
Expanding Monomers as Anti-Shrinkage Additives



Abstract
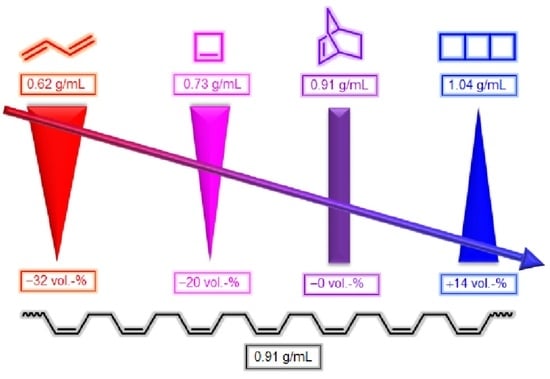
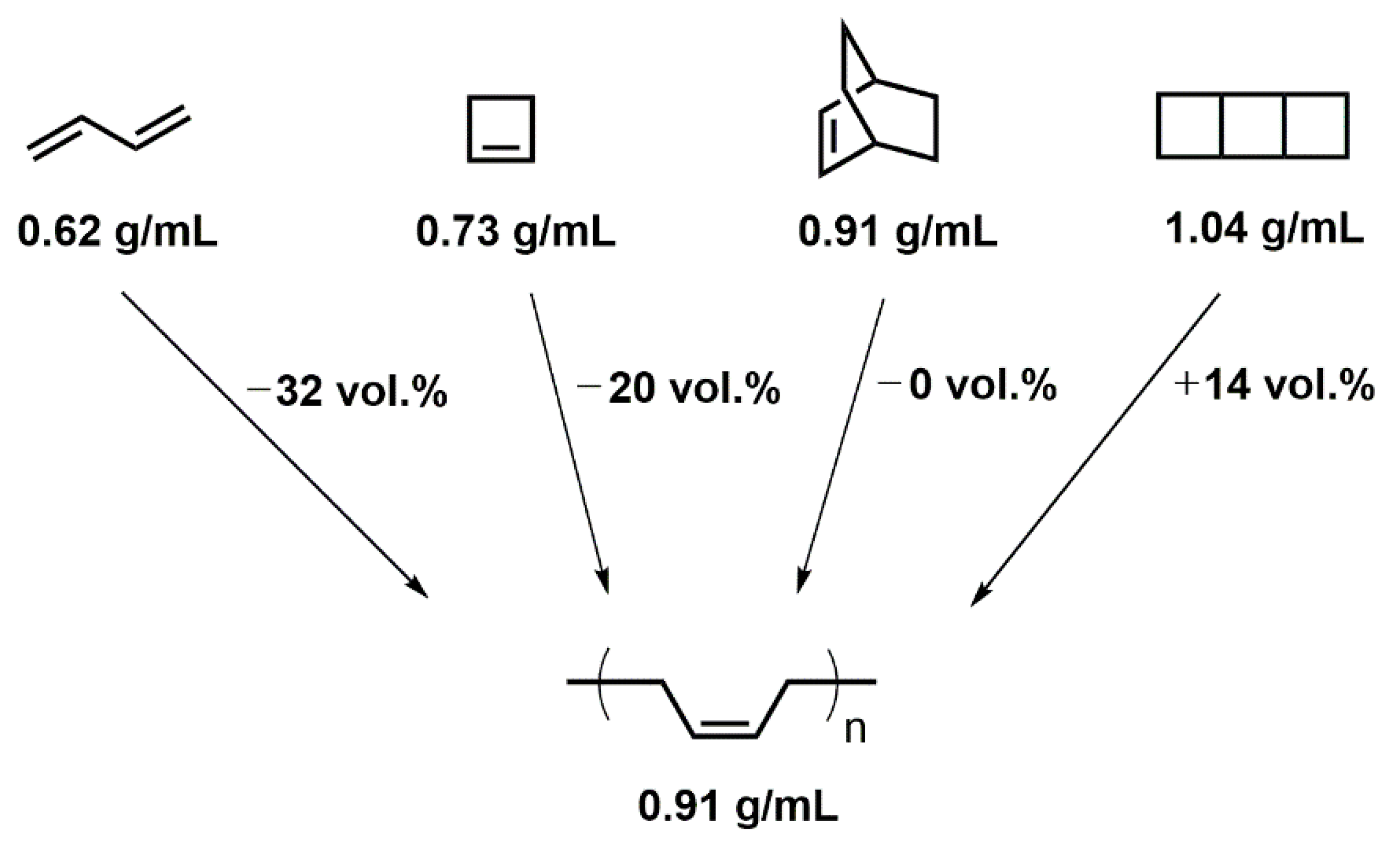
:
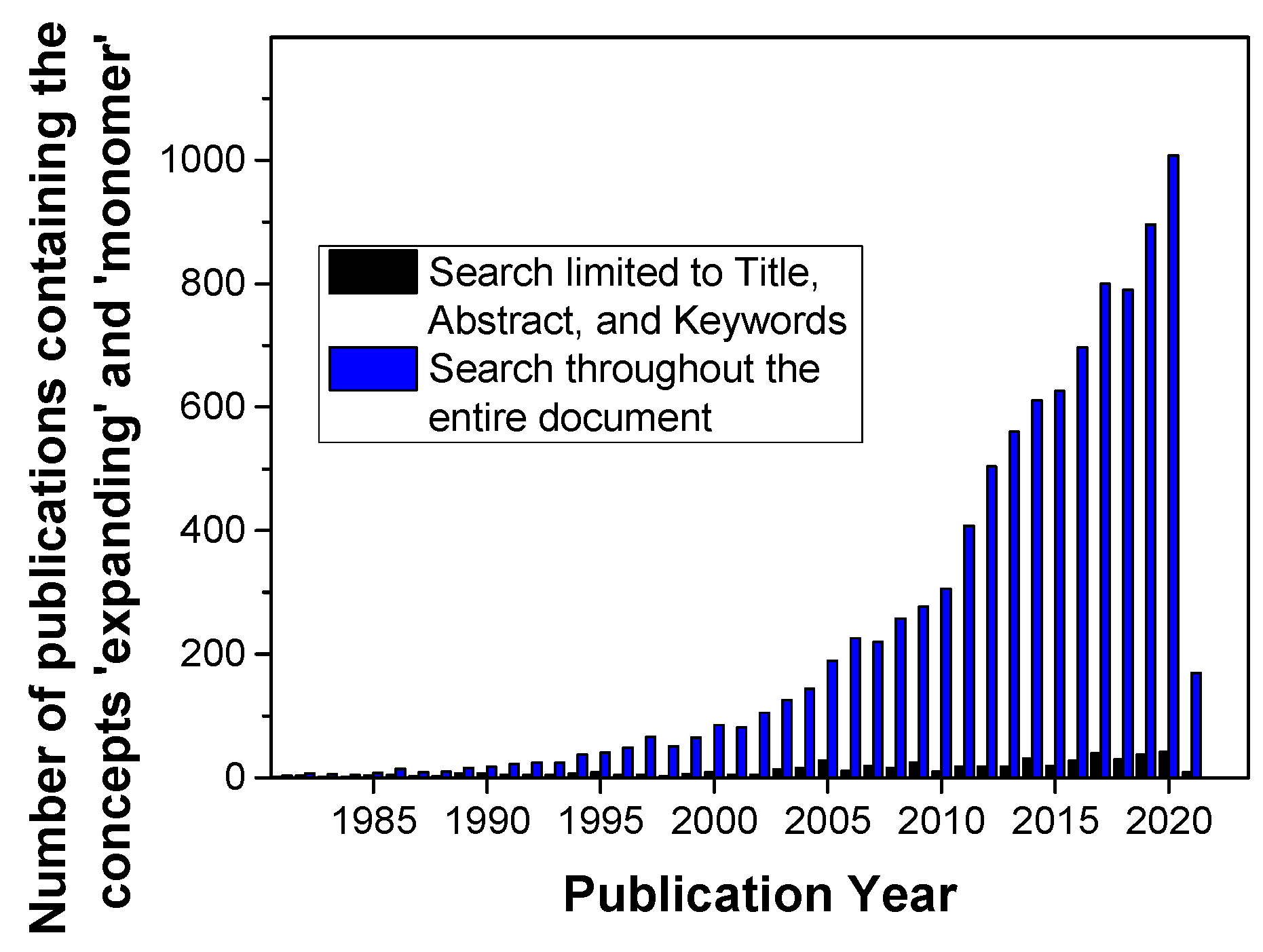
