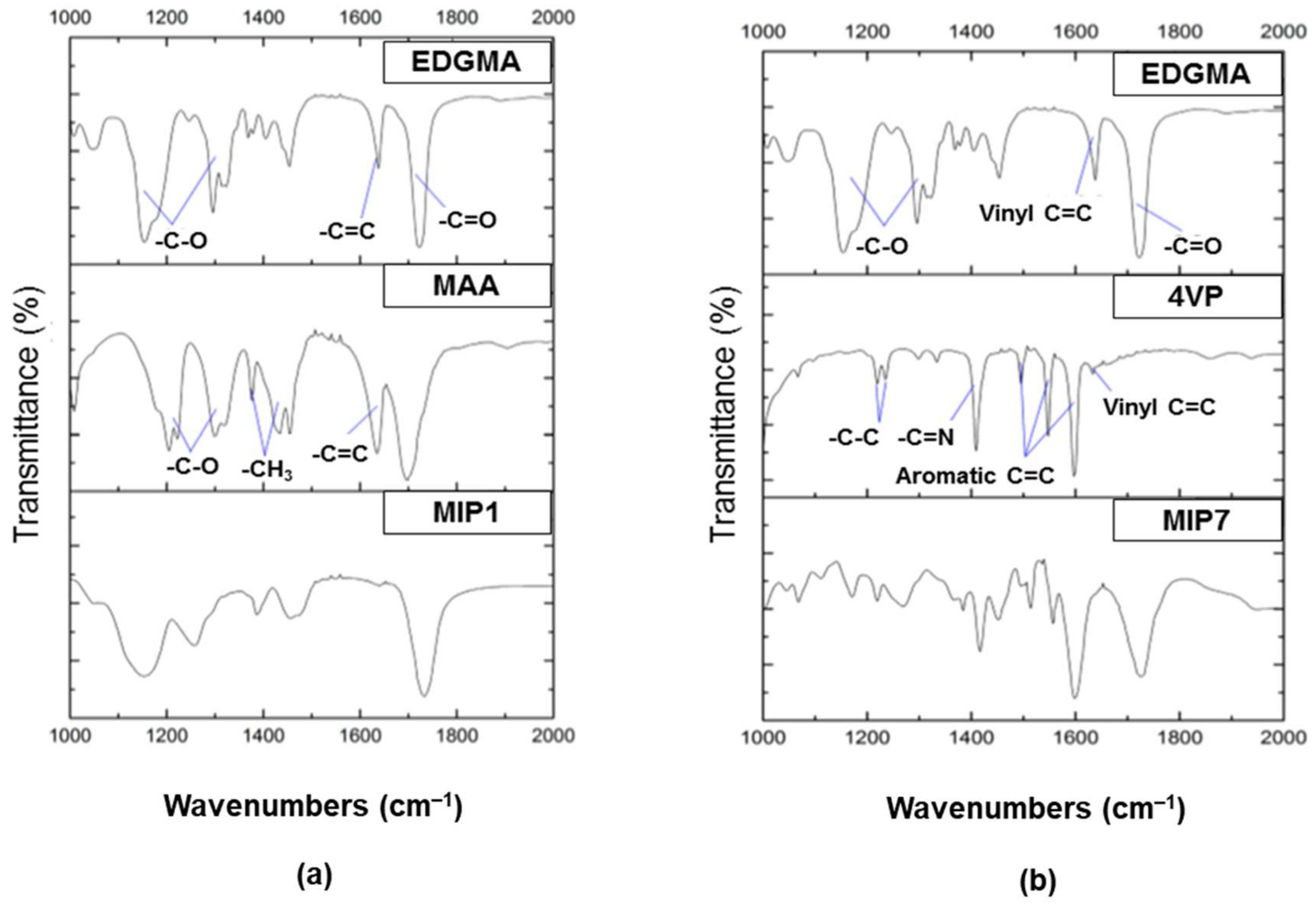
2.1.1. Functional Monomers
2.1.2. Template Molecule
2.1.3. Crosslinker
2.1.4. Porogenic Solvent
2.1.5. Initiator
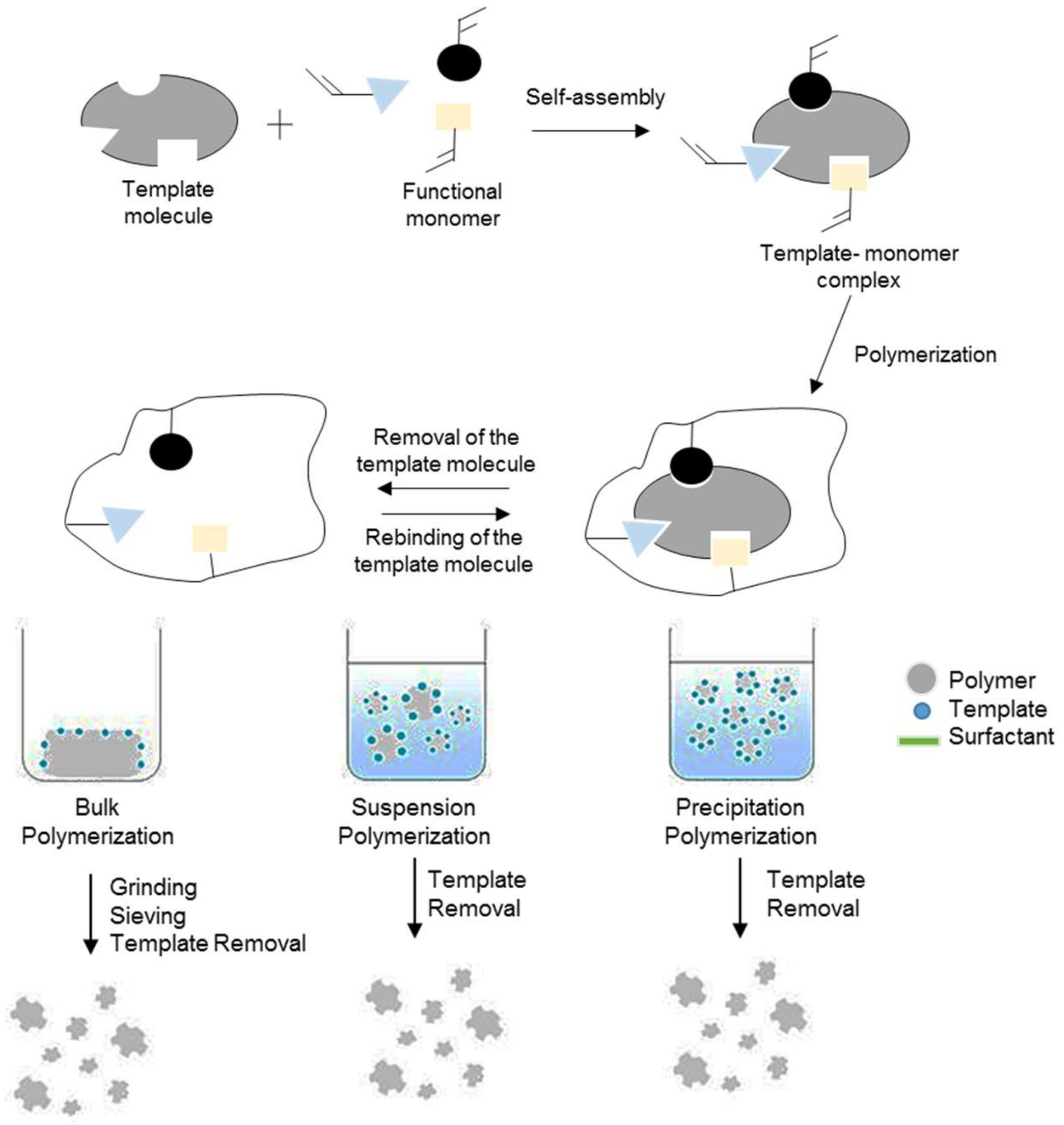
2.2. Polymerization Technique
2.2.1. Bulk Polymerization
2.2.2. In Situ Polymerization
2.2.3. Precipitation Polymerization
2.2.4. Suspension Polymerization
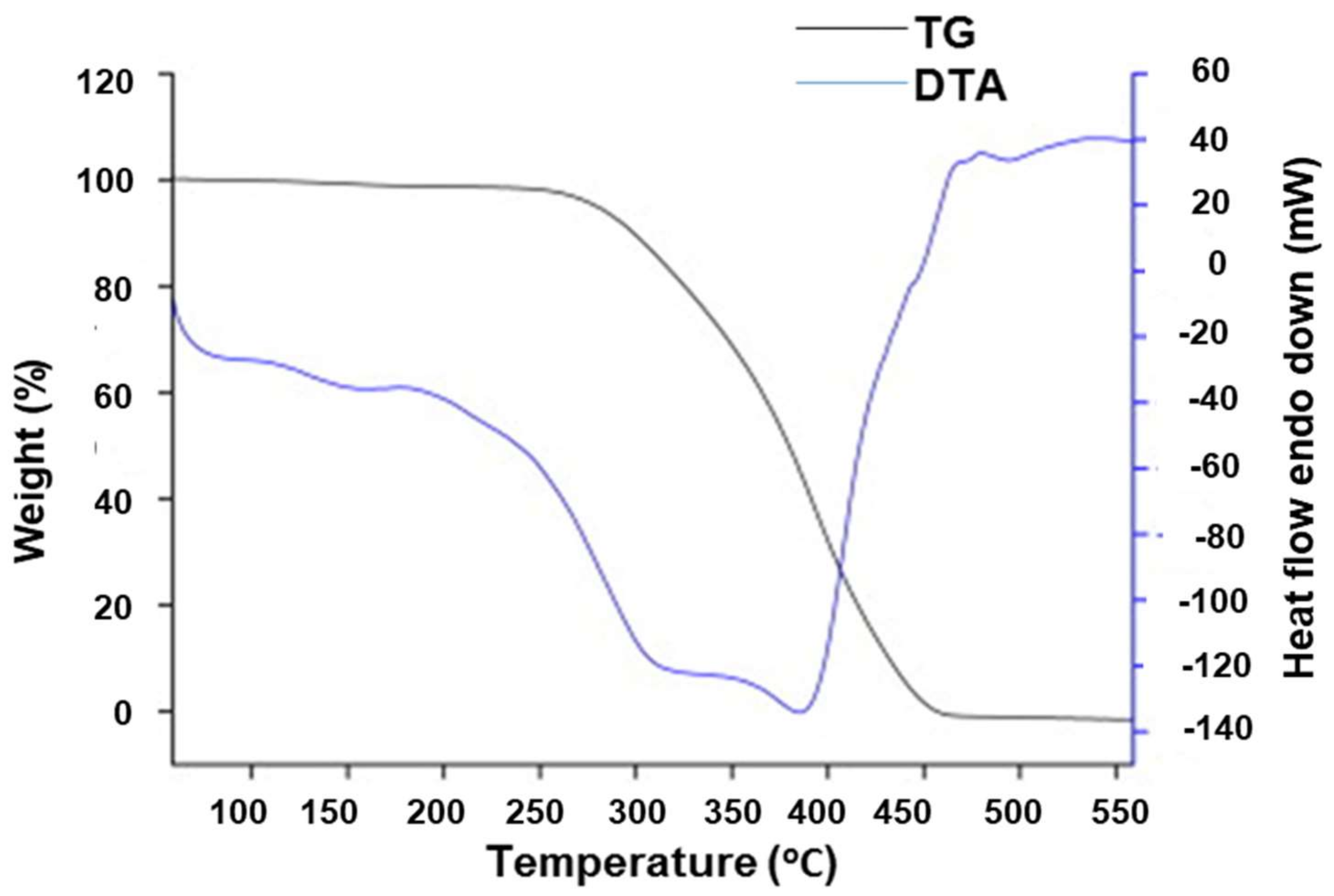
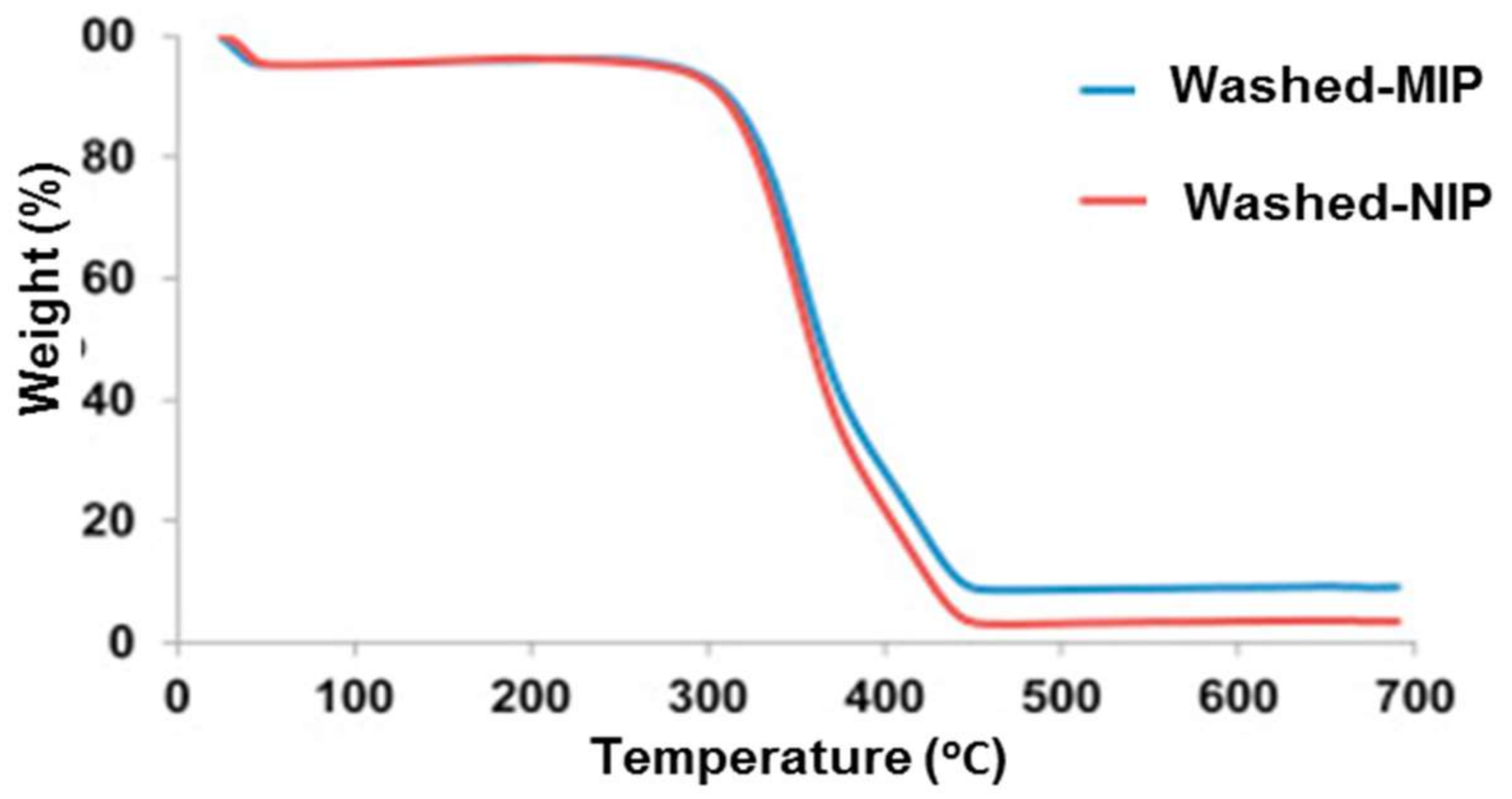
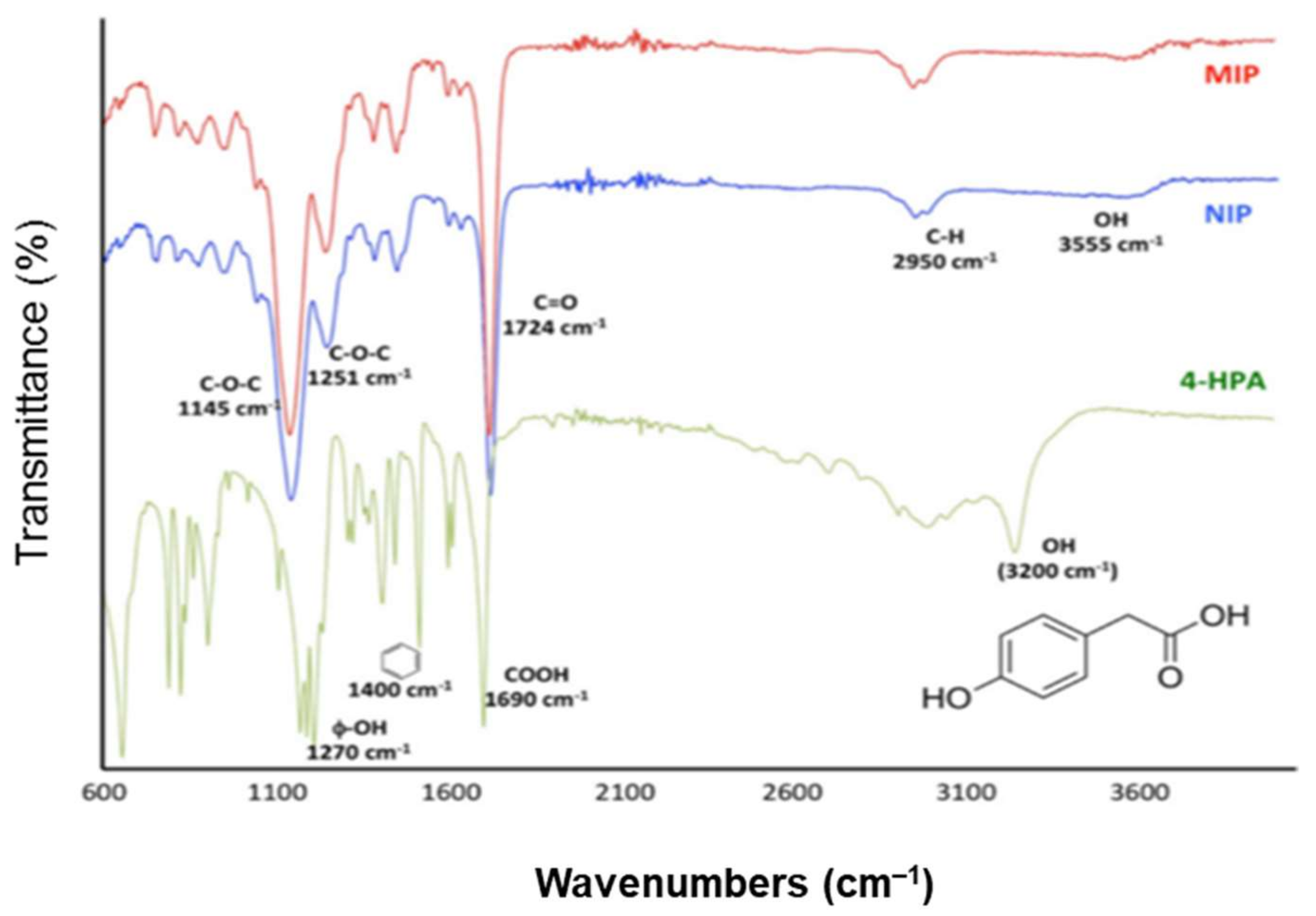
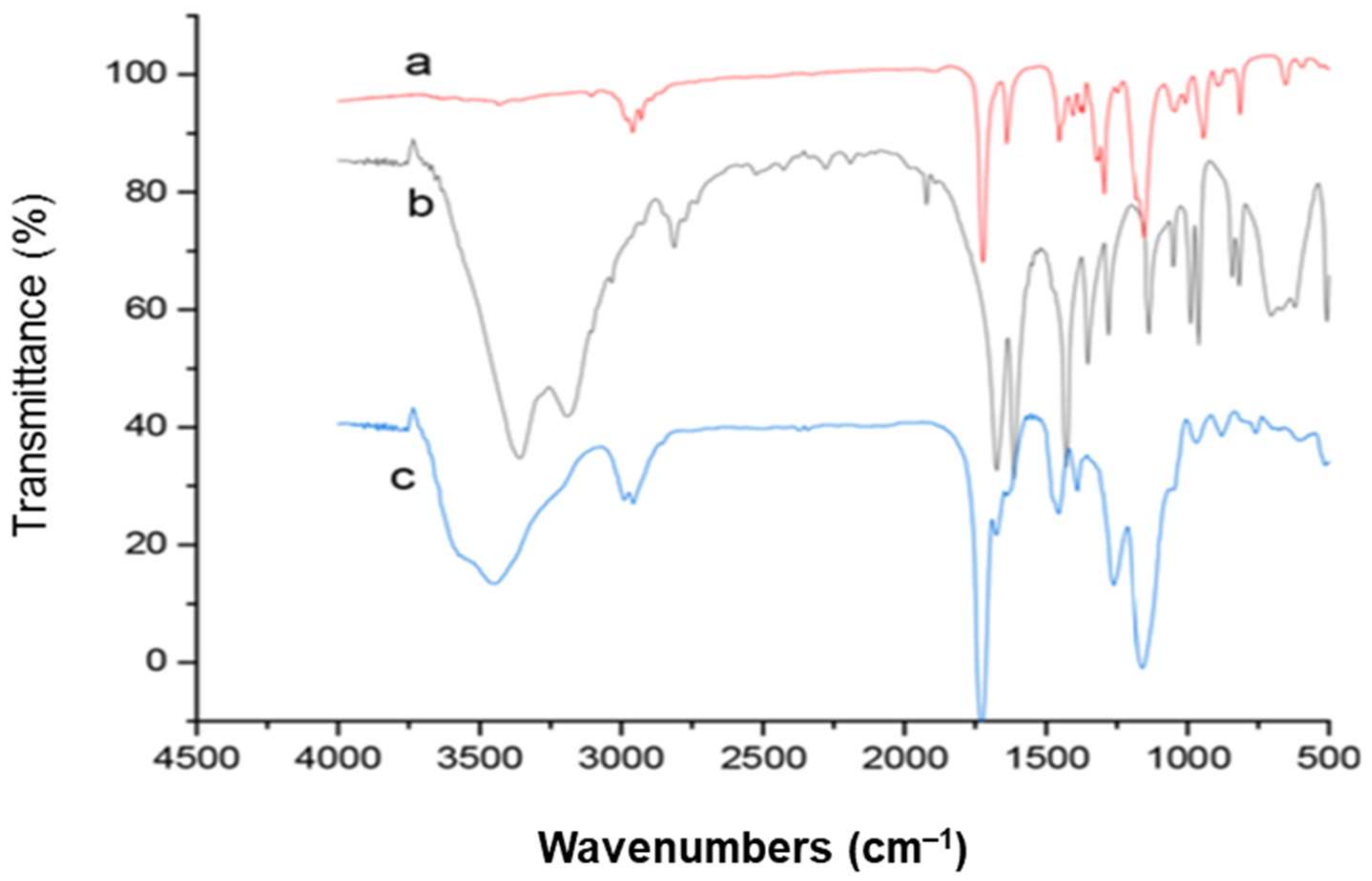
3. Characterization of Molecularly Imprinted Polymer
4. Molecular Imprinted Polymer in Solid Phase Extraction (MISPE)
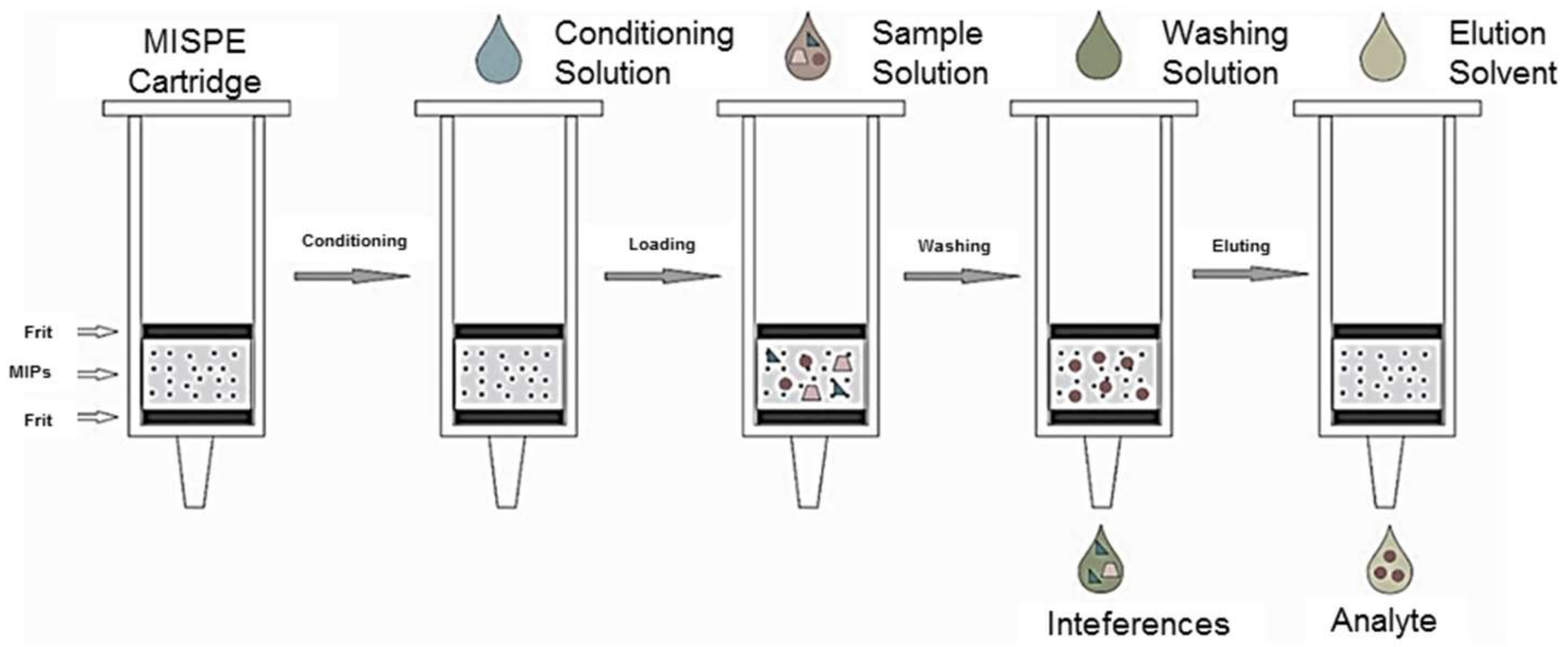
4.1. Offline Modes
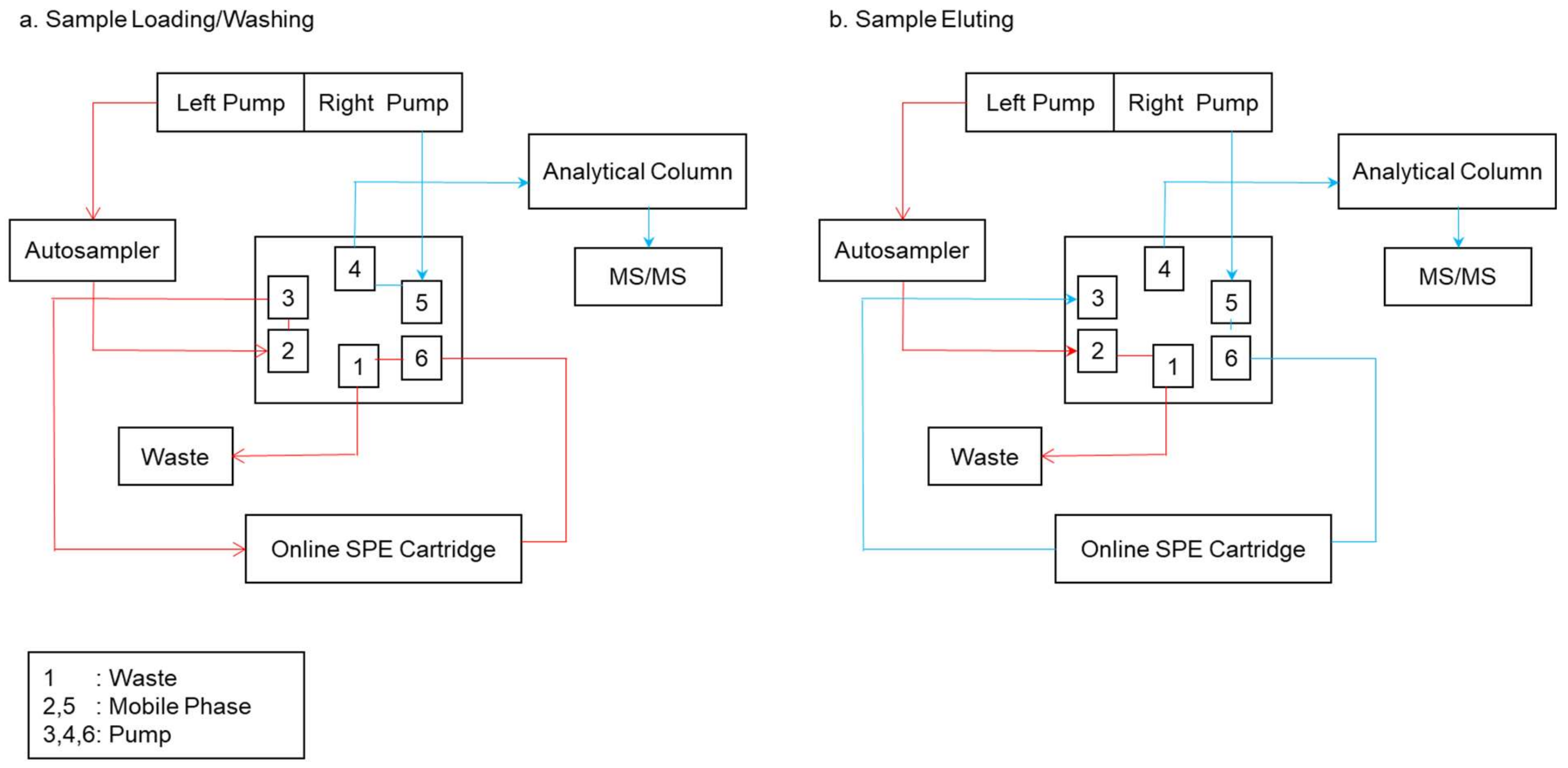
4.2. Online Modes
5. Future Perspective and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability
Conflicts of Interest
Abbreviations
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.W.-H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Fabricant, D.S.; Farnsworth, N.R. The value of plants used in traditional medicine for drug discovery. Environ. Health Perspect. 2001, 109, 69–75. [Google Scholar] [CrossRef]
- Yu, M.; Littlefield, B.; Kishi, Y. Discovery of E7389, a fully synthetic macrocyclic ketone analog of halichondrin, B. Anticancer Agents Nat. Prod. 2005, 23, 241–265. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Kingston, D.G. (Eds.) Anticancer Agents from Natural Products; Taylor & Francis: Boca Raton, FL, USA, 2005; pp. 137–150. [Google Scholar]
- Newman, D.J. The bryostatins 8. In Anticancer Agents from Natural Products; CRC Press: Boca Raton, FL, USA, 2012; pp. 199–218. [Google Scholar]
- Buss, A.D.; Cox, B.; Waigh, R.D. Natural products as leads for new pharmaceuticals. In Burger’s Medicinal Chemistry and Drug Discovery; Wiley-Interscience: Hoboken, NJ, USA, 1995; Volume 1, pp. 983–1033. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Olivon, F.; Allard, P.-M.; Koval, A.; Righi, D.; Genta-Jouve, G.; Neyts, J.; Apel, C.; Pannecouque, C.; Nothias, L.-F.; Cachet, X.; et al. Bioactive natural products prioritization using massive multi-informational molecular networks. ACS Chem. Biol. 2017, 12, 2644–2651. [Google Scholar] [CrossRef]
- Vasapollo, G.; del Sole, L.R.; Mergola, M.R.; Lazzoi, A.; Scardino, S.; Scorrano, G. Mele, molecularly imprinted polymers: Present and future prospective. Int. J. Mol. Sci. 2011, 12, 5908–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Cai, C.; Yang, H.; Chen, X. Synthesis and application of molecularly imprinted polymer on selective solid-phase extraction for the determination of artemisinin in Artemisia annua, L. Anal. Lett. 2013, 46, 107–119. [Google Scholar] [CrossRef]
- Bhawani, S.A.; Sen, T.S.; Ibrahim, M.N.M. Synthesis of molecular imprinting polymers for extraction of gallic acid from urine. Chem. Cent. J. 2018, 12, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xiao, R.; Tang, J.; Zhu, Q.; Li, X.; Xiong, Y.; Wu, X. Preparation and adsorption properties of molecularly imprinted polymer via RAFT precipitation polymerization for selective removal of aristolochic acid I. Talanta 2017, 162, 415–422. [Google Scholar] [CrossRef]
- Verma, K.S.; Xia, K. Analysis of triclosan and triclocarban in soil and biosolids using molecularly imprinted solid phase extraction coupled with HPLC-UV. J. AOAC Int. 2010, 93, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandimalla, V.B.; Ju, H. Molecular imprinting: A dynamic technique for diverse applications in analytical chemistry. ChemInform 2005, 380, 587–605. [Google Scholar] [CrossRef]
- Cirillo, G.; Curcio, M.; Parisi, O.I.; Puoci, F.; Iemma, F.; Spizzirri, U.G.; Restuccia, D.; Picci, N. Molecularly imprinted polymers for the selective extraction of glycyrrhizic acid from liquorice roots. Food Chem. 2011, 125, 1058–1063. [Google Scholar] [CrossRef]
- Karasová, G.; Lehotay, J.; Sádecká, J.; Skačáni, I.; Lachová, M. Selective extraction of derivates ofp-hydroxy-benzoic acid from plant material by using a molecularly imprinted polymer. J. Sep. Sci. 2005, 28, 2468–2476. [Google Scholar] [CrossRef]
- Yu, C.; Mosbach, K. Molecular imprinting utilizing an amide functional group for hydrogen bonding leading to highly efficient polymers. J. Org. Chem. 1997, 62, 4057–4064. [Google Scholar] [CrossRef]
- Xie, J.; Zhu, L.; Luo, H.; Zhou, L.; Li, C.; Xu, X. Direct extraction of specific pharmacophoric flavonoids from gingko leaves using a molecularly imprinted polymer for quercetin. J. Chromatogr. A 2001, 934, 1–11. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, A.L.; Xu, X. Application of a molecularly imprinted polymer for the effective recognition of different anti-epidermal growth factor receptor inhibitors. Anal. Chem. 2003, 75, 6381–6387. [Google Scholar] [CrossRef]
