
Cross-Linking Strategies for Fluorine-Containing Polymer Coatings for Durable Resistant Water- and Oil-Repellency

 ,
,  and
and
Abstract
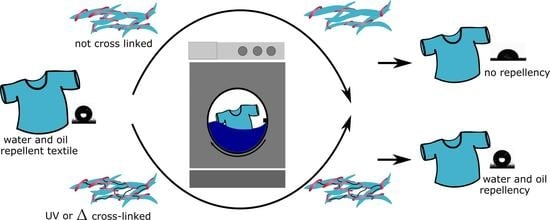
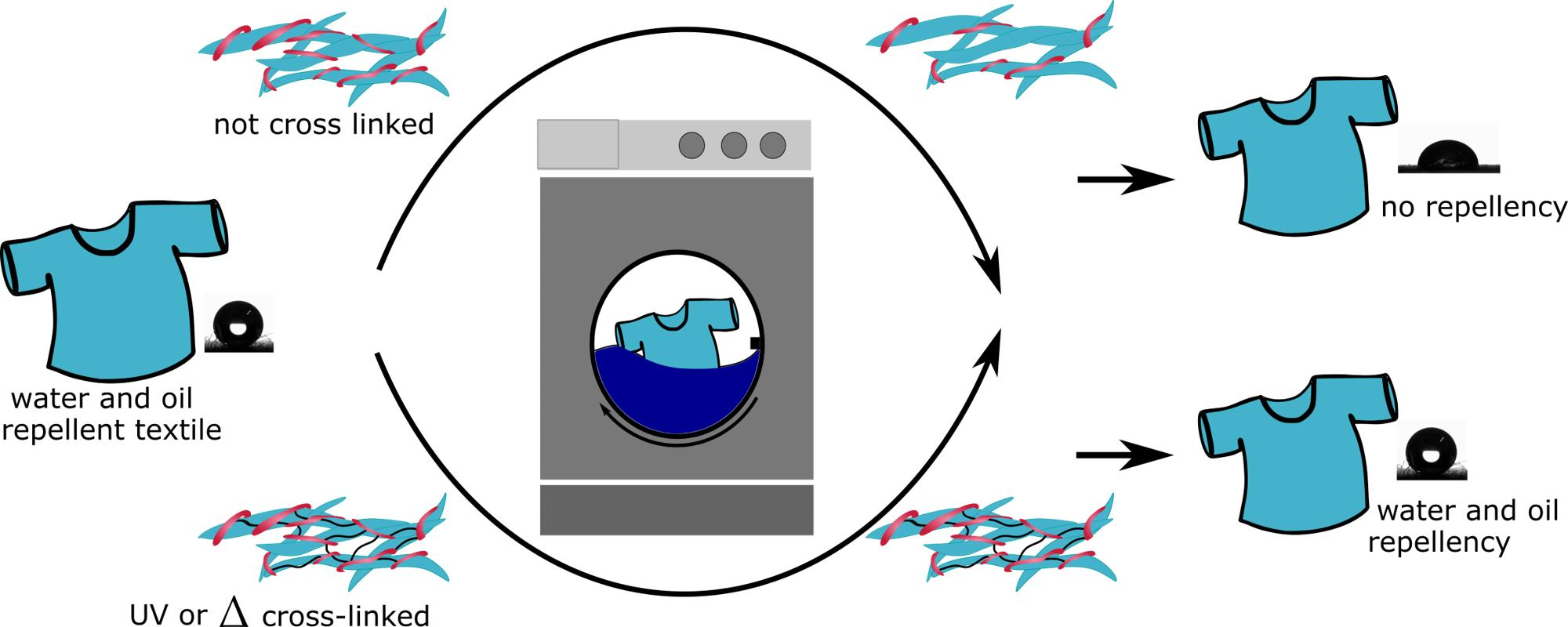
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Instrumentation
2.3. Exemplary Synthesis of Poly(fluoroacrylate-co-Stearyl Methacrylate) via Emulsion Polymerization
2.4. Application of Polymer-Dispersion on Paper Substrates
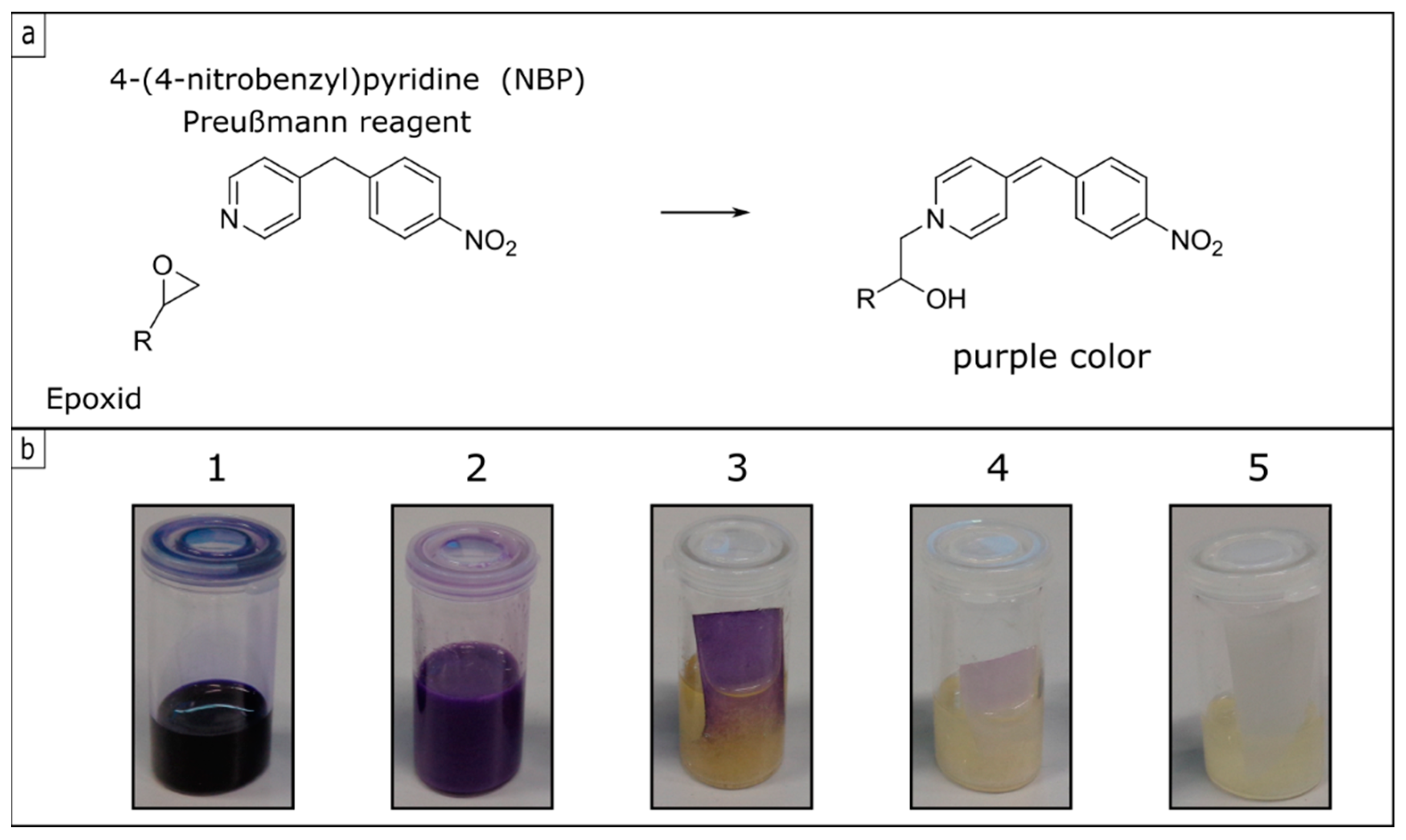
2.5. Model Experiment to Investigate the Chemical Stability
3. Results and Discussion
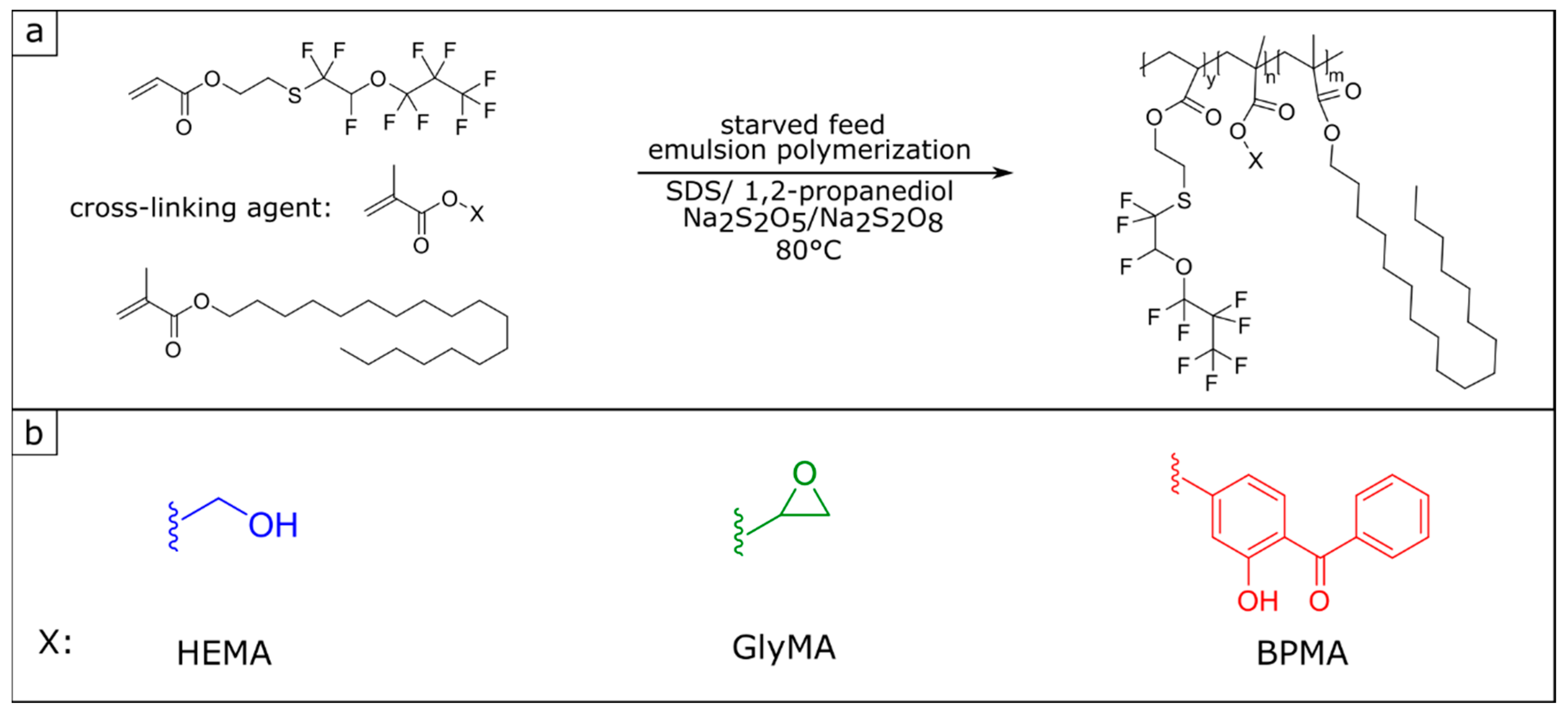
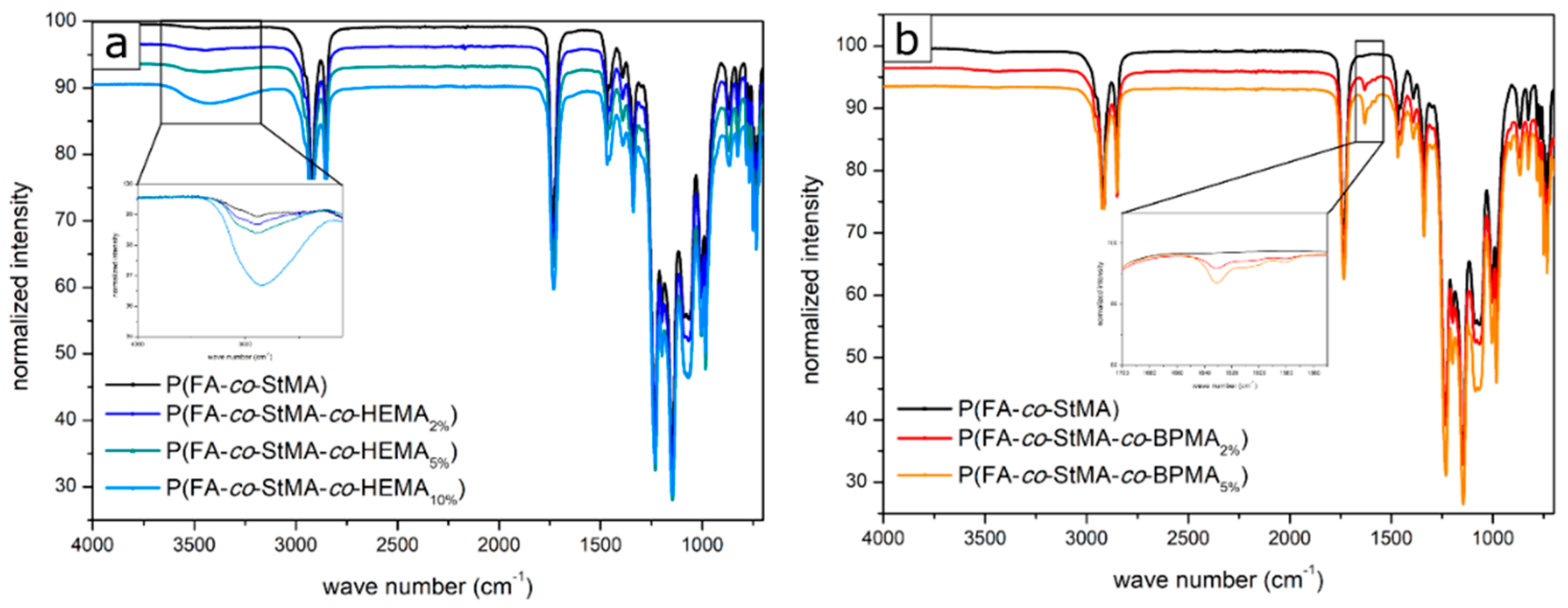
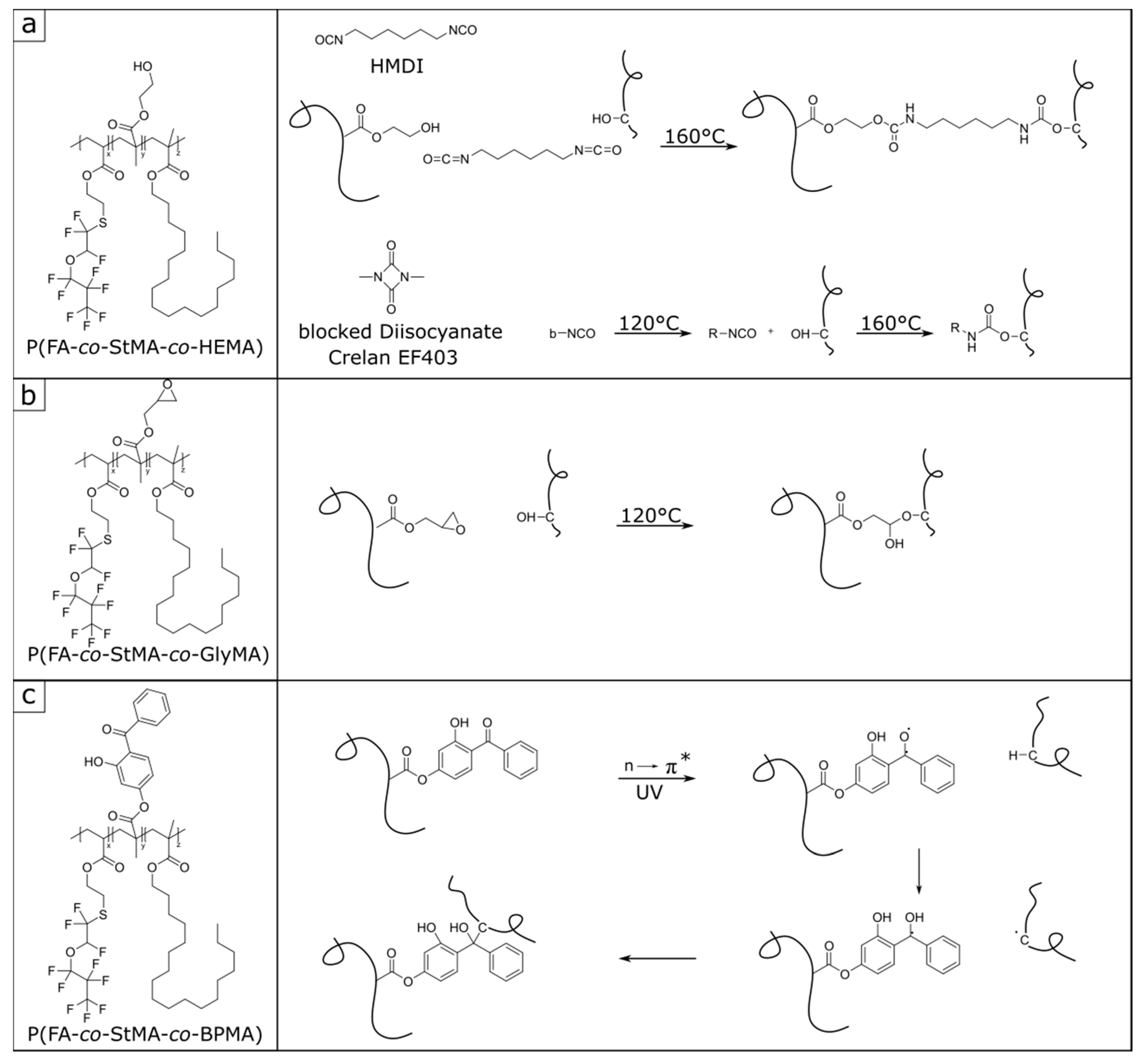
3.1. Synthesis and Characterization of Poly(fluoroacrylate-co-Stearyl Methacrylate) P(FA-co-StMA) with Cross-Linking Reagents
3.2. Application of Polymers on Cellulose Fibers
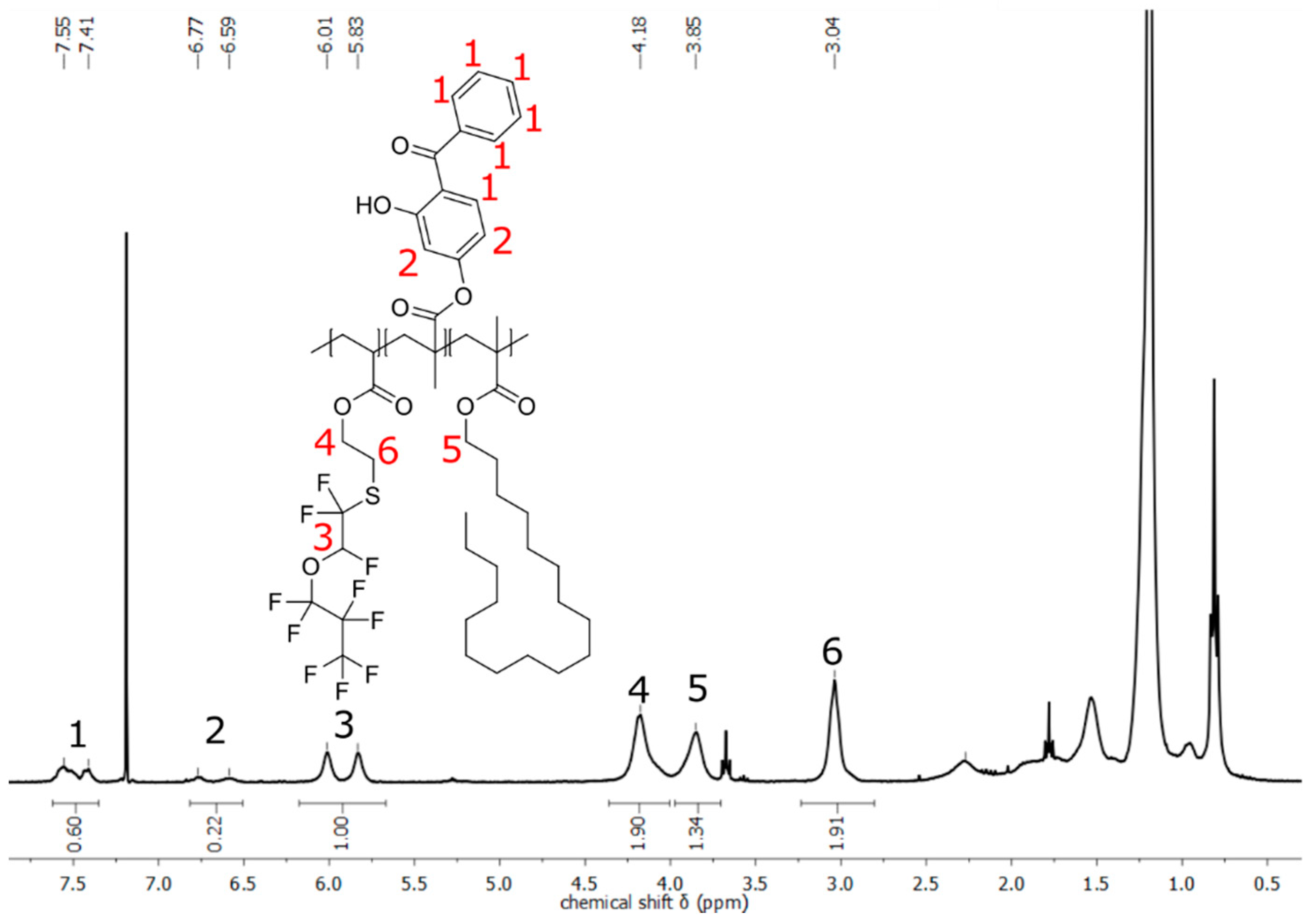
3.3. Investigation and Quantification of Cross-Linking Reactions
3.4. Chemical Integrity under Various Forces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blossey, R. Self-cleaning surfaces—Virtual realities. Nat. Mater. 2003, 2, 301. [Google Scholar] [CrossRef]
- Dodiuk, H.; Rios, P.F.; Dotan, A.; Kenig, S. Hydrophobic and self-cleaning coatings. Polym. Adv. Technol. 2007, 18, 746. [Google Scholar] [CrossRef]
- Ameduri, B. Fluoropolymers: The Right Material for the Right Applications. Chem. Eur. J. 2018, 24, 18830. [Google Scholar] [CrossRef]
- Puts, G.J.; Crouse, P.; Ameduri, B. Polytetrafluoroethylene: Synthesis and Characterization of the Original Extreme Polymer. Chem. Rev. 2019, 119, 1763. [Google Scholar] [CrossRef]
- Davison, G.; Lane, B. Additives in Water-borne Coatings: The Way Forward. In Additives in Water-Borne Coatings; Royal Society of Chemistry: Cambridge, UK, 2003. [Google Scholar]
- Sundararajan, N.; Yang, S.; Ogino, K.; Valiyaveettil, S.; Zhou, X.; Ober, C.K.; Obendorf, S.K.; Allen, R.D. Supercritical CO2 Processing for Submicron Imaging of Fluoropolymers. Chem. Mater. 2000, 12, 41. [Google Scholar] [CrossRef]
- Boutevin, B.; Mouanda, J.; Pietrasanta, Y.; Taha, M. Synthesis of block cotelomers involving a perfluorinated chain and a hydrophilic chain. II. Use of fluorinated telogens with iodine or thiol end groups. J. Polym. Sci. A Polym. Chem. 1986, 24, 2891. [Google Scholar] [CrossRef]
