
Preclinical Evaluation of Polymeric Nanocomposite Containing Pregabalin for Sustained Release as Potential Therapy for Neuropathic Pain

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Obtaining Pregabalin-Loaded Polymeric Nanocomposite (PG-PN)
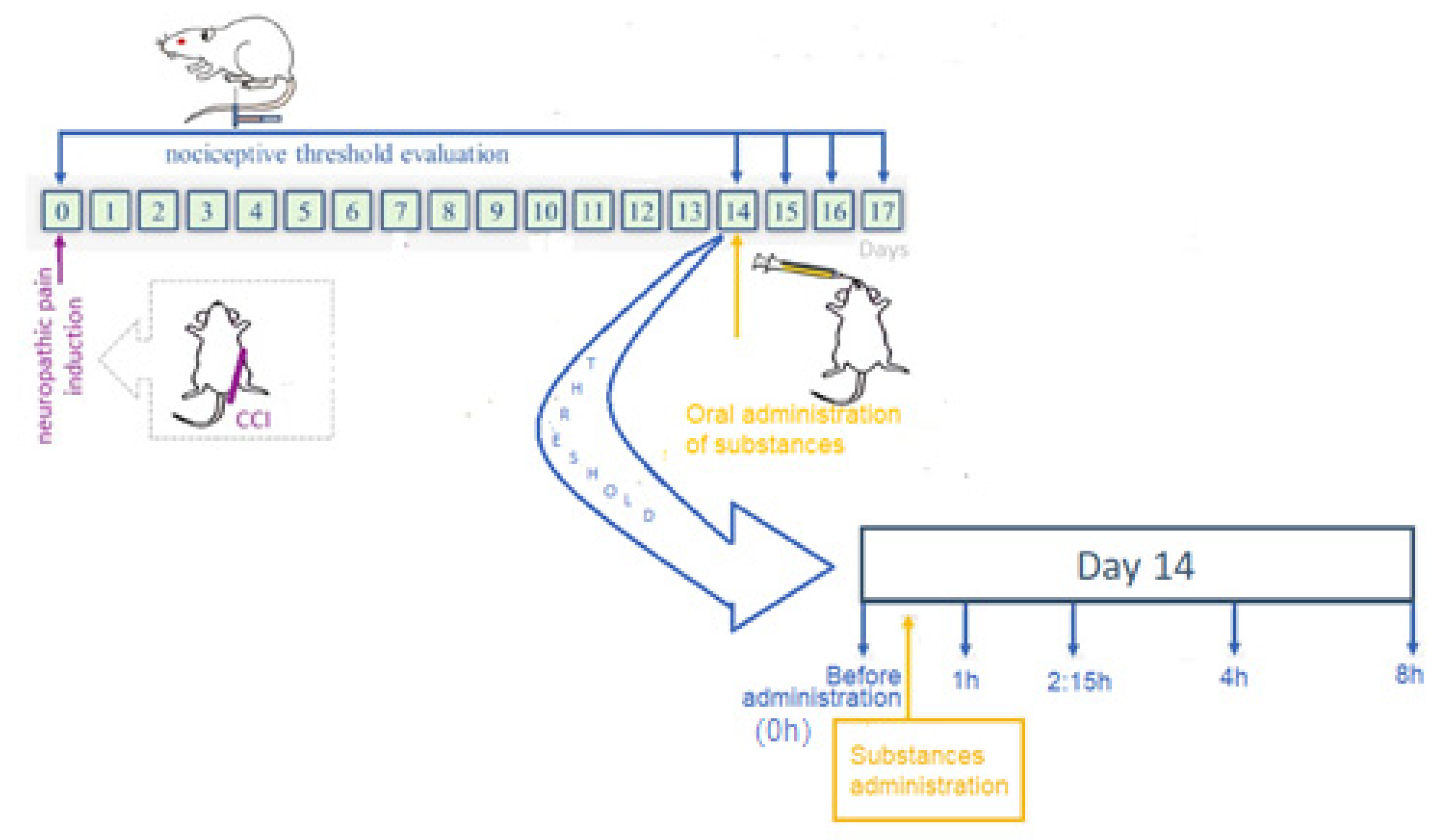
2.3. Experimental Animals
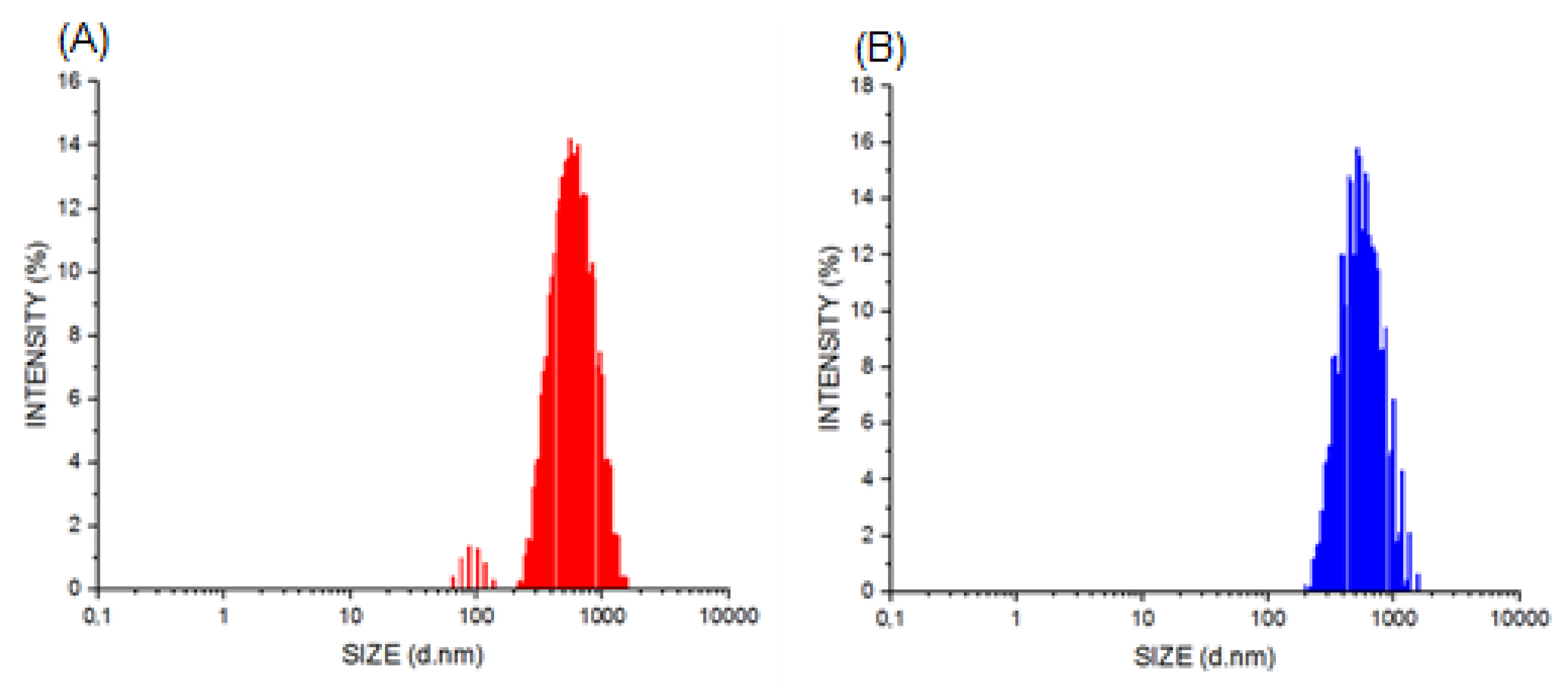
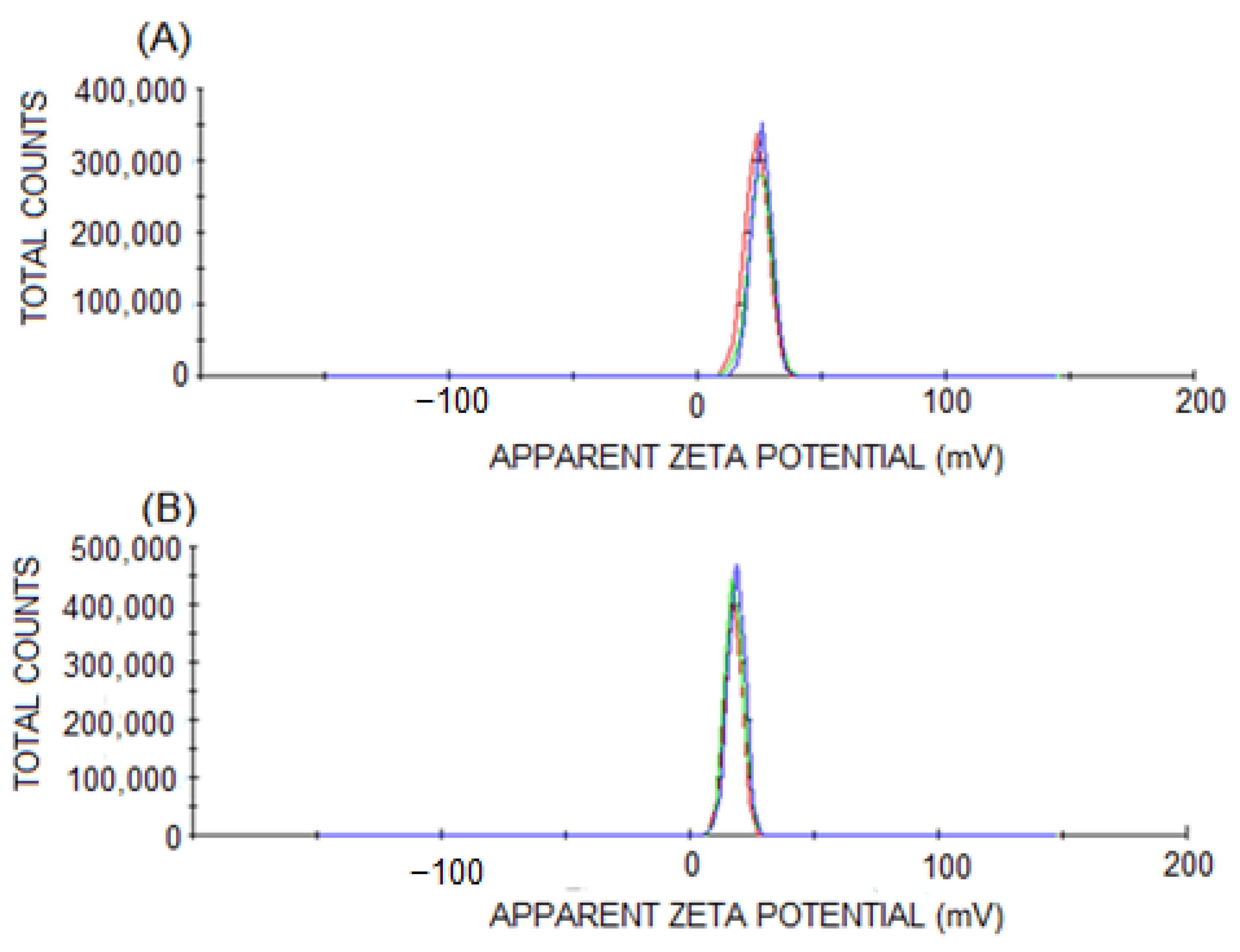
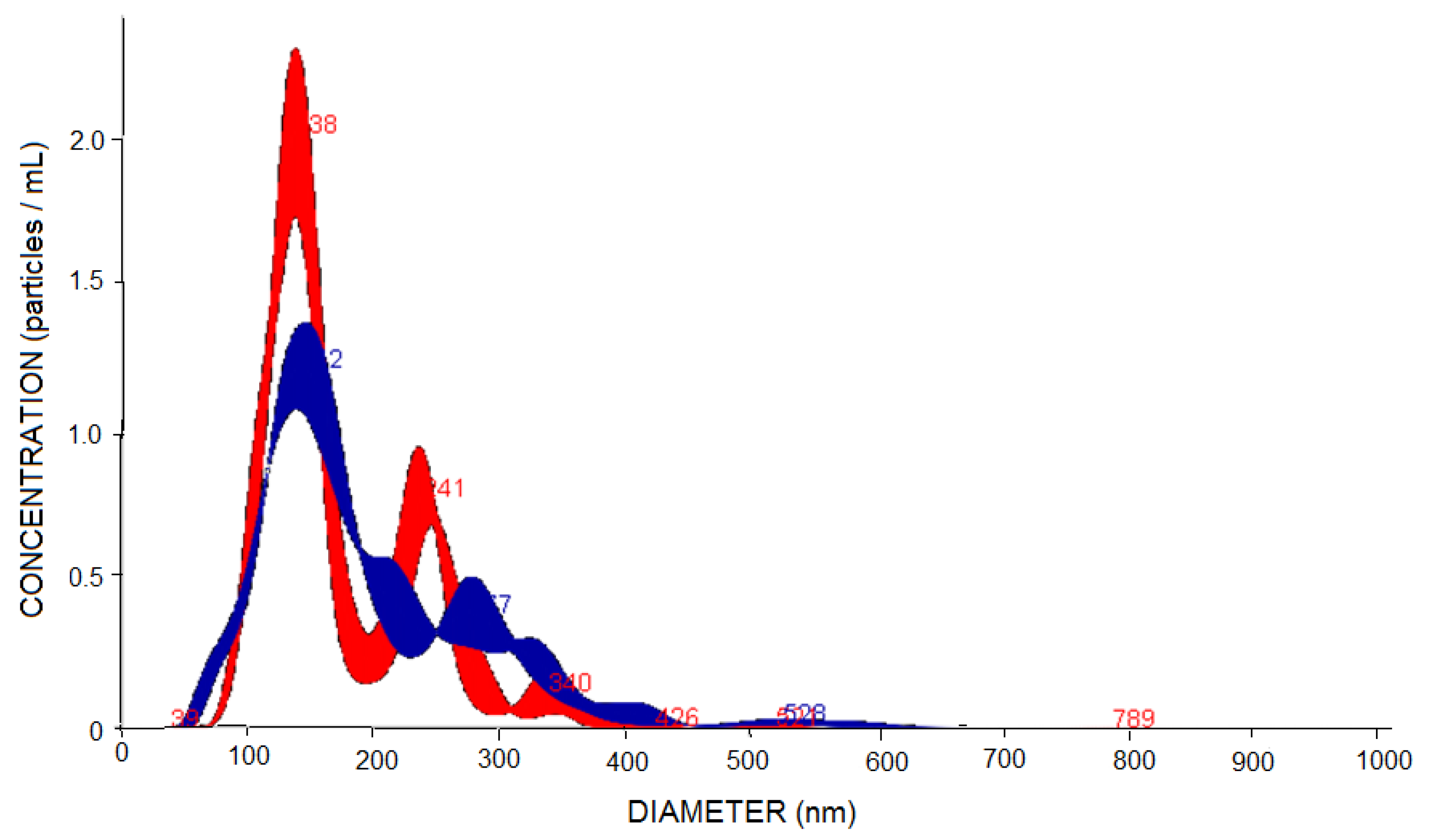
2.4. Characterization of PNs
2.5. Pharmacokinetic Study