- Hu, S.-G.; Li, L.; He, X.-W. Solid-phase extraction of esculetin from the ash bark of Chinese traditional medicine by using molecularly imprinted polymers. J. Chromatogr. A 2005, 1062, 31–37. [Google Scholar] [CrossRef]
- Cao, H.; Xiao, J.B.; Xu, M. Evaluation of new selective molecularly imprinted polymers for the extraction of resveratrol from Polygonum cuspidatum. Macromol. Res. 2006, 14, 324–330. [Google Scholar] [CrossRef]
- Lucci, P.; Nuñez, O.; Galceran, M. Solid-phase extraction using molecularly imprinted polymer for selective extraction of natural and synthetic estrogens from aqueous samples. J. Chromatogr. A 2011, 1218, 4828–4833. [Google Scholar] [CrossRef] [Green Version]
- Mlunguza, N.Y.; Ncube, S.; Mahlambi, P.N.; Chimuka, L.; Madikizela, L.M. Adsorbents and removal strategies of non-steroidal anti-inflammatory drugs from contaminated water bodies. J. Environ. Chem. Eng. 2019, 7, 103142. [Google Scholar] [CrossRef]
- Tanabe, K.; Takeuchi, T.; Matsui, J.; Ikebukuro, K.; Yano, K.; Karube, I. Recognition of barbiturates in molecularly imprinted copolymers using multiple hydrogen bonding. J. Chem. Soc. Chem. Commun. 1995, 2303–2304. [Google Scholar] [CrossRef]
- Manesiotis, P.; Hall, A.J.; Courtois, J.; Irgum, K.; Sellergren, B. An artificial riboflavin receptor prepared by a template analogue imprinting strategy. Angew. Chem. Int. Ed. 2005, 44, 3902–3906. [Google Scholar] [CrossRef]
- Sellergren, B.; Andersson, L.I. Application of imprinted synthetic polymers in binding assay development. Methods 2000, 22, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Molecular imprinting in cross-linked materials with the aid of molecular templates—A way towards artificial antibodies. Angew. Chem. Int. Ed. 1995, 34, 1812–1832. [Google Scholar] [CrossRef]
- Simon, R.; Collins, M.E.; Spivak, D.A. Shape selectivity versus functional group pre-organization in molecularly imprinted polymers. Anal. Chim. Acta 2007, 591, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Spivak, D.A.; Simon, R.; Campbell, J. Evidence for shape selectivity in non-covalently imprinted polymers. Anal. Chim. Acta 2004, 504, 23–30. [Google Scholar] [CrossRef]
- Kempe, M. Antibody-mimicking polymers as chiral stationary phases in HPLC. Anal. Chem. 1996, 68, 1948–1953. [Google Scholar] [CrossRef]
- Andersson, H.S.; Karlsson, J.G.; Piletsky, S.; Koch-Schmidt, A.-C.; Mosbach, K.; Nicholls, I.A. Study of the nature of recognition in molecularly imprinted polymers, II: Influence of monomer–template ratio and sample load on retention and selectivity. J. Chromatogr. A 1999, 848, 39–49. [Google Scholar] [CrossRef]
- Kim, H.; Spivak, D.A. New insight into modeling non-covalently imprinted polymers. J. Am. Chem. Soc. 2003, 125, 11269–11275. [Google Scholar] [CrossRef] [PubMed]
- Spivak, D.A. Optimization, evaluation, and characterization of molecularly imprinted polymers. Adv. Drug Deliv. Rev. 2005, 57, 1779–1794. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, A.; Karlsson, L.; Owens, P.K.; Berggren, C.; Crecenzi, C.; Ensing, K.; Bayoudh, S.; Cormack, P.; Sherrington, D.; Sellergren, B. Evaluation of methods aimed at complete removal of template from molecularly imprinted polymers. Analyst 2001, 126, 784–792. [Google Scholar] [CrossRef]
- Lorenzo, R.A.; Carro, A.M.; Alvarez-Lorenzo, C.; Concheiro, A. To remove or not to remove? The Challenge of extracting the template to make the cavities available in molecularly imprinted polymers (MIPs). Int. J. Mol. Sci. 2011, 12, 4327–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.-F.; Zhang, Y.-J.; Zhang, Y.-P.; Chen, J.; Zhou, X.-M.; Bai, L.-Y. Synthesis and evaluation of molecularly imprinted polymer for the determination of the phthalate esters in the bottled beverages by HPLC. J. Chem. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Osman, R.; Saim, N.; Anuar, N.M.; Subari, S.N.M. Application of molecular imprinted polymer solid phase extraction (MISPE) in the extraction of caffeine from coffee. Open Conf. Proc. J. 2014, 4, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Crescenzi, C.; Bayoudh, S.; Cormack, P.A.G.; Klein, T.; Ensing, K. Determination of clenbuterol in bovine liver by combining matrix solid-phase dispersion and molecularly imprinted solid-phase extraction followed by liquid chromatography/electrospray ion trap multiple-stage mass spectrometry. Anal. Chem. 2001, 73, 2171–2177. [Google Scholar] [CrossRef] [PubMed]