- Kako, T.; Nakajima, A.; Irie, H.; Kato, Z.; Uematsu, K.; Watanabe, T.; Hashimoto, K. Adhesion and sliding of wet snow on a super-hydrophobic surface with hydrophilic channels. J. Mater. Sci. 2004, 39, 547. [Google Scholar] [CrossRef]
- Saito, H.; Takai, K.; Takazawa, H.; Yamauchi, G. A Study on Snow Sticking Weight to Water-Repellent Coatings. J. Soc. Mater. Sci. Jpn. 1997, 46, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Lafuma, A.; Quéré, D. Superhydrophobic states. Nat. Mater. 2003, 2, 457. [Google Scholar] [CrossRef]
- Barthlott, W.; Neinhuis, C. Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 1997, 202, 1. [Google Scholar] [CrossRef]
- Pan, N.; Sun, G. Functional Textiles for Improved Performance, Protection and Health; Woodhead Pub: Cambridge, UK, 2011. [Google Scholar]
- Fürstner, R.; Barthlott, W.; Neinhuis, C.; Walzel, P. Wetting and Self-Cleaning Properties of Artificial Superhydrophobic Surfaces. Langmuir 2005, 21, 956. [Google Scholar] [CrossRef]
- Parker, A.R.; Lawrence, C.R. Water capture by a desert beetle. Nature 2001, 414, 33. [Google Scholar] [CrossRef]
- Gao, X.; Jiang, L. Water-repellent legs of water striders. Nature 2004, 432, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Cong, Q.; Qi, Y.; Jin, J.; Choy, K.-L. Hydrophobic durability characteristics of butterfly wing surface after freezing cycles towards the design of nature inspired anti-icing surfaces. PLoS ONE 2018, 13, e0188775. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Feng, L.; Gao, X.; Jiang, L. Bioinspired Surfaces with Special Wettability. Acc. Chem. Res. 2005, 38, 644. [Google Scholar] [CrossRef]
- Nau, M.; Seelinger, D.; Biesalski, M. Functional surface coatings from tailor-made long-chain hydroxypropyl cellulose ester nanoparticles. Cellulose 2018, 25, 5769. [Google Scholar] [CrossRef]
- Cordt, C.; Geissler, A.; Biesalski, M. Regenerative Superhydrophobic Paper Coatings by In Situ Formation of Waxy Nanostructures. Adv. Mater. Interfaces 2020, 8, 2001265. [Google Scholar] [CrossRef]