2.6. Antinociceptive Effect Study
2.7. Motor Coordination and Balance Evaluation
2.8. Assessment of Barbiturate-Induced Sleep
2.9. Statistical Analysis
3. Results
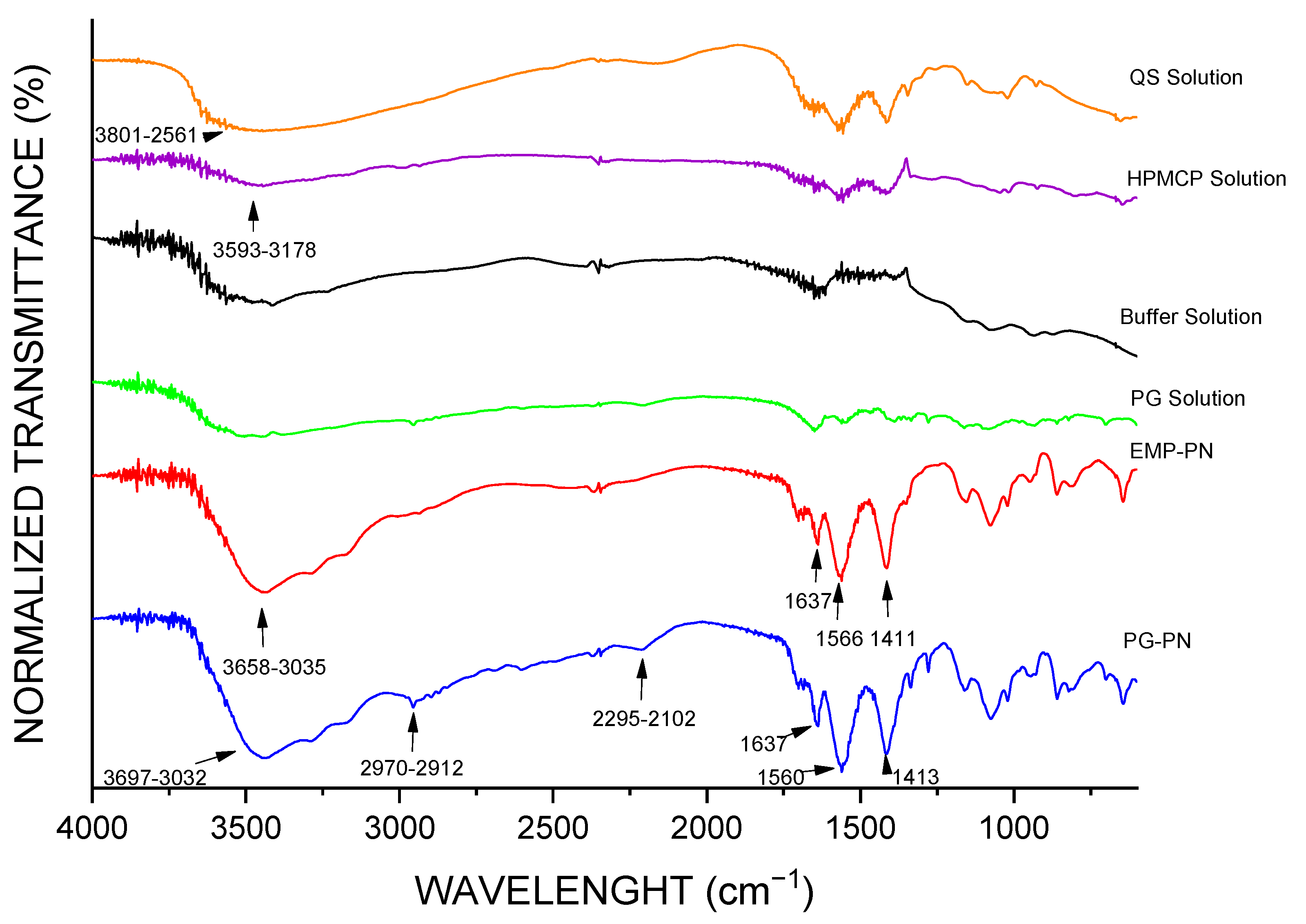
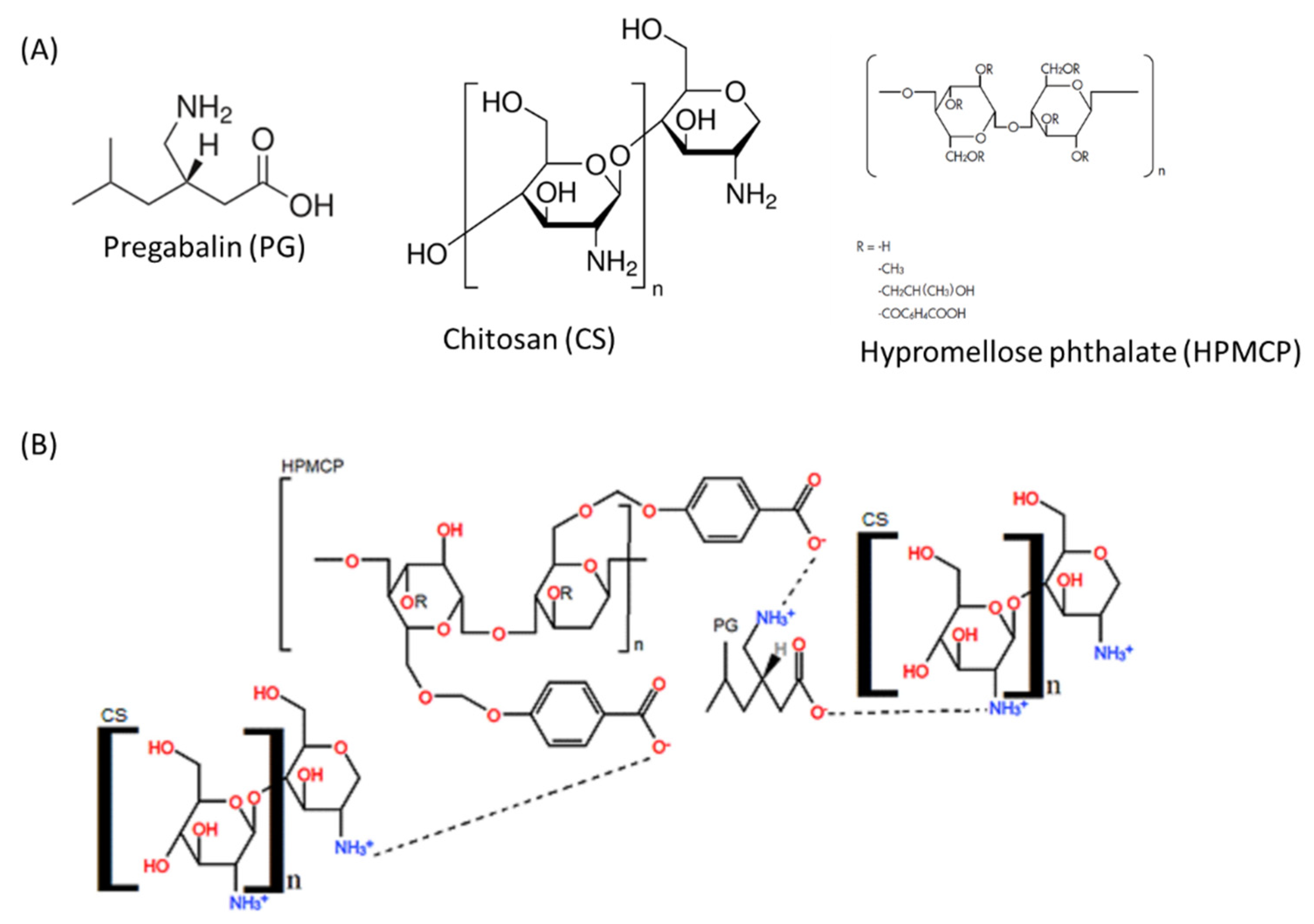
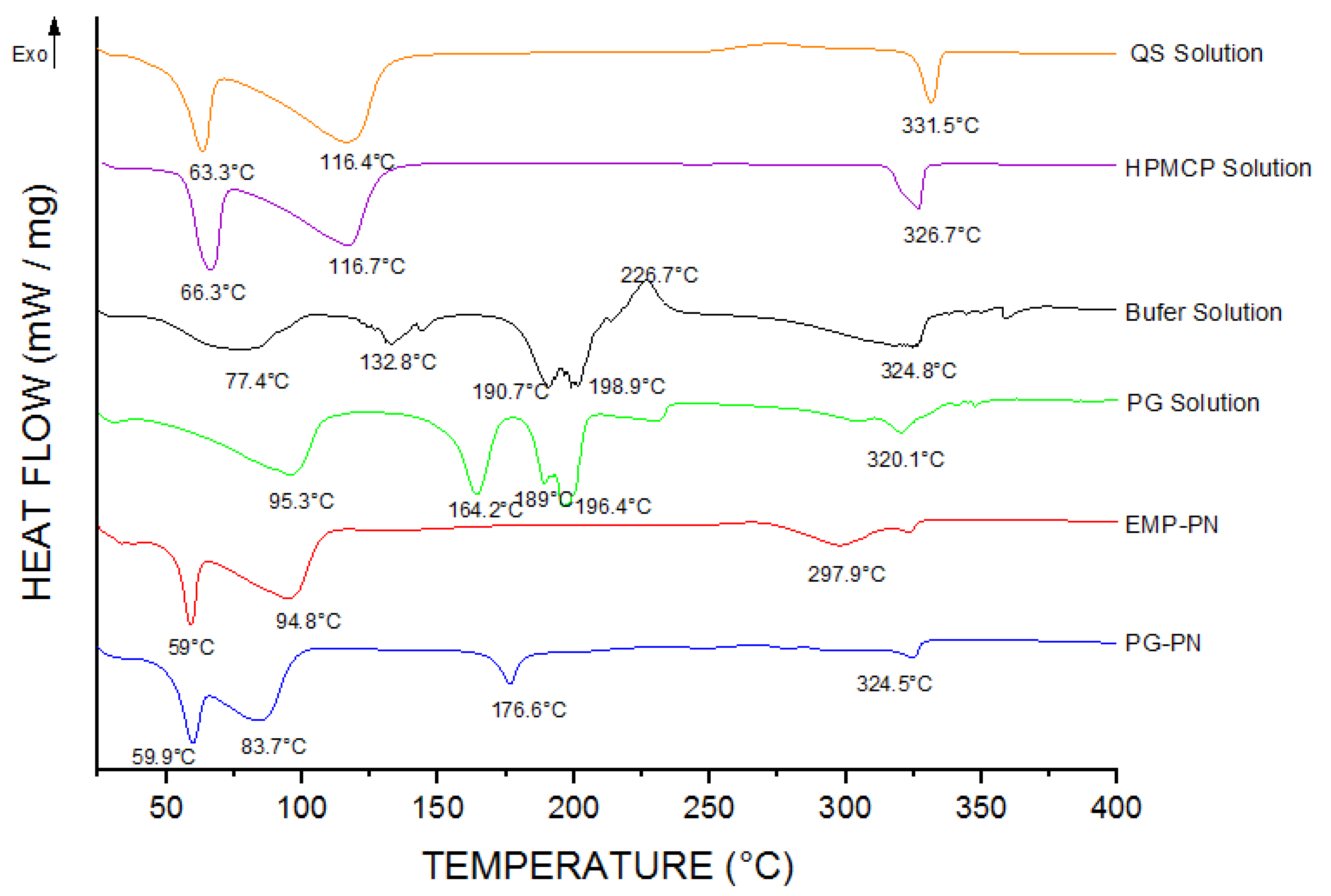
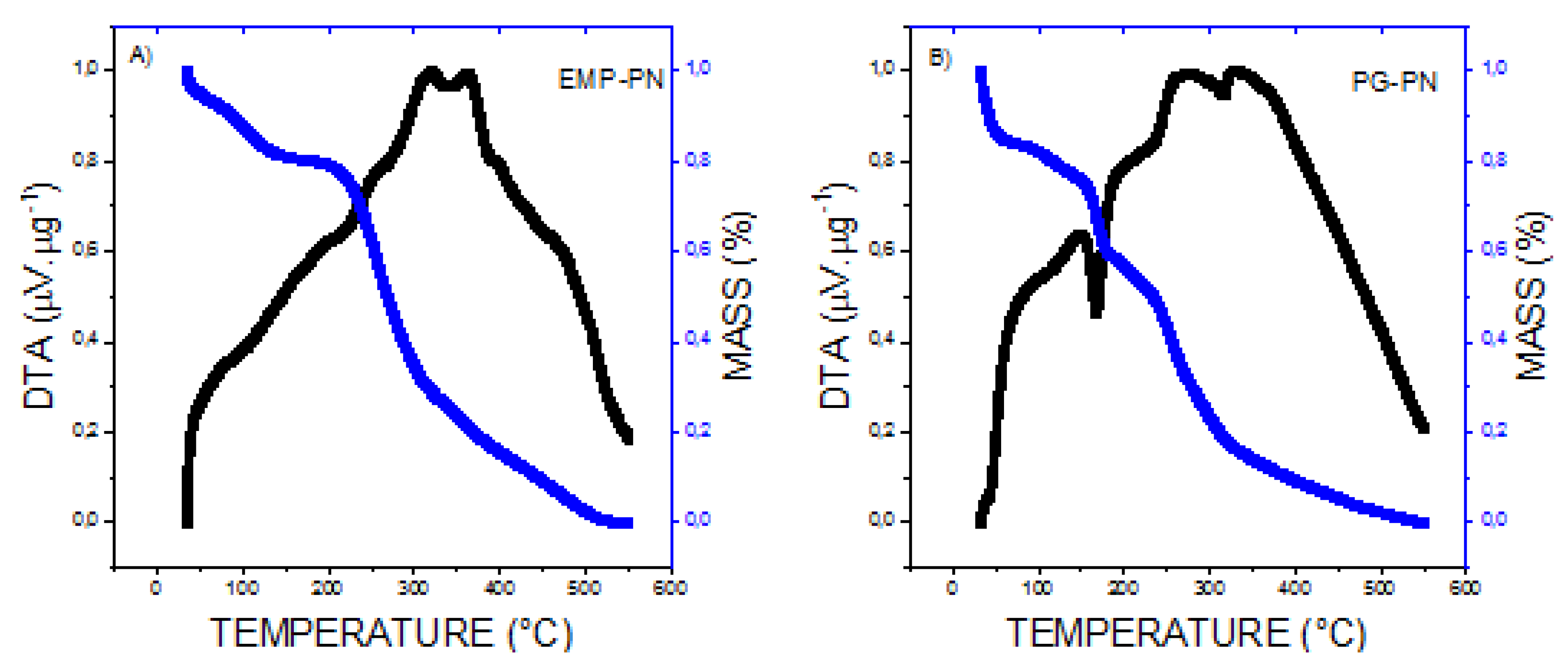
3.1. Characterization of PNs
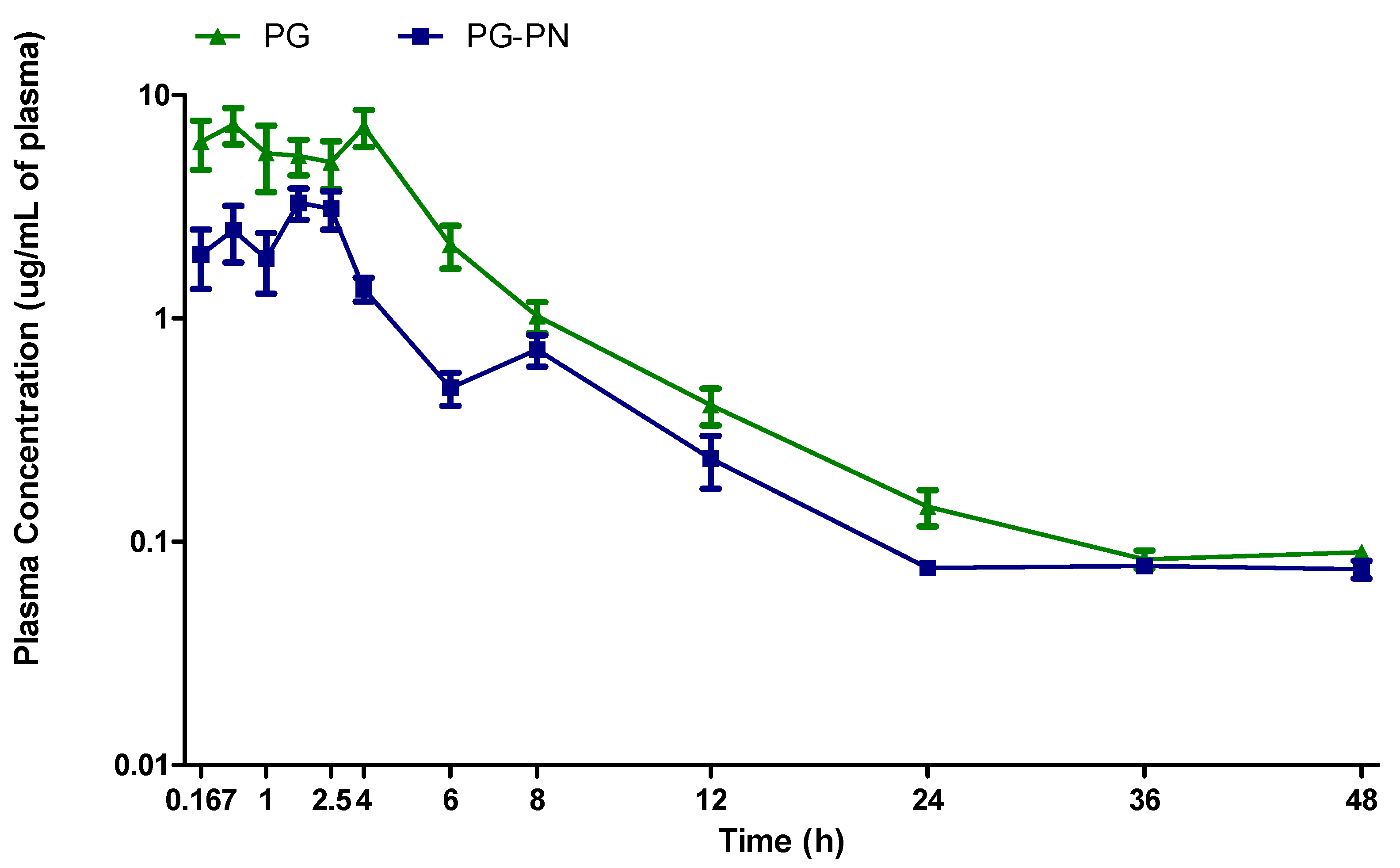
3.2. Pharmacokinetic Study
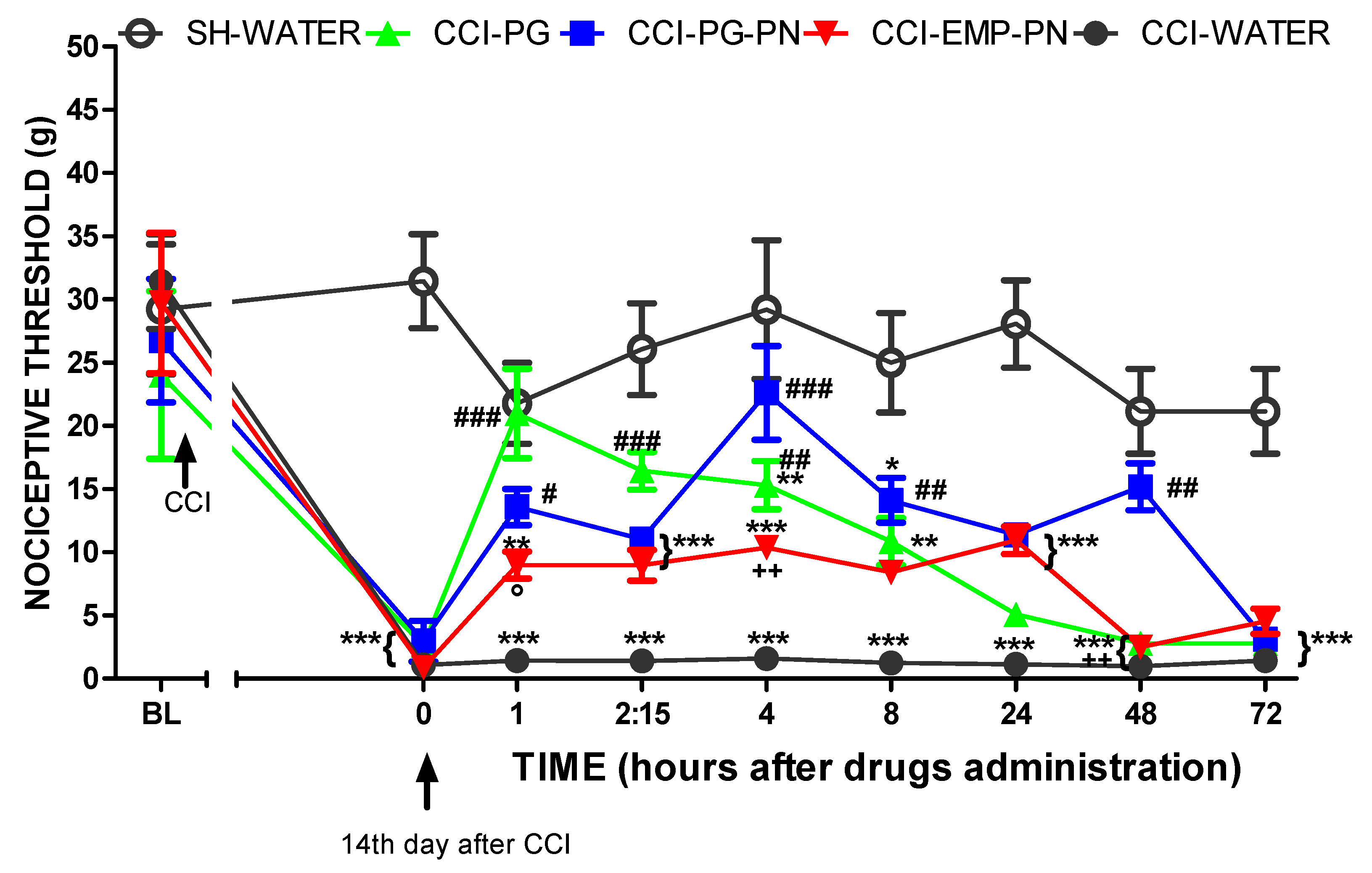