- Zander, Å.; Findlay, P.; Renner, A.T.; Sellergren, B.; Swietlow, A. Analysis of nicotine and its oxidation products in nicotine chewing gum by a molecularly imprinted solid-phase extraction. Anal. Chem. 1998, 70, 3304–3314. [Google Scholar] [CrossRef]
- BelBruno, J.J. Molecularly imprinted polymers. Chem. Rev. 2019, 119, 94–119. [Google Scholar] [CrossRef]
- Marty, J.-D.; Tizra, M.; Mauzac, M.; Rico-Lattes, I.; Lattes, A. New molecular imprinting materials: Liquid crystalline networks. Macromolecules 1999, 32, 8674–8677. [Google Scholar] [CrossRef]
- Song, X.; Li, J.; Wang, J.; Chen, L. Quercetin molecularly imprinted polymers: Preparation, recognition characteristics and properties as sorbent for solid-phase extraction. Talanta 2009, 80, 694–702. [Google Scholar] [CrossRef]
- Shea, K.J.; Sasaki, D.Y.; Stoddard, G.J. Fluorescence probes for evaluating chain solvation in network polymers. An analysis of the solvatochromic shift of the dansyl probe in macroporous styrene-divinylbenzene and styrene-diisopropenylbenzene copolymers. Macromolecules 1989, 22, 1722–1730. [Google Scholar] [CrossRef]
- Gavrilović, I.; Mitchell, K.; Brailsford, A.D.; Cowan, D.A.; Kicman, A.T.; Ansell, R.J. A molecularly imprinted receptor for separation of testosterone and epitestosterone, based on a steroidal cross-linker. Steroids 2011, 76, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A. A note about crosslinking density in imprinting polymerization. Molecules 2021, 26, 5139. [Google Scholar] [CrossRef] [PubMed]
- Guyot, A.; Bartholin, M. Design and properties of polymers as materials for fine chemistry. Prog. Polym. Sci. 1982, 8, 277–331. [Google Scholar] [CrossRef]
- Horák, D.; Švec, F.; Bleha, M.; Kálal, J. Reactive polymers, XXXV. The effect of polymerization conditions on the specific surface area of macroporous copolymers from glycidylmethacrylate-ethylenedimethacrylate. Angew. Makromol. Chemie 1981, 95, 109–115. [Google Scholar] [CrossRef]
- Sellergren, B.; Shea, K.J. Influence of polymer morphology on the ability of imprinted network polymers to resolve enantiomers. J. Chromatogr. A 1993, 635, 31–49. [Google Scholar] [CrossRef]
- Cormack, P.A.; Elorza, A.Z. Molecularly imprinted polymers: Synthesis and characterisation. J. Chromatogr. B 2004, 804, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Piletsky, S.A.; Piletskaya, E.V.; Panasyuk, T.L.; El’Skaya, A.V.; Levi, R.; Karube, A.I.; Wulff, G. Imprinted membranes for sensor technology: Opposite behavior of covalently and noncovalently imprinted membranes. Macromolecules 1998, 31, 2137–2140. [Google Scholar] [CrossRef]
- Kugimiya, A.; Takeuchi, T.; Matsuib, J.; Ikebukuro, K.; Yano, K.; Karube, I. Recognition in novel molecularly imprinted polymer sialic acid receptors in aqueous media. Anal. Lett. 1996, 29, 1099–1107. [Google Scholar] [CrossRef]
- Janczura, M.; Luliński, P.; Sobiech, M. Imprinting technology for effective sorbent fabrication: Current state-of-art and future prospects. Materials 2021, 14, 1850. [Google Scholar] [CrossRef]
- He, C.; Long, Y.; Pan, J.; Li, K.; Liu, F. Application of molecularly imprinted polymers to solid-phase extraction of analytes from real samples. J. Biochem. Biophys. Methods 2007, 70, 133–150. [Google Scholar] [CrossRef] [PubMed]
- He, J.-X.; Pan, H.-Y.; Xu, L.; Tang, R.-Y. Application of molecularly imprinted polymers for the separation and detection of aflatoxin. J. Chem. Res. 2021, 45, 400–410. [Google Scholar] [CrossRef]
- Mohajeri, S.A.; Karimi, G.; Aghamohammadian, J.; Khansari, M.R. Clozapine recognition via molecularly imprinted polymers; bulk polymerization versus precipitation method. J. Appl. Polym. Sci. 2011, 121, 3590–3595. [Google Scholar] [CrossRef]
- Gornik, T.; Shinde, S.; Lamovsek, L.; Koblar, M.; Heath, E.; Sellergren, B.; Kosjek, T. Molecularly imprinted polymers for the removal of antidepressants from contaminated wastewater. Polymers 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Sergeyeva, T.A.; Piletsky, S.A.; Piletska, E.V.; Brovko, O.O.; Karabanova, L.V.; Sergeeva, L.M.; El’Skaya, A.V.; Turner, A.P.F. In situ formation of porous molecularly imprinted polymer membranes. Macromolecules 2003, 36, 7352–7357. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, Y.; Zhang, Y.; Wang, K. Synthesizing vitamin E molecularly imprinted polymers via precipitation polymeriza-tion. J. Chem. Eng. Data 2019, 64, 1045–1050. [Google Scholar] [CrossRef]
- Gomes, C.; Sadoyan, G.; Dias, R.C.S.; Costa, M.R. Development of molecularly imprinted polymers to target polyphenols present in plant extracts. Processes 2017, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Zhang, L.; Song, C.; Zhang, X. Molecularly imprinted solid-phase extraction of matrine from radix Sophorae tonkinensis. Analyst 2011, 136, 3016–3022. [Google Scholar] [CrossRef]