- Distler, D.; Neto, W.S.; Machado, F. Emulsion Polymerization. In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Dechant, J. Handbook of fiber science and technology. Vol. II. Chemical processing of fibers and fabrics. Functional finishes. Acta Polym. 1985, 36, 242. [Google Scholar] [CrossRef]
- Holmquist, H.; Schellenberger, S.; van der Veen, I.; Peters, G.M.; Leonards, P.E.G.; Cousins, I.T. Properties, performance and associated hazards of state-of-the-art durable water repellent (DWR) chemistry for textile finishing. Environ. Int. 2016, 91, 251. [Google Scholar] [CrossRef]
- Schindler, W.D.; Hauser, P.J. Chemical Finishing of Textiles; Elsevier Science: Cambridge, UK, 2004. [Google Scholar]
- Roach, P.; Shirtcliffe, N.J.; Newton, M.I. Progess in superhydrophobic surface development. Soft Matter 2008, 4, 224. [Google Scholar] [CrossRef] [PubMed]
- Bernett, M.K.; Zisman, W.A. Relation of Wettability by Aqueous Solutions to the Surface Constitution of Low-energy Solids. J. Phys. Chem. 1959, 63, 1241. [Google Scholar] [CrossRef]
- Kissa, E. Wetting and Wicking. Text. Res. J. 1996, 66, 660. [Google Scholar] [CrossRef]
- Forsythe, J.; Hill, D. The radiation chemistry of fluoropolymers. Prog. Polym. Sci. 2000, 25, 101. [Google Scholar] [CrossRef]
- Kredel, J.; Gallei, M. Compression-Responsive Photonic Crystals Based on Fluorine-Containing Polymers. Polymers 2019, 11, 2114. [Google Scholar] [CrossRef] [Green Version]
- Zisman, W.A. Influence of constitution on adhesion. Ind. Eng. Chem. 1963, 55, 18. [Google Scholar] [CrossRef]
- Graham, P.; Stone, M.; Thorpe, A.; Nevell, T.G.; Tsibouklis, J. Fluoropolymers with very low surface energy characteristics. J. Fluor. Chem. 2000, 104, 29. [Google Scholar] [CrossRef]
- Kredel, J.; Dietz, C.; Gallei, M. Fluoropolymer-Containing Opals and Inverse Opals by Melt-Shear Organization. Molecules 2019, 24, 333. [Google Scholar] [CrossRef] [Green Version]
- Kredel, J.; Gallei, M. Ozone-Degradable Fluoropolymers on Textile Surfaces for Water and Oil Repellency. ACS Appl. Polym. Mater. 2020, 2, 2867. [Google Scholar] [CrossRef]
- Krafft, M.P.; Riess, J.G. Selected physicochemical aspects of poly- and perfluoroalkylated substances relevant to performance, environment and sustainability-part one. Chemosphere 2015, 129, 4. [Google Scholar] [CrossRef]