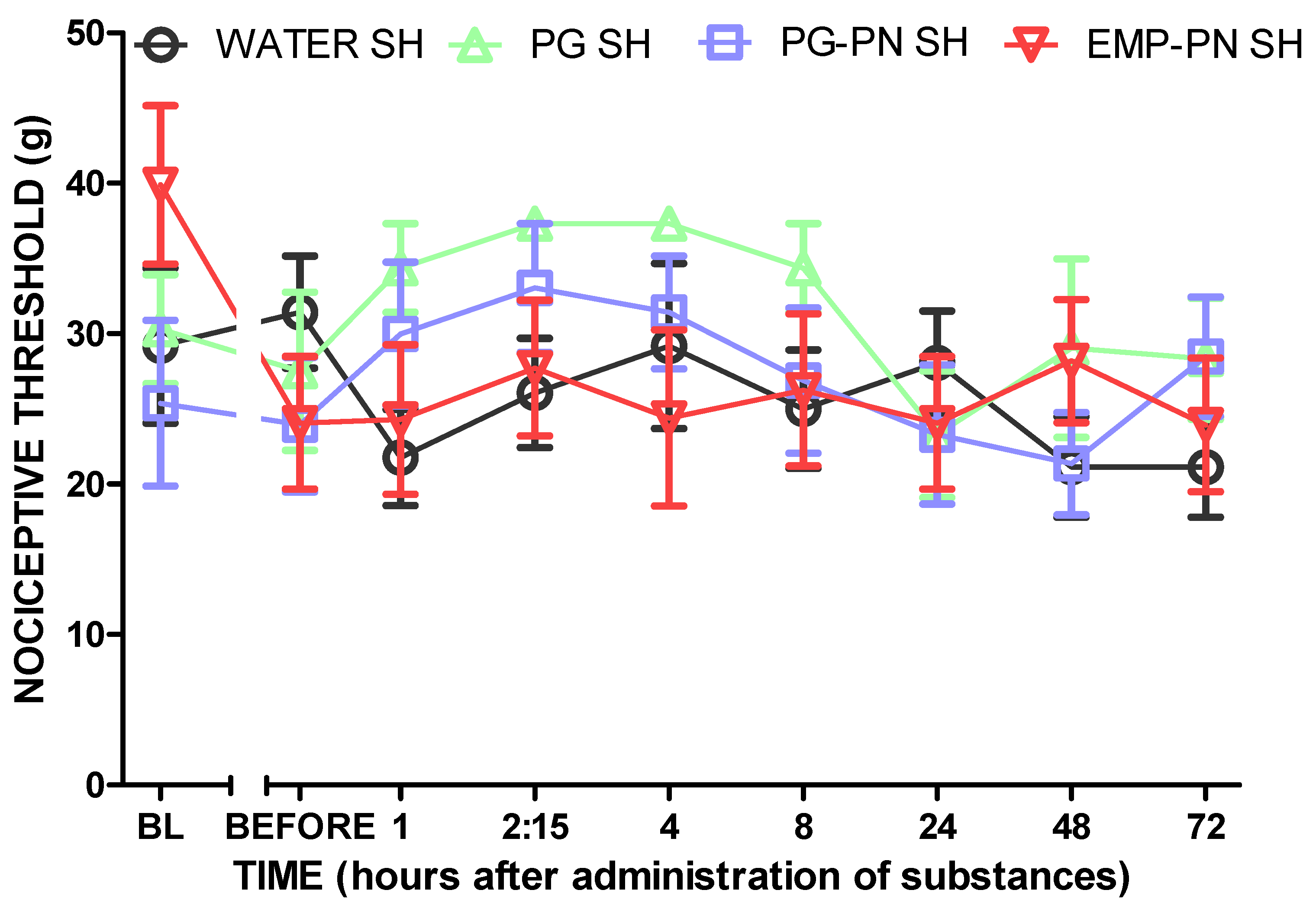
3.3. Antinociceptive Effect Study
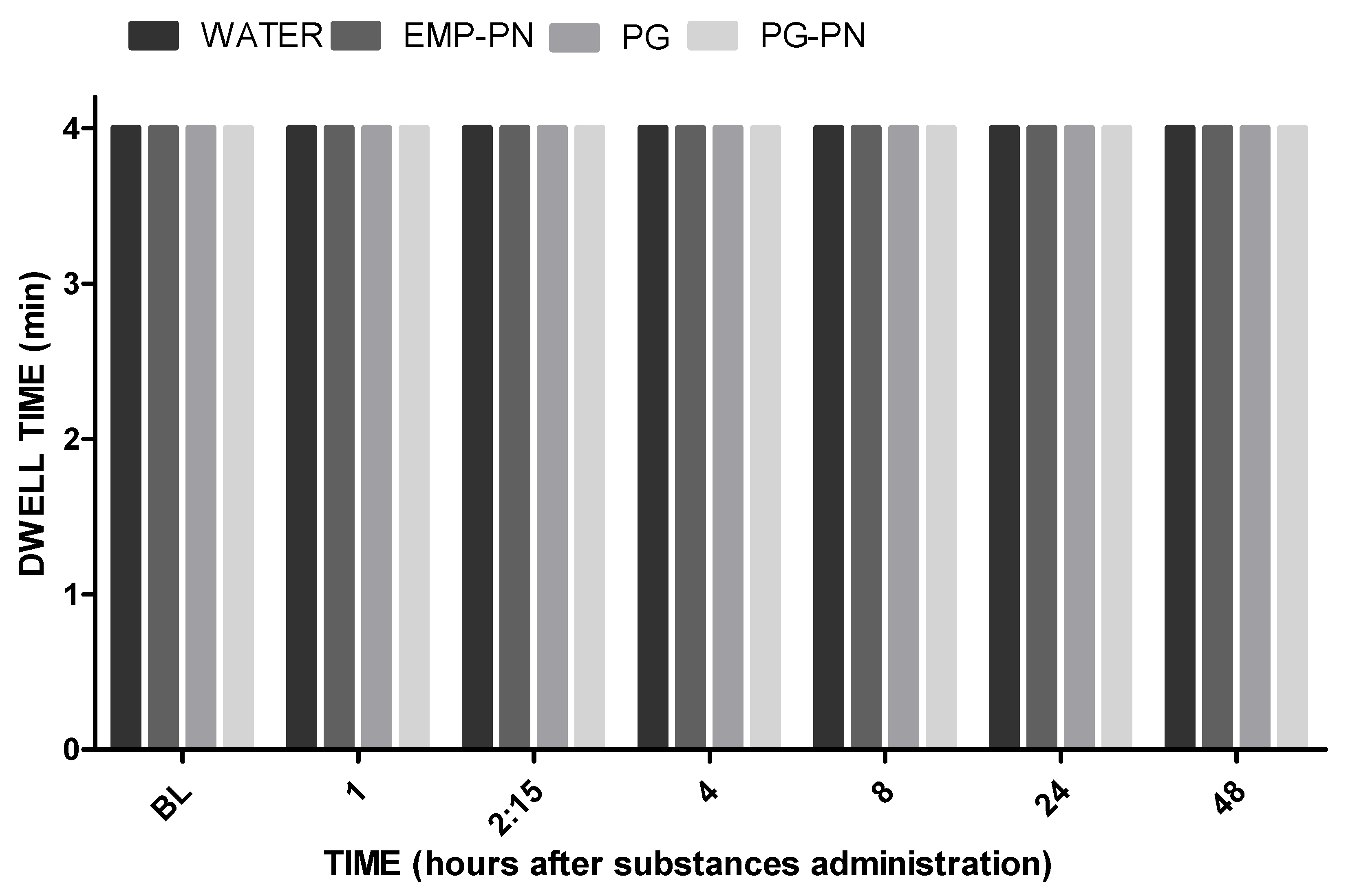
3.4. Motor Coordination and Balance Evaluation
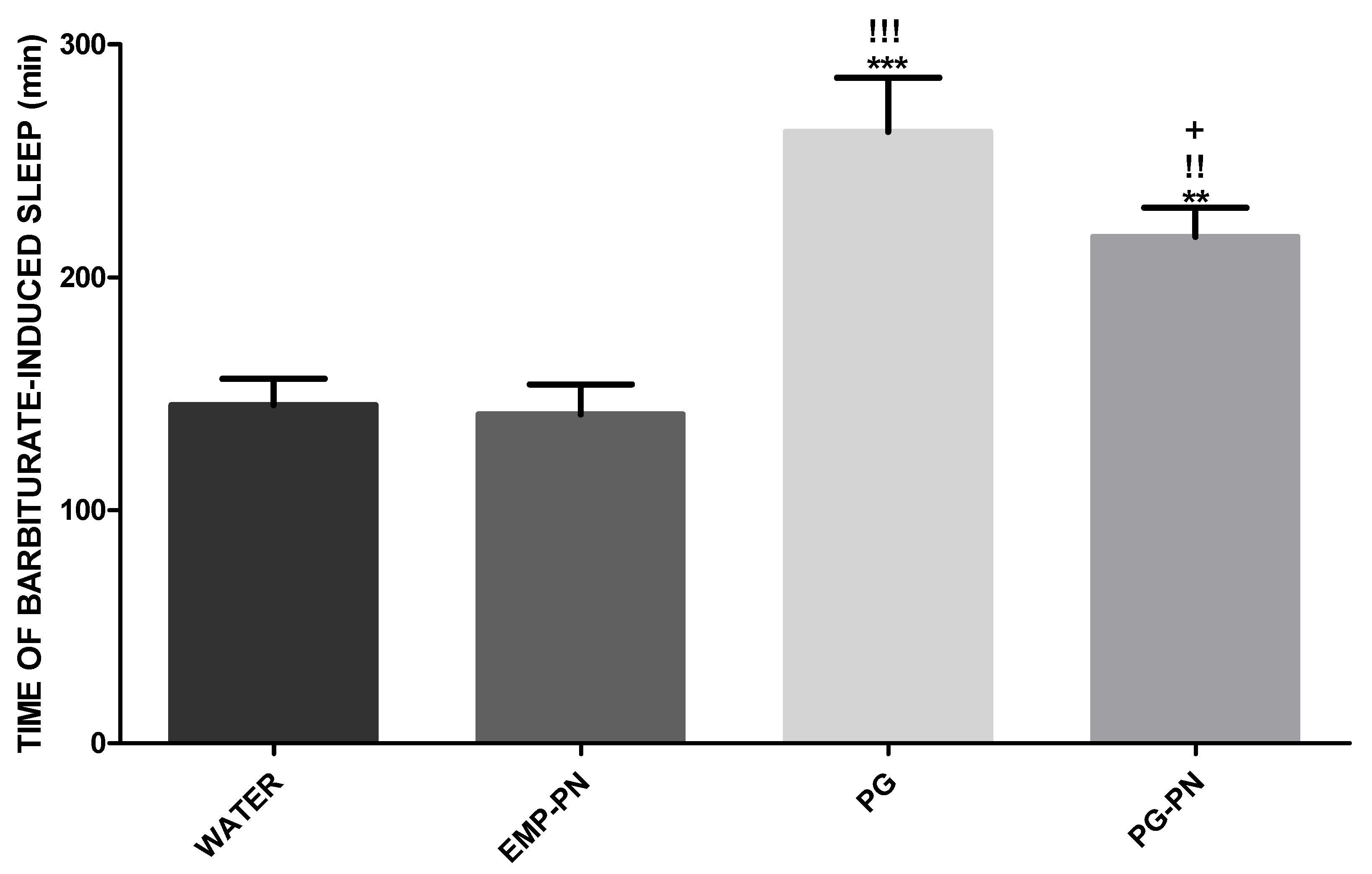
3.5. Evaluation of Barbiturate-Induced Sleep
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dworkin, R.H.; O’Connor, A.B.; Backonja, M.; Farrar, J.T.; Finnerup, N.B.; Jensen, T.S.; Kalso, E.A.; Loeser, J.D.; Miaskowski, C.; Nurmikko, T.J.; et al. Pharmacologic management of neuropathic pain: Evidence-based recommendations. Pain 2007, 132, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Approval of Lyrica ® (Pregabalin). 2004. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/021446_Lyrica%20Capsules_approv.PDF (accessed on 1 February 2021).
- Buoli, M.; CaldirolI, A.; Serati, M. Pharmacokinetic evaluation of pregabalin for the treatment of generalized anxiety disorder. Expert Opin. Drug Metab. Toxicol. 2017, 13, 351–359. [Google Scholar] [CrossRef]
- Echterhoff, R.R. A eficácia da pregabalina no tratamento do transtorno de ansiedade generalizada. In Course Conclusion Paper (Psychiatry Specialization); Universidade Federal do Paraná: Curitiba, Brazil, 2012. [Google Scholar]
- Moore, R.A.; Straube, S.; Wiffen, P.J.; Derry, S.; McQuay, H.J. Pregabalin for acute and chronic pain in adults. Cochrane Database Syst. Rev. 2009, 8, CD007076. [Google Scholar] [CrossRef]
- Qin, C.; Wu, M.; Xu, S.; Wang, X.; Shi, W.; Dong, Y.; Yang, L.; He, W.; Han, X.; Yin, L. Design and optimization of gastro-floating sustained-release tablet of pregabalin: In vitro and in vivo evaluation. Int. J. Pharm. 2018, 545, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (FDA). Approval of Lyrica CR ® (Pregabalin). 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209501Orig1s000Approv.pdf (accessed on 1 February 2021).
- Pfizer. Lyrica Package Insert CR®. 2018. Available online: Labeling.pfizer.com/ShowLabeling.aspx?id=9678 (accessed on 20 January 2021).