- Guć, M.; Schroeder, G. Molecularly imprinted polymers and magnetic molecularly imprinted polymers for selective determination of estrogens in water by ESI-MS/FAPA-MS. Biomolecules 2020, 10, 672. [Google Scholar] [CrossRef]
- Bitas, D.; Samanidou, V. Molecularly imprinted polymers as extracting media for the chromatographic determination of antibiotics in milk. Molecules 2018, 23, 316. [Google Scholar] [CrossRef] [Green Version]
- Viveiros, R.; Rebocho, S.; Casimiro, T. Green strategies for molecularly imprinted polymer development. Polymers 2018, 10, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Serpa, A.; González-Martín, R.; Sajid, M.; Pino, V. Greenness of magnetic nanomaterials in miniaturized extraction techniques: A review. Talanta 2021, 225, 122053. [Google Scholar] [CrossRef]
- Orowitz, T.E.; Ana Sombo, P.; Rahayu, D.; Hasanah, A.N. Microsphere polymers in molecular imprinting: Current and future perspectives. Molecules 2020, 25, 3256. [Google Scholar] [CrossRef]
- Sajid, M.; Płotka-Wasylka, J. Combined extraction and microextraction techniques: Recent trends and future perspectives. TrAC Trends Anal. Chem. 2018, 103, 74–86. [Google Scholar] [CrossRef]
- Kalinowska, K.; Lenartowicz, P.; Namieśnik, J.; Marć, M. Analytical procedures for short chain chlorinated paraffins determination—How to make them greener? Sci. Total Environ. 2019, 671, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Sobiech, M.; Luliński, P.; Wieczorek, P.P.; Marć, M. Quantum and carbon dots conjugated molecularly imprinted polymers as advanced nanomaterials for selective recognition of analytes in environmental, food and biomedical applications. TrAC Trends Anal. Chem. 2021, 142, 116306. [Google Scholar] [CrossRef]
- Mullett, W.M.; Lai, E.P.C. Determination of theophylline in serum by molecularly imprinted solid-phase extraction with pulsed elution. Anal. Chem. 1998, 70, 3636–3641. [Google Scholar] [CrossRef] [PubMed]
- Le Noir, M.; Lepeuple, A.-S.; Guieysse, B.; Mattiasson, B. Selective removal of 17β-estradiol at trace concentration using a molecularly imprinted polymer. Water Res. 2007, 41, 2825–2831. [Google Scholar] [CrossRef]
- Yusof, N.A.; Zakaria, N.D.; Maamor, N.A.M.; Abdullah, A.H.; Haron, J. Synthesis and characterization of molecularly imprinted polymer membrane for the removal of 2,4-dinitrophenol. Int. J. Mol. Sci. 2013, 14, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Asman, S.; Mohamad, S.; Sarih, N.M. Exploiting β-cyclodextrin in molecular imprinting for achieving recognition of benzylparaben in aqueous media. Int. J. Mol. Sci. 2015, 16, 3656–3676. [Google Scholar] [CrossRef]
- Abu Samah, N.; Sánchez-Martín, M.-J.; Sebastián, R.M.; Valiente, M.; López-Mesas, M. Molecularly imprinted polymer for the removal of diclofenac from water: Synthesis and characterization. Sci. Total Environ. 2018, 631–632, 1534–1543. [Google Scholar] [CrossRef]
- Piletska, E.V.; Burns, R.; Terry, L.A.; Piletsky, S.A. Application of a molecularly imprinted polymer for the extraction of kukoamine a from potato peels. J. Agric. Food Chem. 2011, 60, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qin, C.; Li, D.; Hou, Y.; Li, S.; Sun, J. Molecularly imprinted polymer for specific extraction of hypericin from Hypericum perforatum L. herbal extract. J. Pharm. Biomed. Anal. 2014, 98, 210–220. [Google Scholar] [CrossRef]
- Xu, Z.; Gao, C. In situ polymerization approach to graphene-reinforced nylon-6 composites. Macromolecules 2010, 43, 6716–6723. [Google Scholar] [CrossRef]
- Amut, E.; Fu, Q.; Fang, Q.; Liu, R.; Xiao, A.; Zeng, A.; Chang, C. In situ polymerization preparation of chiral molecular imprinting polymers monolithic column for amlodipine and its recognition properties study. J. Polym. Res. 2009, 17, 401–409. [Google Scholar] [CrossRef]
- Dai, C.-M.; Zhang, J.; Zhang, Y.-L.; Zhou, X.-F.; Duan, Y.-P.; Liu, S.-G. Removal of carbamazepine and clofibric acid from water using double templates–molecularly imprinted polymers. Environ. Sci. Pollut. Res. 2013, 20, 5492–5501. [Google Scholar] [CrossRef]
- Dai, C.-M.; Geissen, S.-U.; Zhang, Y.; Zhang, Y.-J.; Zhou, X.-F. Selective removal of diclofenac from contaminated water using molecularly imprinted polymer microspheres. Environ. Pollut. 2011, 159, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Hemmati, K.; Sahraei, R.; Ghaemy, M. Synthesis and characterization of a novel magnetic molecularly imprinted polymer with incorporated graphene oxide for drug delivery. Polymer 2016, 101, 257–268. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, T.; Xu, W.; Huang, W.; Wang, N.; Yang, W. Synthesis and characterization of fluorescence molecularly imprinted polymers as sensor for highly sensitive detection of dibutyl phthalate from tap water samples. Sens. Actuators B Chem. 2017, 240, 1114–1122. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, W.; Xu, W.; Li, H.; Huang, W.; Jiang, B.; Zhou, Z.; Yan, Y. Synthesis and characterization of a surface molecular imprinted polymer as a new adsorbent for the removal of dibenzothiophene. J. Chem. Eng. Data 2012, 57, 1713–1720. [Google Scholar] [CrossRef]