- Prevedouros, K.; Cousins, I.T.; Buck, R.C.; Korzeniowski, S.H. Sources, fate and transport of perfluorocarboxylates. Environ. Sci. Technol. 2006, 40, 32. [Google Scholar] [CrossRef]
- Butt, C.M.; Muir, D.C.; Mabury, S.A. Biotransformation pathways of fluorotelomer-based polyfluoroalkyl substances: A review. Environ. Toxicol. Chem. 2014, 33, 243. [Google Scholar] [CrossRef]
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; de Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; van Leeuwen, S. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integ. Environ. Asses. 2011, 7, 513. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.H.; Berti, W.R.; Szostek, B.; Buck, R.C. Investigation of the Biodegradation Potential of a Fluoroacrylate Polymer Product in Aerobic Soils. Environ. Sci. Technol. 2008, 42, 800. [Google Scholar] [CrossRef]
- Benskin, J.P.; Muir, D.C.G.; Scott, B.F.; Spencer, C.; de Silva, A.O.; Kylin, H.; Martin, J.W.; Morris, A.; Lohmann, R.; Tomy, G.; et al. Perfluoroalkyl acids in the Atlantic and Canadian Arctic Oceans. Environ. Sci. Technol. 2012, 46, 5815. [Google Scholar] [CrossRef]
- Jensen, A.A.; Leffers, H. Emerging endocrine disrupters: Perfluoroalkylated substances. Int. J. Androl. 2008, 31, 161. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.W.; Burris, J.M.; Ehresman, D.J.; Froehlich, J.W.; Seacat, A.M.; Butenhoff, J.L.; Zobel, L.R. Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ. Health Perspect. 2007, 115, 1298. [Google Scholar] [CrossRef]
- Midasch, O.; Drexler, H.; Hart, N.; Beckmann, M.W.; Angerer, J. Transplacental exposure of neonates to perfluorooctanesulfonate and perfluorooctanoate: A pilot study. Int. Arch. Occup. Environ. Health 2007, 80, 643. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, Y.; Liao, C.; Cai, Y.; Jiang, G. Perspectives on the Inclusion of Perfluorooctane Sulfonate into the Stockholm Convention on Persistent Organic Pollutants. Environ. Sci. Technol. 2009, 43, 5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Official Journal of the European Union. 2010. Available online: http://www.epa. gov/oppt/pfoa/pubs/pfoastewardship.htm (accessed on 23 February 2020).