- Fukasawa, H.; Muratake, H.; Nagae, M.; Sugiyama, K.; Shudo, K. Transdermal administration of aqueous pregabalin solution as a potential treatment option for patients with neuropathic pain to avoid central nervous system-mediated side effects. Biol. Pharm. Bull. 2014, 37, 1816–1819. [Google Scholar] [CrossRef] [Green Version]
- Aydogan, E.; Comoglu, T.; Pehlivanoglu, B.; Dogan, M.; Comoglu, S.; Dogan, A.; Basci, N. Process and formulation variables of pregabalin microspheres prepared by w/o/o double emulsion solvent diffusion method and their clinical application by animal modeling studies. Drug Dev. Ind. Pharm. 2015, 41, 1311–1320. [Google Scholar] [CrossRef]
- Taksande, J.B.; Umekar, M.J. Preparation of intranasal pregabalin microspheres: In vitro, ex vivo and in vivo pharmacodynamic evaluation. J. Pharm. Res. 2018, 12, 112–121. [Google Scholar]
- Ghumman, S.A.; Bashir, S.; Noreen, S.; Khan, A.M.; Malik, M.Z. Taro-corms mucilage-alginate microspheres for the sustained release of pregabalin: In vitro &in vivo evaluation. Int. J. Biol. Macromol. 2019, 139, 1191–1202. [Google Scholar] [CrossRef]
- Jeong, K.H.; Woo, H.S.; Kim, C.J.; Lee, K.H.; Jeon, J.Y.; Lee, S.Y.; Kang, J.H.; Lee, S.; Choi, Y.W. Formulation of a modified-release pregabalin tablet using hot-melt coating with glyceryl behenate. Int. J. Pharm. 2015, 495, 1–8. [Google Scholar] [CrossRef]
- Cevik, O.; Gidon, D.; Kizilel, S. Visible-light-induced synthesis of pH-responsive composite hydrogels for controlled delivery of the anticonvulsant drug pregabalin. Acta Biomater. 2015, 11, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Madan, J.R.; Adokar, B.R.; Dua, K. Development and evaluation of in situ gel of pregabalin. Int. J. Pharm. Investig. 2015, 5, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, N.; Kumar, R.; Sarwal, A.; Sinha, V.R. Preparation and evaluation of floating tablets of pregabalin. Drug Dev. Ind. Pharm. 2016, 42, 654–660. [Google Scholar] [CrossRef]
- Cinay, G.E.; Erkoc, P.; Alipour, M.; Hashimoto, Y.; Sasaki, Y.; Akiyoshi, K.; Kizilel, S. Nanogel-integrated pH-responsive composite hydrogels for controlled drug delivery. ACS Biomater. Sci. Eng. 2017, 3, 370–380. [Google Scholar] [CrossRef]
- Arafa, M.G.; Ayoub, B.M. DOE optimization of nano-based carrier of pregabalin as hydrogel: New therapeutic &chemometric approaches for controlled drug delivery systems. Sci. Rep. 2017, 7, 41503. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Hwang, K.M.; Park, Y.S.; Nguyen, T.T.; Park, E.S. Preparation and evaluation of non-effervescent gastroretentive tablets containing pregabalin for once-daily administration and dose proportional pharmacokinetics. Int. J. Pharm. 2018, 550, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Lim, S.H.; Shim, C.R.; Park, J.; Song, W.H.; Kwon, M.C.; Lee, J.H.; Park, J.S.; Choi, H.G. Development of a novel controlled-release tablet of pregabalin: Formulation variation and pharmacokinetics in dogs and humans. Drug Des. Devel. Ther. 2020, 14, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Yajima, R.; Matsumoto, K.; Yokono, K.; Watabe, Y.; Enoki, Y.; Taguchi, K.; Ise, Y.; Katayama, S.; Kizu, J. Pharmacokinetic and pharmacodynamic studies of pregabalin suppositories based on pharmacological research. J. Pharm. Pharmacol. 2019, 71, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.; Anil, S.; Kim, S.K.; Shim, M.S. Seaweed polysaccharide-based nanoparticles: Preparation and applications for drug delivery. Polymers 2016, 26, 30. [Google Scholar] [CrossRef] [Green Version]
- Thambi, T.; Li, Y.; Lee, D.S. Injectable hydrogels for sustained release of therapeutic agents. J. Control. Release 2017, 267, 57–66. [Google Scholar] [CrossRef]
- Thambi, T.; Phan, V.H.G.; Kim, S.H.; Le, T.M.D.; Duong, H.T.T.; Lee, D.S. Smart injectable biogels based on hyaluronic acid bioconjugates finely substituted with poly(β-amino ester urethane) for cancer therapy. Biomater. Sci. 2019, 7, 5424–5437. [Google Scholar] [CrossRef]
- Phan, V.H.G.; Duong, H.T.T.; Tran, P.T.; Thambi, T.; Ho, D.K.; Murgia, X. Self-assembled amphiphilic starch based drug delivery platform: Synthesis, preparation, and interactions with biological barriers. Biomacromolecules 2021, 22, 572–585. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Jafari, S.M.; Tong, Q.; Riaz, T.; Assadpour, E.; Aadil, R.M.; Niazi, S.; Khan, I.M.; Shehzad, Q.; Ali, A.; et al. Drug nanodelivery systems based on natural polysaccharides against different diseases. Adv. Colloid Interface Sci. 2020, 284, 102251. [Google Scholar] [CrossRef]
- Shah, B.M.; Palakurthi, S.S.; Khare, T.; Khare, S.; Palakurthi, S. Natural proteins and polysaccharides in the development of micro/nano delivery systems for the treatment of inflammatory bowel disease. Int. J. Biol. Macromol. 2020, 165, 722–737. [Google Scholar] [CrossRef]
- Liu, Z.; Jiao, Y.; Wang, Y.; Zhou, C.; Zhang, Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv. Drug Deliv. Rev. 2008, 60, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, B.Z.; Liu, Y.J.; Wu, Z.M.; Guo, X.D. Application of mesoscale simulation to explore the aggregate morphology of pH-sensitive nanoparticles used as the oral drug delivery carriers under different conditions. Colloids Surf. B. 2017, 151, 280–286. [Google Scholar] [CrossRef]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Arachchige, M.C.M.; Reshetnyak, Y.K.; Andreev, O.A. Advanced targeted nanomedicine. J. Biotechnol. 2015, 202, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Calvo, P.; Remuñán-López, C.; Vila-Jato, J.L.; Alonso, M.J. Novel hydrophilic chitosan-polyethylene oxide nanoparticles as protein carriers. J. Appl. Polym. Sci. 1997, 63, 125–132. [Google Scholar] [CrossRef]
- Barbi, M.D.S.; Carvalho, F.C.; Kiill, C.P.; Barud, H.D.S.; Santagneli, S.H.; Ribeiro, S.J.; Gremião, M.P. Preparation and Characterization of Chitosan Nanoparticles for Zidovudine Nasal Delivery. J. Nanosci. Nanotechnol. 2015, 15, 865–874. [Google Scholar] [CrossRef]
- Zimmermann, M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983, 16, 109–110. [Google Scholar] [CrossRef]
- Rodrigues, R.F.; Kawano, T.; Placido, R.V.; Costa, L.H.; Podestá, M.H.M.C.; Santos, R.S.; Galdino, G.; Barros, C.M.; Boralli, V.B. Investigation of the combination of pregabalin with duloxetine or amitriptyline on the pharmacokinetics and antiallodynic effect during neuropathic pain in rats. Pain Physician 2021, 24, E511–E520. [Google Scholar] [PubMed]
- Harms, P.G.; Ojeda, S.R. A rapid and simple procedure for chronic cannulation of the rat jugular vein. J. Appl. Physiol. 1974, 36, 391–392. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Guidance for industry: Bioanalytical method Validation. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 10 May 2020).
- Boroujerdi, M. Noncompartmental approach in pharmacokinetics based on statistical moments. In Pharmacokinetics: Principles and Applications; Mc Graw-Hill: New York, NY, USA, 2002; pp. 331–341. [Google Scholar]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Dixon, W.J. Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 1980, 20, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Dunham, N.W.; Miya, T.S. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Nassis, C.Z.; Lago, L.C.; Mory, S.B.; Raquel, M.K.S.; Figueiredo, C.R.; Lebre, A.T.; Giesbrecht, A.M. Estudo da ação depressora inespecífica do suco extraído das folhas de Bryophyllum calcynum Salisb. (Crassulaceae) sobre o sistema nervoso central. Comparação com efeitos da difenidramina. Arq. Méd. ABC 1991, 14, 64–68. [Google Scholar]
- Mello, F.B.; Langeloh, A.; Mello, J.R.B. Study of the toxicity and efficacy in Wistar rats of a phytoterapic used as sedative and/or hypnotic. Lat. Am. J. Pharm. 2007, 26, 38–44. [Google Scholar]
- European Patent Organization. Pregabalin Patent. Available online: www.escavador.com.br/patentes/383391/composicao-PG (accessed on 11 November 2016).
- Tscharnuter, W. Photon Correlation Spectroscopy in Particle Sizing. In Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation, 1st ed.; Meyers, R.A., Ed.; John Wiley & Sons: Chichester, UK, 2006; pp. 5469–5485. [Google Scholar] [CrossRef]
- Boyle, J.; Eriksson, M.E.; Gribble, L.; Gouni, R.; Johnsen, S.; Coppini, D.V.; Kerr, D. Randomized, placebo-controlled comparison of amitriptyline, duloxetine, and pregabalin in patients with chronic diabetic peripheral neuropathic pain: Impact on pain, polysomnographic sleep, daytime functioning, and quality of life. Diabetes Care. 2012, 35, 2451–2458. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; Mao, J. Neuropathic pain: Mechanisms and their clinical implications. BMJ. 2014, 348, f7656. [Google Scholar] [CrossRef] [Green Version]
- Zilliox, L.A. Neuropathic Pain. Continuum 2017, 23, 512–532. [Google Scholar] [CrossRef]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Silva, H.S.R.C.; dos Santos, K.S.C.R.; Ferreira, E.I. Chitosan: Hydrossoluble derivatives, pharmaceutical applications, and recent advances. Quim. Nova 2006, 29, 776–785. [Google Scholar] [CrossRef]
- Ergun, R.; Guo, J.; Huebner-Keese, B. Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Elsevier: Waltham, MA, USA, 2016; pp. 694–702. [Google Scholar] [CrossRef]
- Naves, V.M.L. Development of chitosan and hydroxypropylmethyl cellulose phthalate nanoparticles containing methotrexate for potential treatment of glioblastoma. Ph.D. Thesis, Universidade Federal de Alfenas, Alfenas, Brazil, 22 February 2018. Available online: https://bdtd.unifal-mg.edu.br:8443 (accessed on 30 October 2021).