- Liu, X.; Wu, F.; Au, C.; Tao, Q.; Pi, M.; Zhang, W. Synthesis of molecularly imprinted polymer by suspension polymerization for selective extraction of p-hydroxybenzoic acid from water. J. Appl. Polym. Sci. 2018, 136, 46984. [Google Scholar] [CrossRef]
- Bi, W.; Tian, M.; Row, K.H. Separation of phenolic acids from natural plant extracts using molecularly imprinted anion-exchange polymer confined ionic liquids. J. Chromatogr. A 2012, 1232, 37–42. [Google Scholar] [CrossRef]
- Arabi, M.; Ghaedi, M.; Ostovan, A. Synthesis and application of in-situ molecularly imprinted silica monolithic in pipette-tip solid-phase microextraction for the separation and determination of gallic acid in orange juice samples. J. Chromatogr. B 2017, 1048, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mora-Granados, M.; González-Gómez, D.; Jeong, J.; Gallego-Picó, A. A molecularly imprinted polymer for selective extraction of phenolic acids from human urine. Appl. Sci. 2021, 11, 1577. [Google Scholar] [CrossRef]
- Cantarella, M.; Carroccio, S.C.; Dattilo, S.; Avolio, R.; Castaldo, R.; Puglisi, C.; Privitera, V. Molecularly imprinted polymer for selective adsorption of diclofenac from contaminated water. Chem. Eng. J. 2019, 367, 180–188. [Google Scholar] [CrossRef]
- Huang, M.; Pang, W.; Zhang, J.; Lin, S.; Hu, J. A target analogue imprinted polymer for the recognition of antiplatelet active ingredients in Radix Salviae Miltiorrhizae by LC/MS/MS. J. Pharm. Biomed. Anal. 2012, 58, 12–18. [Google Scholar] [CrossRef]
- Sanagi, M.M.; Salleh, S.; Ibrahim, W.A.W.; Abu Naim, A.; Hermawan, D.; Miskam, M.; Hussain, I.; Aboul-Enein, H.Y. Molecularly imprinted polymer solid-phase extraction for the analysis of organophosphorus pesticides in fruit samples. J. Food Compos. Anal. 2013, 32, 155–161. [Google Scholar] [CrossRef]
- He, X.; Mei, X.; Wang, J.; Lian, Z.; Tan, L.; Wu, W. Determination of diethylstilbestrol in seawater by molecularly imprinted solid-phase extraction coupled with high-performance liquid chromatography. Mar. Pollut. Bull. 2016, 102, 142–147. [Google Scholar] [CrossRef]
- Li, X.; Cao, X.; Zhang, Z.; Zhang, Z.; Jiang, Z.; Yin, J. Synthesis of molecularly imprinted polymer adsorbents for solid-phase extraction of strobilurin fungicides from agricultural products. J. Sep. Sci. 2020, 43, 2133–2141. [Google Scholar] [CrossRef]
- Zunngu, S.S.; Madikizela, L.M.; Chimuka, L.; Mdluli, P. Synthesis and application of a molecularly imprinted polymer in the solid-phase extraction of ketoprofen from wastewater. Comptes Rendus Chim. 2017, 20, 585–591. [Google Scholar] [CrossRef]
- Maranata, G.J.; Surya, N.O.; Hasanah, A.N. Optimising factors affecting solid phase extraction performances of molecular imprinted polymer as recent sample preparation technique. Heliyon 2021, 7, e05934. [Google Scholar] [CrossRef]
- Yi, L.-X.; Fang, R.; Chen, G.-H. Molecularly imprinted solid-phase extraction in the analysis of agrochemicals. ChemInform 2014, 45, 608–618. [Google Scholar] [CrossRef]
- Chen, F.-F.; Wang, R.; Shi, Y.-P. Molecularly imprinted polymer for the specific solid-phase extraction of kirenol from Siegesbeckia pubescens herbal extract. Talanta 2012, 89, 505–512. [Google Scholar] [CrossRef]
- Theodoridis, G.; Zacharis, C.K.; Tzanavaras, P.D.; Themelis, D.G.; Economou, A. Automated sample preparation based on the sequential injection principle: Solid-phase extraction on a molecularly imprinted polymer coupled on-line to high-performance liquid chromatography. J. Chromatogr. A 2004, 1030, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Caro, E.; Marcé, R.M.; Cormack, P.A.; Sherrington, D.C.; Borrull, F. On-line solid-phase extraction with molecularly imprinted polymers to selectively extract substituted 4-chlorophenols and 4-nitrophenol from water. J. Chromatogr. A 2003, 995, 233–238. [Google Scholar] [CrossRef]
- Bjarnason, B.; Chimuka, L.; Ramström, O. On-line solid-phase extraction of triazine herbicides using a molecularly imprinted polymer for selective sample enrichment. Anal. Chem. 1999, 71, 2152–2156. [Google Scholar] [CrossRef]