- Muthu, S.S. Roadmap to Sustainable Textiles and Clothing: Environmental and Social Aspects of Textiles and Clothing Supply Chain; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Parsons, J.R.; Sáez, M.; Dolfing, J.; de Voogt, P. Biodegradation of perfluorinated compounds. Rev. Environ. Contam. Toxicol. 2008, 196, 53. [Google Scholar] [CrossRef]
- Simon, C.M.; Kaminsky, W. Chemical recycling of polytetrafluoroethylene by pyrolysis. Polym. Degrad. Stab. 1998, 62, 1. [Google Scholar] [CrossRef]
- Wang, N.; Szostek, B.; Buck, R.C.; Folsom, P.W.; Sulecki, L.M.; Capka, V.; Berti, W.R.; Gannon, J.T. Fluorotelomer Alcohol BiodegradationDirect Evidence that Perfluorinated Carbon Chains Breakdown. Environ. Sci. Technol. 2005, 39, 7516. [Google Scholar] [CrossRef]
- Amedro, D.; Vereecken, L.; Crowley, J.N. Kinetics and mechanism of the reaction of perfluoro propyl vinyl ether with OH: Assessment of its fate in the atmosphere. Phys. Chem. Chem. Phys. 2015, 17, 18558. [Google Scholar] [CrossRef]
- Lyons, B.J. Radiation crosslinking of fluoropolymers—A review. Radiat. Phys. Chem. 1995, 45, 159. [Google Scholar] [CrossRef]
- Alishiri, M.; Shojaei, A.; Abdekhodaie, M.J.; Yeganeh, H. Synthesis and characterization of biodegradable acrylated polyurethane based on poly(ε-caprolactone) and 1,6-hexamethylene diisocyanate. Mater. Sci. Eng. C 2014, 42, 763. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Khan, A.; Stuparu, M.C. Post-polymerization modification reactions of poly(glycidyl methacrylate)s. RSC Adv. 2017, 7, 55874. [Google Scholar] [CrossRef] [Green Version]
- Viel, B.; Ruhl, T.; Hellmann, G.P. Reversible Deformation of Opal Elastomers. Chem. Mater. 2007, 19, 5673. [Google Scholar] [CrossRef]
- Schäfer, C.G.; Gallei, M.; Zahn, J.T.; Engelhardt, J.; Hellmann, G.P.; Rehahn, M. Reversible Light-, Thermo-, and Mechano-Responsive Elastomeric Polymer Opal Films. Chem. Mater. 2013, 25, 2309. [Google Scholar] [CrossRef]
- Zhang, X.; Shiraishi, Y.; Hirai, T. Cu(II)-Selective Green Fluorescence of a Rhodamine−Diacetic Acid Conjugate. Org. Lett. 2007, 9, 5039. [Google Scholar] [CrossRef]
- Shabbir, M. Textiles and Clothing: Environmental Concerns and Solutions; Wiley-Scrivener: Hoboken, NJ, USA, 2019. [Google Scholar]