- Apolinário, A.C.; Salata, G.C.; Bianco, A.F.R.; Fukumori, C.; Lopes, L.B. Opening the pandora’s box of nanomedicine: There is indeed “plenty of room at the bottom”. Quim. Nova 2020, 43, 212–225. [Google Scholar]
- Masserini, M. Nanoparticles for brain drug delivery. ISRN Biochem. 2013, 2013, 238428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.M.; Maria, D.N.; Mishra, S.R.; Guragain, D.; Wang, X.; Jablonski, M.M. Once daily pregabalin eye drops for management of glaucoma. ACS Nano. 2019, 13, 13728–13744. [Google Scholar] [CrossRef] [PubMed]
- Külkamp, I.; Paese, K.; Guterres, S.S.; Pohlmann, A.R. Stabilization of lipoic acid by encapsulation in polymeric nanocapsules designed for cutaneous administration. Quim. Nova 2009, 32, 2078–2084. [Google Scholar] [CrossRef]
- Liu, C.H.; Wu, C.T. Optimization of nanostructured lipid carriers for lutein delivery. Colloid Surf. A Physicochem. Eng. Asp. 2010, 353, 149–156. [Google Scholar] [CrossRef]
- Ruela, A.L.; de Figueiredo, E.C.; de Araújo, M.B.; Carvalho, F.C.; Pereira, G.R. Molecularly imprinted microparticles in lipid-based formulations for sustained release of donepezil. Eur. J. Pharm. Sci. 2016, 93, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, A.A.; Araújo, C.R.M.; Soares, N.A.; Goulart, M.O.F.; de Abreu, F.C. Different strategies for crosslinking of chitosan. Quim. Nova 2011, 34, 1215–1223. [Google Scholar] [CrossRef]
- Powers, K.W.; Brown, S.C.; Krishna, V.B.; Wasdo, S.C.; Moudgil, B.M.; Roberts, S.M. Research strategies for safety evaluation of nanomaterials. Part VI. Characterization of nanoscale particles for toxicological evaluation. Toxicol. Sci. 2006, 90, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Sipos, P.; Szucs, M.; Szabó, A.; Eros, I.; Szabó-Révész, P. An assessment of the interactions between diclofenac sodium and ammonia methacrylate copolymer using thermal analysis and Raman spectroscopy. J. Pharm. Biomed. Anal. 2008, 46, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Soares, G.A.; de Castro, A.D.; Cury, B.S.; Evangelista, R.C. Blends of cross-linked high amylose starch/pectin loaded with diclofenac. Carbohydr. Polym. 2013, 91, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Chew, M.L.; Plotka, A.; Alvey, C.W.; Pitman, V.W.; Alebic-Kolbah, T.; Scavone, J.M.; Bockbrader, H.N. Pharmacokinetics of pregabalin controlled-release in healthy volunteers: Effect of food in five single-dose, randomized, clinical pharmacology studies. Clin. Drug Investig. 2014, 34, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Matha, K.; Lollo, G.; Taurino, G.; Respaud, R.; Marigo, I.; Shariati, M.; Bussolati, O.; Vermeulen, A.; Remaut, K.; Benoit, J.P. Bioinspired hyaluronic acid and polyarginine nanoparticles for DACHPt delivery. Eur. J. Pharm. Biopharm. 2020, 150, 1–13. [Google Scholar] [CrossRef] [PubMed]
- DiPiro, J.T.; Spruill, W.J.; Wade, W.E.; Blouin, R.A.; Pruemer, J.M. Concepts in Clinical Pharmacokinetics, 4th ed.; American Society of Health-System Pharmacists: Bethesda, MD, USA, 2005; pp. 29–44. [Google Scholar]
- Bee, L.A.; Dickenson, A.H. Descending facilitation from the brainstem determines behavioral and neuronal hypersensitivity following nerve injury and efficacy of pregabalin. Pain 2008, 140, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Field, M.J.; McCleary, S.; Hughes, J.; Singh, L. Gabapentin and pregabalin, but not morphine and amitriptyline, block both static and dynamic components of mechanical allodynia induced by streptozocin in the rat. Pain 1999, 80, 391–398. [Google Scholar] [CrossRef]
- Nakai, K.; Nakae, A.; Hashimoto, R.; Mashimo, T.; Hosokawa, K. Antinociceptive effects of mirtazapine, pregabalin, and gabapentin after chronic constriction injury of the infraorbital nerve in rats. J. Oral Facial Pain Headache 2014, 28, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Farias, J.B.; Caram-Salas, N.L.; Salinas-Abarca, A.B.; Ocampo, J.; Granados-Soto, V. Ultra-low doses of naltrexone enhance the antiallodynic effect of pregabalin or gabapentin in neuropathic rats. Drug Dev. Res. 2017, 78, 371–380. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kawakami, K.; Miyatake, K.; Morimoto, M.; Shigemasa, Y.; Minami, S. Analgesic effects of chitin and chitosan. Carbohydr. Polym. 2002, 49, 249–252. [Google Scholar] [CrossRef]
- Organization for Economic Co-operation and Development (OECD). Test No. 423: Acute Oral toxicity—Acute Toxic Class Method, in OECD Guidelines for the Testing of Chemicals; Section 4. Available online: https://doi.org/10.1787/9789264071001-en (accessed on 22 October 2019).
- Khan, J.; Noboru, N.; Imamura, Y.; Eliav, E. Effect of pregabalin and diclofenac on tactile allodynia, mechanical hyperalgesia and pro inflammatory cytokine levels (IL-6, IL-1β) induced by chronic constriction injury of the infraorbital nerve in rats. Cytokine 2018, 104, 124–129. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | PG | PG-PN |
|---|---|---|
| AUC (0-∞) (µg/mL h) | 39.60 (82.55–24.25) | 22.08 * (32.16–13.53) |
| Cmax (µg/mL) | 6.56 (5.06–13.88) | 3.48 ** (5.70–1.40) |
| Tmax (h) | 0.75 (0.5–4) | 1.5 (0.5–2.5) |
| Vd (L/kg) | 2.51 (1.82–6.44) | 8.21 * (98.02–3.33) |
| t1/2 (h) | 7.03 (6.68–10.83) | 10.01 (178.35–7.42) |
| Cl (L/kg/h) | 0.25 (0.12–0.41) | 0.46 * (0.31–0.74) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, R.F.; Nunes, J.B.; Agostini, S.B.N.; dos Santos, P.F.; Cancino-Bernardi, J.; Placido, R.V.; Moraes, T.R.; Freitas, J.T.J.; Pereira, G.R.; Carvalho, F.C.; et al. Preclinical Evaluation of Polymeric Nanocomposite Containing Pregabalin for Sustained Release as Potential Therapy for Neuropathic Pain. Polymers 2021, 13, 3837. https://doi.org/10.3390/polym13213837
Rodrigues RF, Nunes JB, Agostini SBN, dos Santos PF, Cancino-Bernardi J, Placido RV, Moraes TR, Freitas JTJ, Pereira GR, Carvalho FC, et al. Preclinical Evaluation of Polymeric Nanocomposite Containing Pregabalin for Sustained Release as Potential Therapy for Neuropathic Pain. Polymers. 2021; 13(21):3837. https://doi.org/10.3390/polym13213837
Chicago/Turabian StyleRodrigues, Rafaela Figueiredo, Juliana Barbosa Nunes, Sandra Barbosa Neder Agostini, Paloma Freitas dos Santos, Juliana Cancino-Bernardi, Rodrigo Vicentino Placido, Thamyris Reis Moraes, Jennifer Tavares Jacon Freitas, Gislaine Ribeiro Pereira, Flávia Chiva Carvalho, and et al. 2021. "Preclinical Evaluation of Polymeric Nanocomposite Containing Pregabalin for Sustained Release as Potential Therapy for Neuropathic Pain" Polymers 13, no. 21: 3837. https://doi.org/10.3390/polym13213837
APA StyleRodrigues, R. F., Nunes, J. B., Agostini, S. B. N., dos Santos, P. F., Cancino-Bernardi, J., Placido, R. V., Moraes, T. R., Freitas, J. T. J., Pereira, G. R., Carvalho, F. C., Galdino, G., & Boralli, V. B. (2021). Preclinical Evaluation of Polymeric Nanocomposite Containing Pregabalin for Sustained Release as Potential Therapy for Neuropathic Pain. Polymers, 13(21), 3837. https://doi.org/10.3390/polym13213837