- Yilmaz, E.; Soylak, M. Type of new generation separation and preconcentration methods. In New Generation Green Solvents for Separation and Preconcentration of Organic and Inorganic Species; Elsevier: Amsterdam, The Netherlands, 2020; pp. 75–148. [Google Scholar]
- Li, H.; Li, H.; Chen, Z.; Lin, J. On-chip solid phase extraction coupled with electrophoresis using modified magnetic microspheres as stationary phase. Sci. China Ser. B Chem. 2009, 52, 2287–2294. [Google Scholar] [CrossRef]
- Turiel, E.; Martín-Esteban, A. Molecularly imprinted polymers-based microextraction techniques. TrAC Trends Anal. Chem. 2019, 118, 574–586. [Google Scholar] [CrossRef]
- Zi-Hui, M.; Qin, L. Determination of degradation products of nerve agents in human serum by solid phase extraction using molecularly imprinted polymer. Anal. Chim. Acta 2001, 435, 121–127. [Google Scholar] [CrossRef]
- Zhu, X.; Yang, J.; Su, Q.; Cai, J.; Gao, Y. Molecularly imprinted polymer for monocrotophos and its binding characteristics for organophosphorus pesticides. Ann. Chim. 2005, 95, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, S.; Jahani, M. Selective solid-phase extraction using molecular imprinted polymer sorbent for the analysis of Florfenicol in food samples. Food Chem. 2013, 141, 1242–1251. [Google Scholar] [CrossRef]
- Jiang, X.; Zhao, C.; Jiang, N.; Zhang, H.; Liu, M. Selective solid-phase extraction using molecular imprinted polymer for the analysis of diethylstilbestrol. Food Chem. 2008, 108, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, E.C.; Tarley, C.R.T.; Kubota, L.T.; Rath, S.; Arruda, M.A.Z. On-line molecularly imprinted solid phase extraction for the selective spectrophotometric determination of catechol. Microchem. J. 2007, 85, 290–296. [Google Scholar] [CrossRef]
- Masqué, N.; Marcé, R.M.; Borrull, F.; Cormack, P.A.G.; Sherrington, D.C. Synthesis and evaluation of a molecularly imprinted polymer for selective on-line solid-phase extraction of 4-nitrophenol from environmental water. Anal. Chem. 2000, 72, 4122–4126. [Google Scholar] [CrossRef]
- He, J.; Wang, S.; Fang, G.; Zhu, H.; Zhang, Y. Molecularly imprinted polymer online solid-phase extraction coupled with high-performance liquid chromatography—UV for the determination of three sulfonamides in pork and chicken. J. Agric. Food Chem. 2008, 56, 2919–2925. [Google Scholar] [CrossRef]
- Wu, S.G.; Lai, E.P.; Mayer, P.M. Molecularly imprinted solid phase extraction-pulsed elution-mass spectrometry for determination of cephalexin and alpha-aminocephalosporin antibiotics in human serum. J. Pharm. Biomed. Anal. 2004, 36, 483–490. [Google Scholar] [CrossRef]
- Moein, M.M.; Javanbakht, M.; Akbari-Adergani, B. Molecularly imprinted polymer cartridges coupled on-line with high performance liquid chromatography for simple and rapid analysis of dextromethorphan in human plasma samples. J. Chromatogr. B 2011, 879, 777–782. [Google Scholar] [CrossRef]
- Fresco-Cala, B.; Batista, A.D.; Cárdenas, S. Molecularly imprinted polymer micro- and nano-particles: A review. Molecules 2020, 25, 4740. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Polymerization | Template Molecules | Functional Monomers | Crosslinkers | Initiator | Porogen Solvent | Washing Solvent | Ref. |
|---|---|---|---|---|---|---|---|
| Bulk | Clozapine | Methacrylic acid | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Chloroform | Acetic acid/methanol | [57] |
| Quercetin | Acrylamide | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Tetrahydrofuran | Acetic acid/methanol | [20] | |
| Propranolol (Racemic) | Methacrylic acid | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Toluene | Acetic acid/methanol | [71] | |
| Estradiol | 4-Vinylpyridine | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetonitrile | Methanol | [72] | |
| Serotonin | Methacrylic acid, 2-Hydroxyethylmethacrylate | Ethyleneglycol dimethacrylate | 2 2’-azobis(2 4-dimethylvaleronitrile) | Acetic acid/methanol/Acetonitrile | Acetic acid/methanol/water | [58] | |
| 2,4-Dinitrophenol | Acrylamide | Ethyleneglycol dimethacrylate | Benzoyl peroxide | Acetonitrile | Methanol/acetic acid | [73] | |
| Benzylparaben | Methacrylic acid | Trimethylolpropane trimethacrylate | Benzoyl peroxide | Toluene | Methanol/acetic acid | [74] | |
| Diclofenec | Allylthiourea | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetonitrile | Methanol/acetic acid | [75] | |
| Kukoamine | Itaconic acid | Ethyleneglycol dimethacrylate | 1, 1’-azobis (cyclohexanecarbonitrile) | Dimethylformamide | Methanol | [76] | |
| Hispidin | Acrylic acid, Methacrylic acid, 4-Vinylpyridine | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Dimethyl sulphate, Tetrahydrofuran | Ethanol/Acetic acid | [77] | |