- Williams, J.T. Waterproof and Water Repellent Textiles and Clothing; Woodhead Pub: Cambridge, UK, 2018. [Google Scholar]
- Kamagata, K.; Toyama, M. Effect of the length of branches on the critical surface tension of poly(n-alkyl methacrylates) and copolymers of stearyl methacrylate with methacrylonitrile. J. Appl. Polym. Sci. 1974, 18, 167. [Google Scholar] [CrossRef]
- Malner, T. Fluorhaltige Polyacrylat-Dispersionen für die Wasser—Und Ölabweisende Textilausruüstung. Ph.D. Thesis, Universität Freiburg, Breisgau, Germany, 2002. [Google Scholar]
- Wallach, J.A.; Huang, S.J. Copolymers of Itaconic Anhydride and Methacrylate-Terminated Poly(lactic acid) Macromonomers. Biomacromolecules 2000, 1, 174. [Google Scholar] [CrossRef]
- Morita, S. Hydrogen-bonds structure in poly(2-hydroxyethyl methacrylate) studied by temperature-dependent infrared spectroscopy. Front. Chem. 2014, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.M.; Lee, J.M.; Ha, M.H.; Lee, K.; Choe, S. Highly crosslinked poly(glycidyl methacrylate-co-divinyl benzene) particles by precipitation polymerization. Polymer 2007, 48, 3107. [Google Scholar] [CrossRef]
- Jang, J.; Bae, J.; Ko, S. Synthesis and curing of poly(glycidyl methacrylate) nanoparticles. J. Polym. Sci. A Polym. Chem. 2005, 43, 2258. [Google Scholar] [CrossRef]
- Prucker, O.; Brandstetter, T.; Rühe, J. Surface-attached hydrogel coatings via C,H-insertion crosslinking for biomedical and bioanalytical applications. Biointerphases 2017, 13, 10801. [Google Scholar] [CrossRef] [Green Version]
- Mehlhase, S.; Schäfer, C.G.; Morsbach, J.; Schmidt, L.; Klein, R.; Frey, H.; Gallei, M. Vinylphenylglycidyl ether-based colloidal architectures: High-functionality crosslinking reagents, hybrid raspberry-type particles and smart hydrophobic surfaces. RSC Adv. 2014, 4, 41348. [Google Scholar] [CrossRef] [Green Version]
- Janko, M.; Jocher, M.; Boehm, A.; Babel, L.; Bump, S.; Biesalski, M.; Meckel, T.; Stark, R.W. Cross-Linking Cellulosic Fibers with Photoreactive Polymers: Visualization with Confocal Raman and Fluorescence Microscopy. Biomacromolecules 2015, 16, 2179. [Google Scholar] [CrossRef]