| In situ | 6-Amino caproic acid | Caprolatam | Graphene Oxide | Potassium persulfate | Sulfuric acid | Boiling water | [78] |
| S-(-)-amlodipine | Acrylamide | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Dodecanol | Methanol/Acetic acid | [79] | |
| Precipitation | Clozapine | Methacrylic acid | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Chloroform | Acetic acid/methanol | [57] |
| Aristolochic acid | Acrylamide | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Dimethylformamide | Methanol/Acetic acid | [14] | |
| Carbamazepine, Clofibric acid | 2-Vinylpyridine | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Toluene | Methanol/Acetic acid | [80] | |
| Diclofenac | 2-Vinylpyridine | Ethyleneglycol diacrylate | Azobisisobutyronitrile | Toluene | Methanol/Acetic acid | [81] | |
| Rivastigmine | Acrylamide-2-methylpropanesulfonic acid | Ethyleneglycol dimethacrylate | Benzoyl peroxide | Acetonitrile | Methanol/Acetic acid | [82] | |
| Dibutylphthalate | Acrylamide | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetonitrile | Ethanol | [83] | |
| Vitamin E (α-tocophenol) | Acrylamide | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetone | Methanol/Acetic acid | [60] | |
| Gallic acid | Acrylic acid | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetonitrile | Methanol | [13] | |
| Suspension | Dibenzothiophene | 4-Vinylpyridine | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Toluene | Ethanol | [84] |
| Polydatin | Acrylic acid, Methacrylic acid, Acrylamide | 2-(Dimethylamino)ethyl methacrylate, N-vinylpyrrolidone, Ethyleneglycol dimethacrylate), Trimethylolpropane triacrylate | Azobisisobutyronitrile | Acetonitrile, Dimethylformamide, Methanol | n-heptane/span 80 | [61] | |
| 4-hydroxybenzoic acid | 4-Vinylpyridine | Ethyleneglycol dimethacrylate | Azobisisobutyronitrile | Acetonitrile | Methanol/Acetic acid | [85] |
| Template | Analyte | Monomer/Porogen Solvent/Cross linker/Initiator | Polymerization Technique | Template Removal | MISPE Mode | SPE Condition | Adsorption Evaluation | Ref. |
|---|---|---|---|---|---|---|---|---|
| Ketoprofen | Wastewater | 2-VP/ACN: Toluene/ EDGMA/ ABCHC | Bulk | Acetic acid: ACN (1:9; v/v) | Offline | Cartridge: 14 mg Sample: 50 mL Conditioned: Methanol (1 mL), Deionized water (1 mL) Washing: 1 mL of 5% (v/v) triethiamine in water Eluted: Methanol (1 mL) | Comparing MIP-SPE efficiency with NIP-SPE | [94] |
| PMPA, EMPA, IMPA, CMPA, BMPA, MPA | Human Serum | MAA/ ACN/ TRIM/ ABCHC | Bulk | Methanol: Acetic acid (9:1; v/v) | Offline | Cartridge: 500 mg Sample: 1 mL Conditioned: 30% Hydrochloric acid (5 mL) Washing: Water, ACN Eluted: Water | The recoveries for degradation products is in range 81.2–90.5% | [104] |
| Monocrotophos | Water, Soil sample | MAA/ DCM/ EDGMA/ AIBN | Bulk | Methanol: 10% Acetic acid | Offline | Cartridge: 200 mg Sample: 1 mL Conditioned: Methanol (1 mL), LC Grade Water (2 mL) Washing: 2 mL CH2Cl2: ACN (95:5, v/v) Eluted: CH2Cl2: Methanol (90:10, v/v) | Extraction of 1 L river water at 100 mg/L spike level at range 77.5–99.1% while 5 g soil sample at 100 μg/kg at range 79.3–93.5% | [105] |
| Artemisinin | Artemisinin (Anti malaria drug) | Styrene/ ACN/ EDGMA/ AIBN | Bulk | Methanol: Acetic acid (9:1; v/v) | Offline | Cartridge: 100 mg Sample: 2 mL Conditioned: ACN (5 mL) Washing: Methanol: Acetic acid (9:1; v/v) Eluted: 5 mL Methanol: Acetic acid (9:1; v/v) | 40 mg in 5 mL ACN solution: QMIP: 8.46 mg g−1 QNIP: 4.49 mg g−1 | [12] |
| Quinalphos | Quinalphos (Organophosphorus pesticides) | MAA/ ACN/ EDGMA/ AIBN | Bulk | Methanol: Acetic acid (9:1; v/v) | Offline | Cartridge: 100 mg Sample: 10 mL Conditioned: Methanol (10 mL), Deionized water (10 mL) Washing: 5% Acetic acid in methanol Eluted: Methanol (6 mL) | High extraction recovery of analyte with MIPs compared to NIPs and C18 | [91] |
| Florfenicol | Florfenicol | 4-VP/ THF/ EDGMA/ AIBN | Precipitation | Methanol | Offline | Cartridge: 120 mg Sample: 10 mL Conditioned: Methanol (10%, v/v), Aceticacid (5%, v/v), water Washing: Aceticacid (1%, v/v) Eluted: ACN: water (25%, v/v) | 120 mg in 10 mL in various concentrations shaken 3 h at RT: QMIP: 4.32 mg g−1 QNIP: 2.88 mg g−1 | [106] |
| Diethylstilbestrol (DES) | DES | APTES/ Methanol/ TEOS/ Activated silica gel | Surface | Methanol: Hydro-Chloric acid (1:1; v/v) | Offline | Cartridge: 200 mg Sample: 1 mL Conditioned: Methanol (10 mL), Water (10 mL) Washing: 2 mL Methanol: Water (98:2; v/v) Eluted: 2 mL Methanol | 50 mg in 10 mL in various concentrations shaken at 1 h at RT: QMIP: 62.58 mg g−1 QNIP: 19.89 mg g−1 | [107] |
| Diethylstilbestrol (DES) | Seawater | MAA/ Chlorofoam/ EDGMA/ AIBN | Suspension | Methanol (5 mL), Water (5 mL) | Offline | Cartridge: 100 mg Sample: 20 mL Conditioned: Methanol (5 mL), Water (5 mL) Washing: 1 mL Methanol: Acetic acid (99:1; v/v) Eluted: 1 mL Methanol: ACN (65:35; v/v) | 20 mg in 20 mL in methanol/water (60:40, v/v) shaken at 300 rpm: QMIP: 8.43 mg g−1 QNIP: 4.43 mg g−1 | [92] |
| Catechol | Catechol | 4-VP/ ACN/ EDGMA/ AIBN | Bulk | Methanol: Acetic acid (4:1; v/v) | Online | Cartridge: 70 mg Sample: 1.3 mL Conditioned: 2% ACN (v/v) Washing: Nitric acid, ACN Eluted: Nitric acid | Permanganate solution used reduced from Mn(VII) to Mn(II) at 528 nm | [108] |
| Kirenol | Kirenol | AA/ THF/ EDGMA/ AIBN | Bulk | Methanol: Acetic acid (9:1; v/v) | Online | Cartridge: 100 mg Sample: 6 mL Conditioned: ACN (5 mL) Washing: Methanol (5 mL), Acetic acid (5 mL) | Comparing MIP recognition ability with NIP. | [97] |
| Naproxen | Urine | 4-VP/ Toluene/ EDGMA/ AIBN | Bulk | - | Offline | Cartridge: 200 mg Sample: 20 μL Conditioned: 6 mL ACN/Water/Acetic acid (60:30:10), 6 mL Mili Q Water (pH3) Washing: 2 mL ACN Eluted: 3 mL ACN/1% Acetic acid | Naproxen selectively extracted by MISPE | [99] |
| Simazine | Simazine | MAA/ DCM/ EDGMA/ AIBN | Bulk | - | Online | Sample: 200 μL Conditioned: 2 mL Water | Large volume sample extract and low detection limit acquired | [99] |
| 4-Nitrophenol | Environmental water | 4-VP/ ACN/ EDGMA/ AIBN | Bulk | - | Online | Sample: 20 μL Conditioned: 2 mL ACN, 2 mL Mili Q Water (pH 2.5) Washing: 0.4 mL DCM, 2 mL Mili Q Water (pH 2.5) Eluted: 1% CAN | Comparing efficiency of MIP-SPE and NIP-SPE | [109] |
| Sulfamethazine | Sulfamethazine | APTES/ ACN/ TEOS/ Activated silica gel | Surface | Methanol | Online | Cartridge: 50 mg Sample: 10 mL Conditioned: Methanol | Comparing efficiency of MIP-SPE and NIP-SPE | [110] |
| 4-Hydroxylphenylacetic acid | Human Urine | 4-VP/ Toluene/ EDGMA/ AIBN | Bulk | 10% Acetic acid/ Methanol (v/v) | Offline | Cartridge: 100 mg Sample: 0.5 mL Conditioned: 2 mL Water, 2 mL ACN Washing: 1 mL Water Eluted: 1.5 mL ACN/1% formic acid | 20 mg in 2.5 mL in solution incubated for 24 h: QMIP > QNIP IF > 3.5 | [88] |
| Cephalexin | Human serum | TFMAA/ ACN/ EDGMA/ AIBN | Bulk | Methanol/20% Acetic acid | Online | Cartridge: 50 mg Sample: 20 μL Conditioned: ACN Washing: 1% TFA Methanol Eluted: Methanol (3 mL) | The detection limit was estimated at 0.04 μg mL−1 of cephalexin | [111] |
| Dextromethorphan | Human plasma | MAA/ Chlorofoam/ EDGMA/ AIBN | Precipitation | Methanol/ Phosphate Buffer | Online | Cartridge: 120 mg Sample: 50 μg−1 Conditioned: Methanol (1 mL), Ultrapure Water (1 mL) Washing: 0.1 M Hydrochloric acid (1 mL), Ultrapure Water (1 mL), DCM (1.5 mL) Eluted: 3 × 1 mL Methanol/Phosphate Buffer (90:10) (0.05 M, pH 5) | 25 mg in 100 mL solution at shaken 30 min: Qmax MIP is 90 mg g−1 | [112] |
| Theophylinne | Serum | MAA/ Chlorofoam/ EDGMA/ AIBN | Bulk | Methano/Acetic acid (9:1) | Online | - | Comparing efficiency of MIP-SPE and NIP-SPE | [71] |
| Endorine | Aqueous sample (river, tap water) | Provided by POIYINTELL | Surface | - | Offline | Cartridge: 100 mg Sample: 100 mL Conditioned: ACN (5 mL), Water (5 mL) Washing: 4 mL (water, ACN; 80:20), 2 mL water Eluted: Methanol (1 mL) | MISPE high recoveries than commercial C18 SPE | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamaruzaman, S.; Nasir, N.M.; Mohd Faudzi, S.M.; Yahaya, N.; Mohamad Hanapi, N.S.; Wan Ibrahim, W.N. Solid-Phase Extraction of Active Compounds from Natural Products by Molecularly Imprinted Polymers: Synthesis and Extraction Parameters. Polymers 2021, 13, 3780. https://doi.org/10.3390/polym13213780
Kamaruzaman S, Nasir NM, Mohd Faudzi SM, Yahaya N, Mohamad Hanapi NS, Wan Ibrahim WN. Solid-Phase Extraction of Active Compounds from Natural Products by Molecularly Imprinted Polymers: Synthesis and Extraction Parameters. Polymers. 2021; 13(21):3780. https://doi.org/10.3390/polym13213780
Chicago/Turabian StyleKamaruzaman, Sazlinda, Najihah Mohammad Nasir, Siti Munirah Mohd Faudzi, Noorfatimah Yahaya, Nor Suhaila Mohamad Hanapi, and Wan Nazihah Wan Ibrahim. 2021. "Solid-Phase Extraction of Active Compounds from Natural Products by Molecularly Imprinted Polymers: Synthesis and Extraction Parameters" Polymers 13, no. 21: 3780. https://doi.org/10.3390/polym13213780
APA StyleKamaruzaman, S., Nasir, N. M., Mohd Faudzi, S. M., Yahaya, N., Mohamad Hanapi, N. S., & Wan Ibrahim, W. N. (2021). Solid-Phase Extraction of Active Compounds from Natural Products by Molecularly Imprinted Polymers: Synthesis and Extraction Parameters. Polymers, 13(21), 3780. https://doi.org/10.3390/polym13213780