- Hageage, G.J.; Harrington, B.J. Use of Calcofluor White in Clinical Mycology. Lab. Med. 1984, 15, 109. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
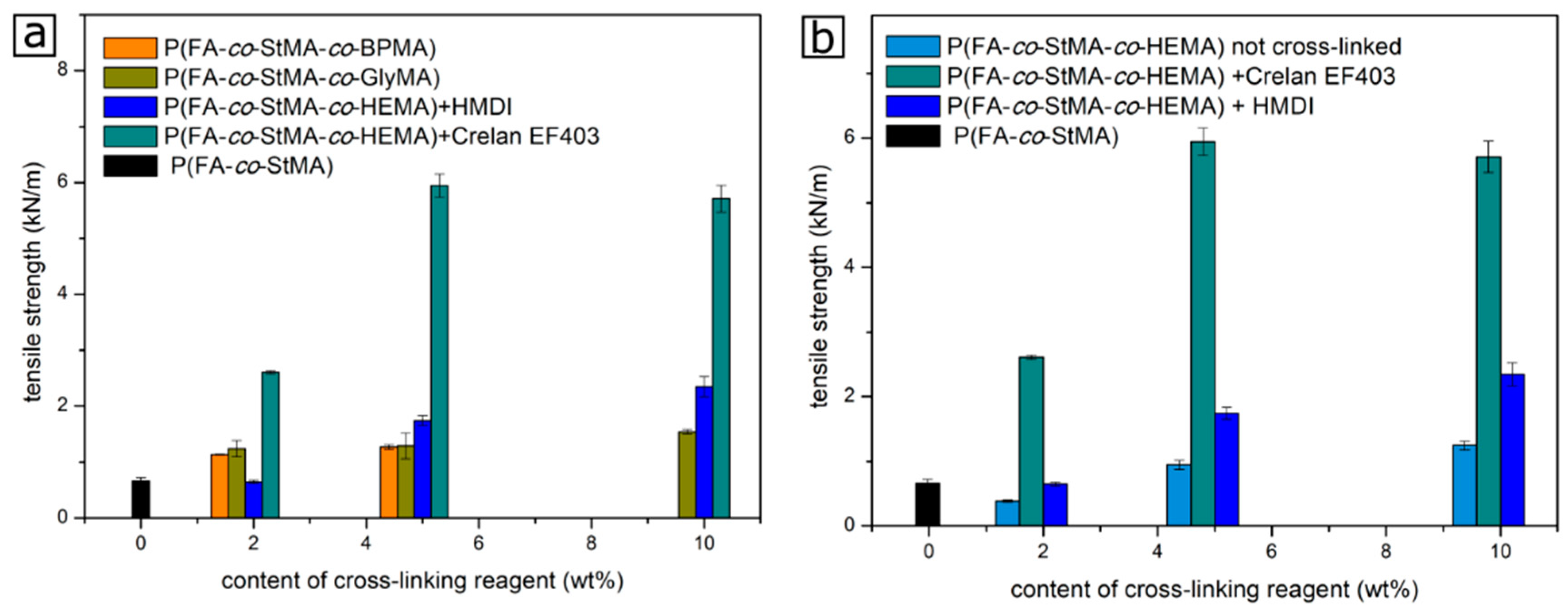{kind=link}
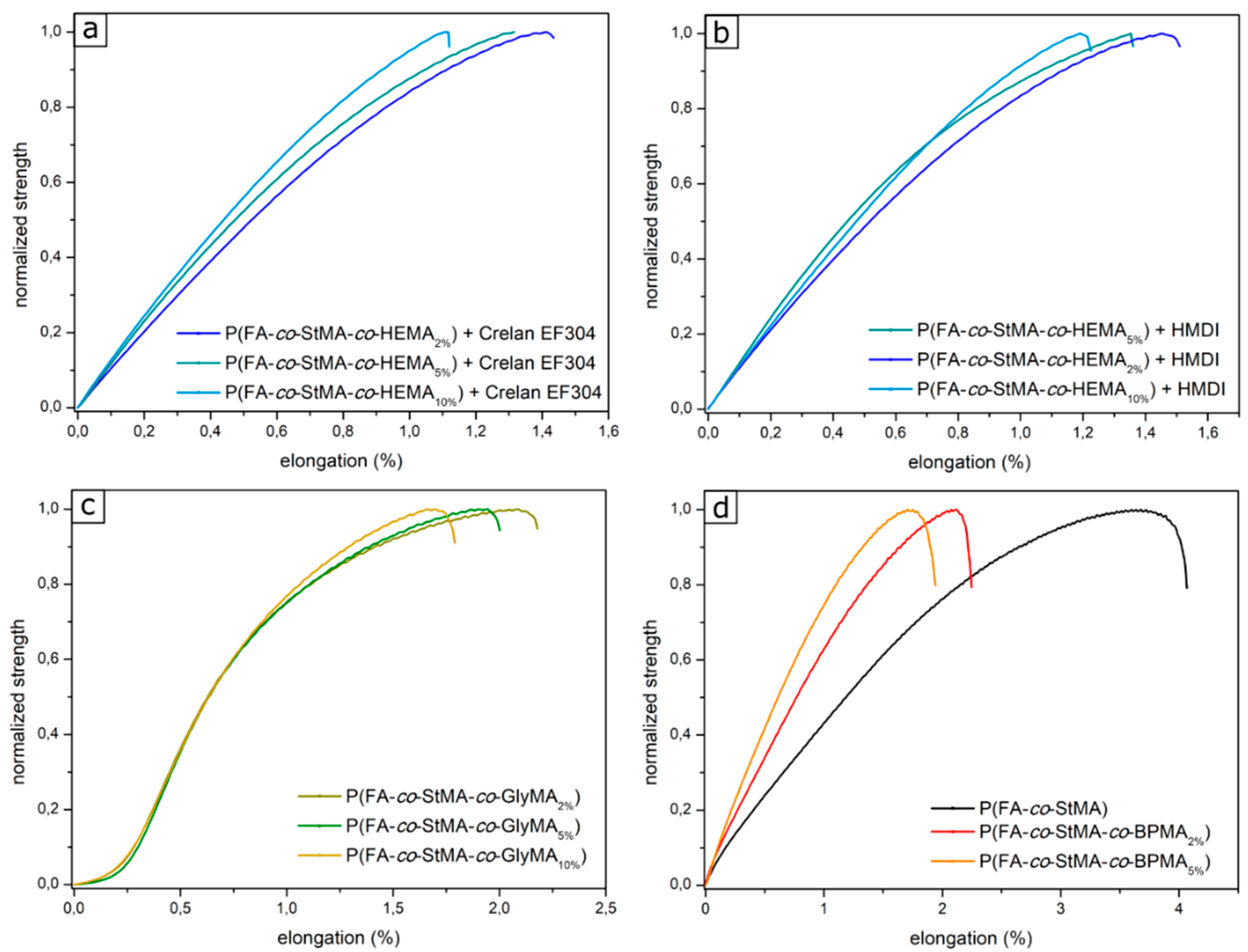{kind=link}
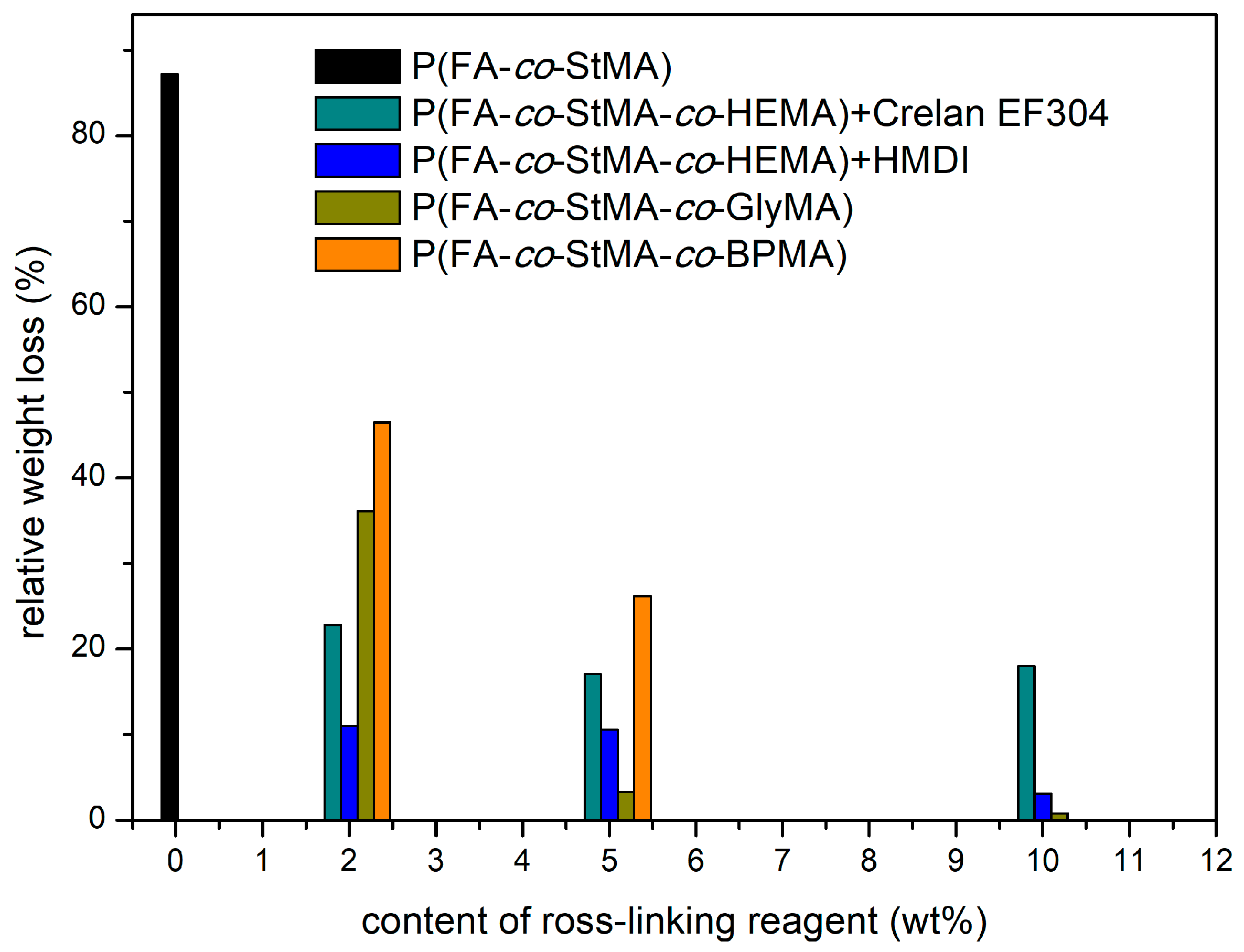{kind=link}
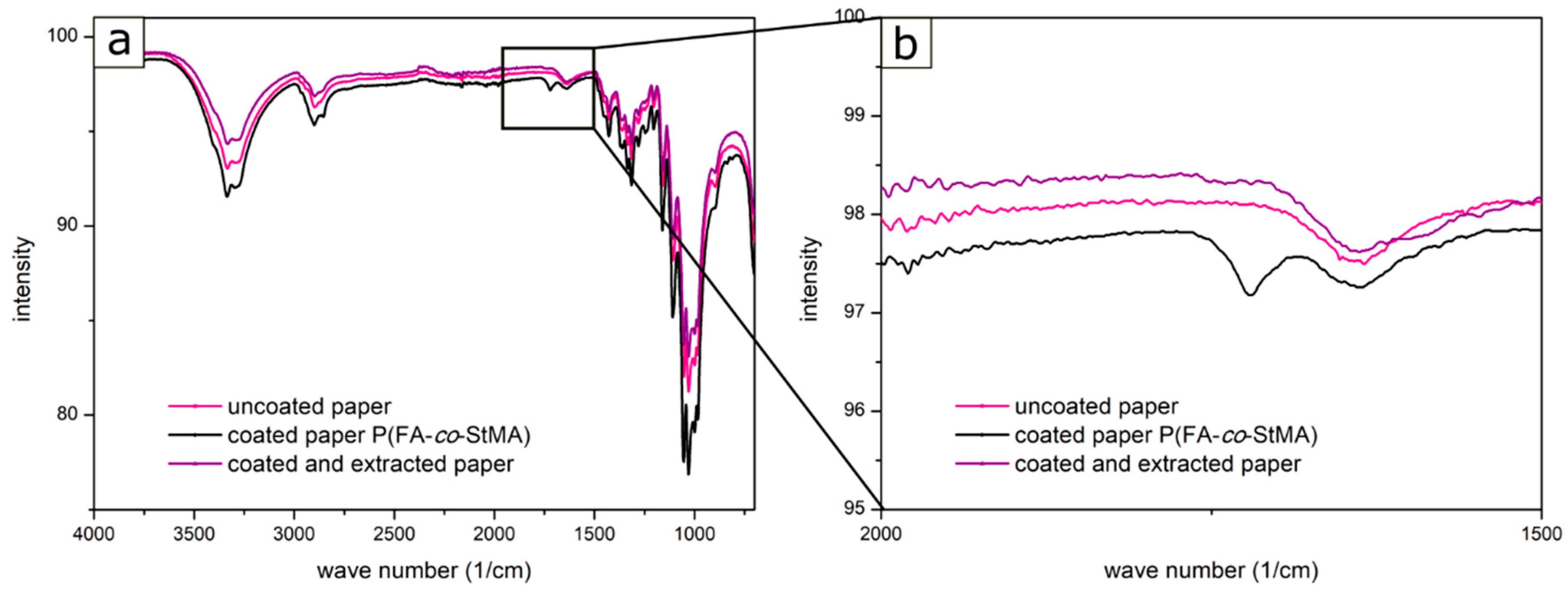{kind=link}
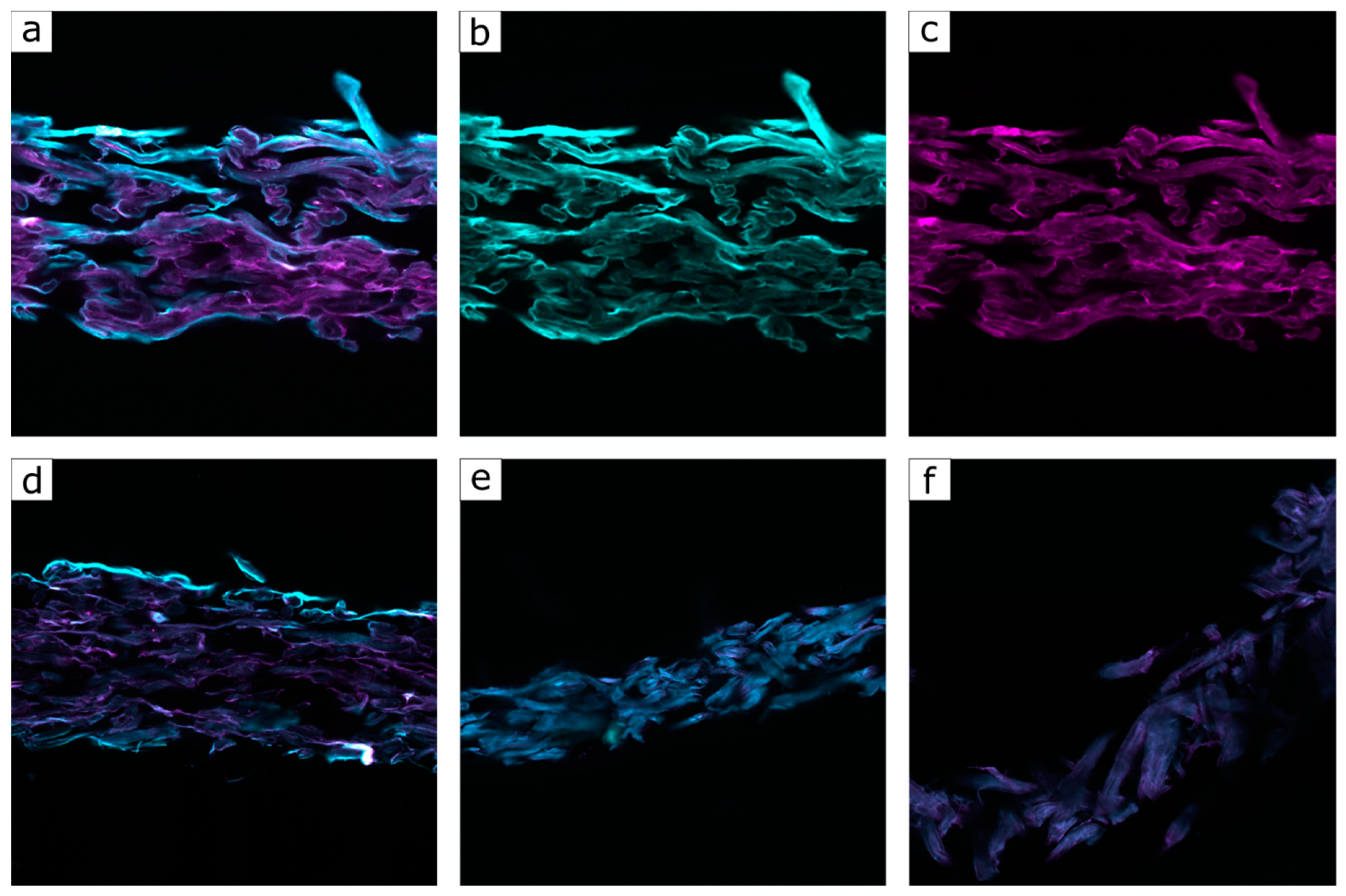{kind=link}
| Polymer | m(FA) (g) | m(StMA) (g) | m(X) (g) | |
|---|---|---|---|---|
| 1 | P(FA-co-StMA) | 20.56 | 13.54 | 0 |
| 2 | P(FA-co-StMA-co-HEMA2%) | 20.22 | 13.20 | 0.68 |
| 3 | P(FA-co-StMA-co-HEMA5%) | 19.70 | 12.69 | 1.71 |
| 4 | P(FA-co-StMA-co-HEMA10%) | 18.85 | 11.85 | 3.41 |
| 5 | P(FA-co-StMA-co-GlyMA2%) | 20.22 | 13.20 | 0.68 |
| 6 | P(FA-co-StMA-co-GlyMA5%) | 19.70 | 12.69 | 1.71 |
| 7 | P(FA-co-StMA-co-GlyMA10%) | 18.85 | 11.85 | 3.41 |
| 8 | P(FA-co-StMA-co-BPMA2%) | 20.22 | 13.20 | 0.68 |
| 9 | P(FA-co-StMA-co-BPMA5%) | 19.70 | 12.69 | 1.71 |
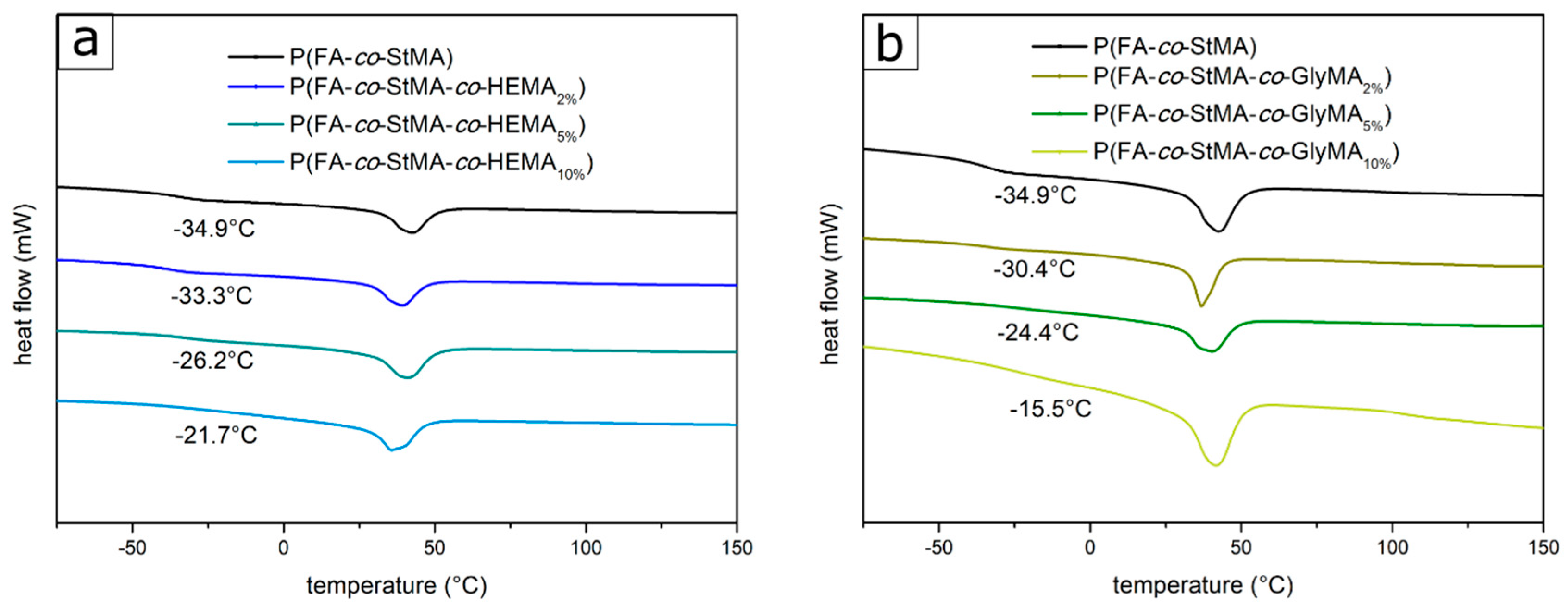
| Polymer | Assigned Fractions FA/StMA/X (%) | Fractions Calculated from 1H-NMR FA/StMA/X (%) | Fractions Calculated from DSC Thermograms FA/StMA/X (%) | |
|---|---|---|---|---|
| 2 | P(FA-co-StMA-co-HEMA2%) | 59.0/39.0/2.0 | / | 59.1/39.1/1.8 |
| 3 | P(FA-co-StMA-co-HEMA5%) | 57.5/37.5/5.0 | / | 57.2/37.1/5.7 |
| 4 | P(FA-co-StMA-co-HEMA10%) | 55.0/35.0/10.0 | 51.1/40.8/8.1 | 55.9/35.8/8.3 |
| 5 | P(FA-co-StMA-co-GlyMA2%) | 59.0/39.0/2.0 | / | 58.4/38.3/3.3 |
| 6 | P(FA-co-StMA-co-GlyMA5%) | 57.5/37.5/5.0 | / | 56.8/37.7/6.5 |
| 7 | P(FA-co-StMA-co-GlyMA10%) | 55.0/35.0/10.0 | / | 54.2/34.1/11.7 |
| 8 | P(FA-co-StMA-co-BPMA2%) | 59.0/39.0/2.0 | 62.2/36.2/1.6 | / |
| 9 | P(FA-co-StMA-co-BPMA5%) | 57.5/37.5/5.0 | 55.3/38.8/5.9 | / |
| Fiber Coated with Polymer | Contact Angle | Oil Repellency |
|---|---|---|
| Uncoated fiber | 0° | 0 |
| P(FA-co-StMA) | 139 ± 3° | 4 |
| P(FA-co-StMA-co-HEMA2%) + HMDI | 138 ± 2° | 4 |
| P(FA-co-StMA-co-HEMA5%) + HMDI | 140 ± 3° | 5 |
| P(FA-co-StMA-co-HEMA10%) + HMDI | 142 ± 4° | 6 |
| P(FA-co-StMA-co-HEMA2%) + Crelan EF403 | 134 ± 3° | 4 |
| P(FA-co-StMA-co-HEMA5%) + Crelan EF403 | 135 ± 1° | 6 |
| P(FA-co-StMA-co-HEMA10%) + Crelan EF403 | 140 ± 3° | 6 |
| P(FA-co-StMA-co-GlyMA2%) | 141 ± 4° | 6 |
| P(FA-co-StMA-co-GlyMA5%) | 145 ± 6° | 6 |
| P(FA-co-StMA-co-GlyMA10%) | 146 ± 4° | 6 |
| P(FA-co-StMA-co-BPMA2%) | 138 ± 1° | 4 |
| P(FA-co-StMA-co-BPMA5%) | 140 ± 3° | 5 |
| Fiber Coated with Polymer | Contact Angle before Extraction | Oil Repellency before Extraction | Contact Angle after Extraction | Oil Repellency after Extraction |
|---|---|---|---|---|
| Uncoated fiber | 0 | 0 | 0 | 0 |
| P(FA-co-StMA) | 139 ± 3 | 4 | 0 | 0 |
| P(FA-co-StMA-co-HEMA2%) + HMDI | 138 ± 2 | 4 | 136 ± 2 | 4 |
| P(FA-co-StMA-co-HEMA5%) + HMDI | 140 ± 3 | 5.5 | 139 ± 4 | 5 |
| P(FA-co-StMA-co-HEMA10%) + HMDI | 142 ± 4 | 6 | 138 ± 1 | 6 |
| P(FA-co-StMA-co-HEMA2%)+Crelan EF403 | 134 ± 3 | 4 | 135 ± 4 | 4 |
| P(FA-co-StMA-co-HEMA5%)+Crelan EF403 | 135 ± 1 | 6 | 135 ± 2 | 5.5 |
| P(FA-co-StMA-co-HEMA10%)+Crelan EF403 | 140 ± 3 | 6 | 137 ± 1 | 5.5 |
| P(FA-co-StMA-co-GlyMA2%) | 141 ± 4 | 6 | 140 ± 6 | 5.5 |
| P(FA-co-StMA-co-GlyMA5%) | 145 ± 6 | 6 | 143 ± 4 | 5.5 |
| P(FA-co-StMA-co-GlyMA10%) | 146 ± 4 | 6 | 144 ± 3 | 5.5 |
| P(FA-co-StMA-co-BPMA2%) | 138 ± 1 | 4 | 133 ± 3 | 4 |
| P(FA-co-StMA-co-BPMA5%) | 140 ± 3 | 5 | 137 ± 2 | 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kredel, J.; Schmitt, D.; Schäfer, J.-L.; Biesalski, M.; Gallei, M. Cross-Linking Strategies for Fluorine-Containing Polymer Coatings for Durable Resistant Water- and Oil-Repellency. Polymers 2021, 13, 723. https://doi.org/10.3390/polym13050723
Kredel J, Schmitt D, Schäfer J-L, Biesalski M, Gallei M. Cross-Linking Strategies for Fluorine-Containing Polymer Coatings for Durable Resistant Water- and Oil-Repellency. Polymers. 2021; 13(5):723. https://doi.org/10.3390/polym13050723
Chicago/Turabian StyleKredel, Julia, Deborah Schmitt, Jan-Lukas Schäfer, Markus Biesalski, and Markus Gallei. 2021. "Cross-Linking Strategies for Fluorine-Containing Polymer Coatings for Durable Resistant Water- and Oil-Repellency" Polymers 13, no. 5: 723. https://doi.org/10.3390/polym13050723
APA StyleKredel, J., Schmitt, D., Schäfer, J. -L., Biesalski, M., & Gallei, M. (2021). Cross-Linking Strategies for Fluorine-Containing Polymer Coatings for Durable Resistant Water- and Oil-Repellency. Polymers, 13(5), 723. https://doi.org/10.3390/polym13050723