1. Introduction
2. Measurement Techniques for Volumetric Shrinkage/Expansion
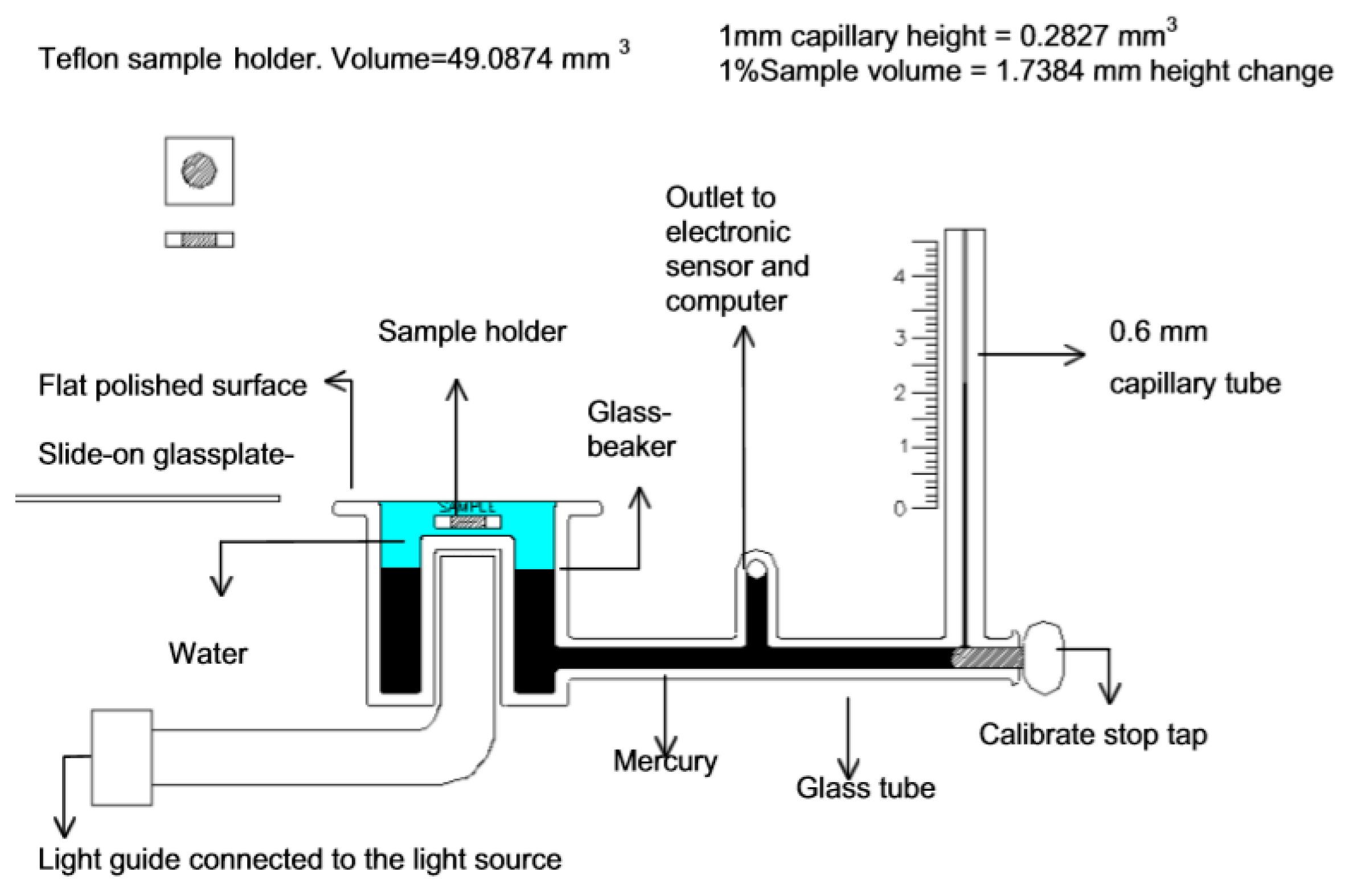
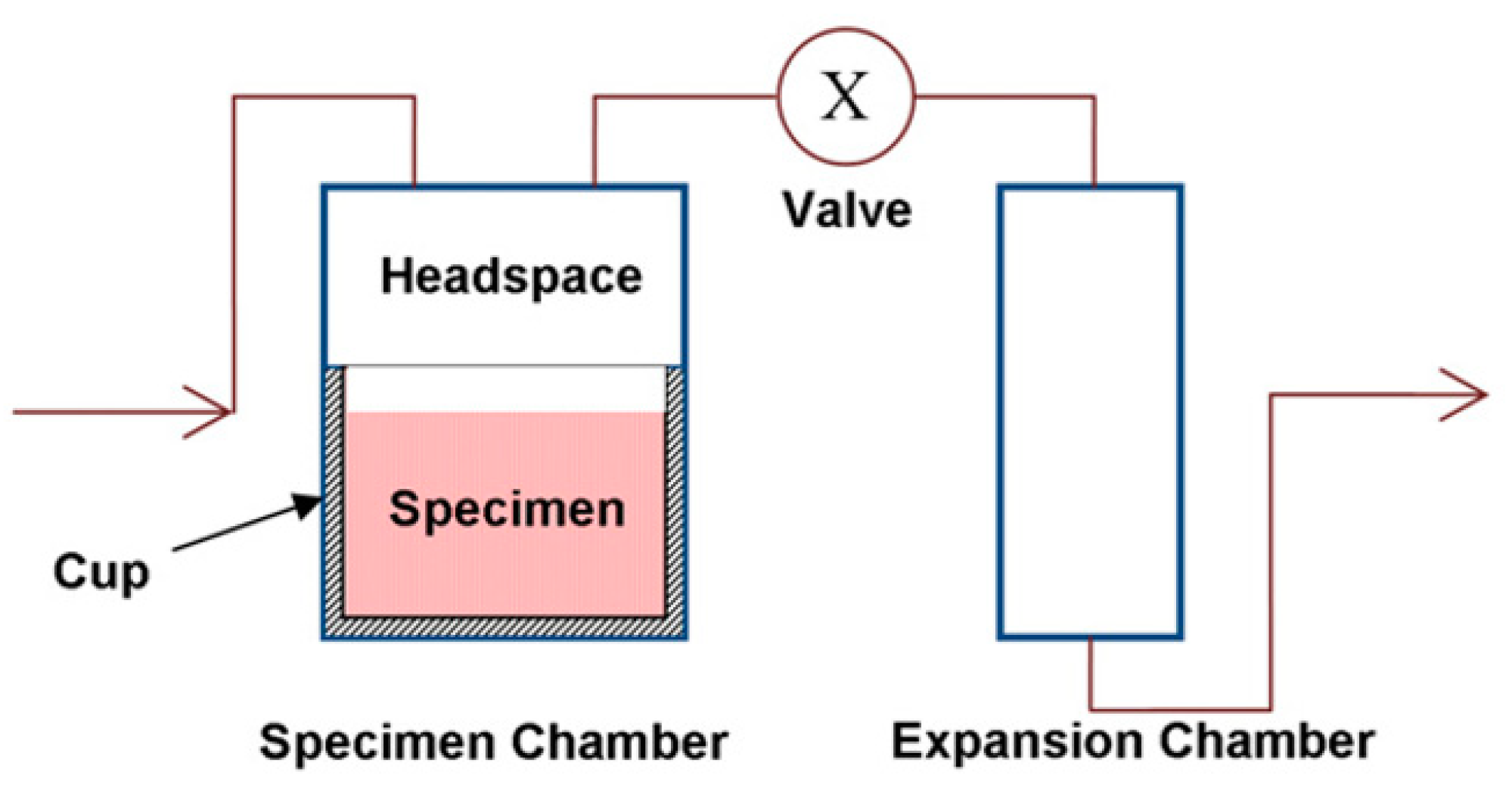
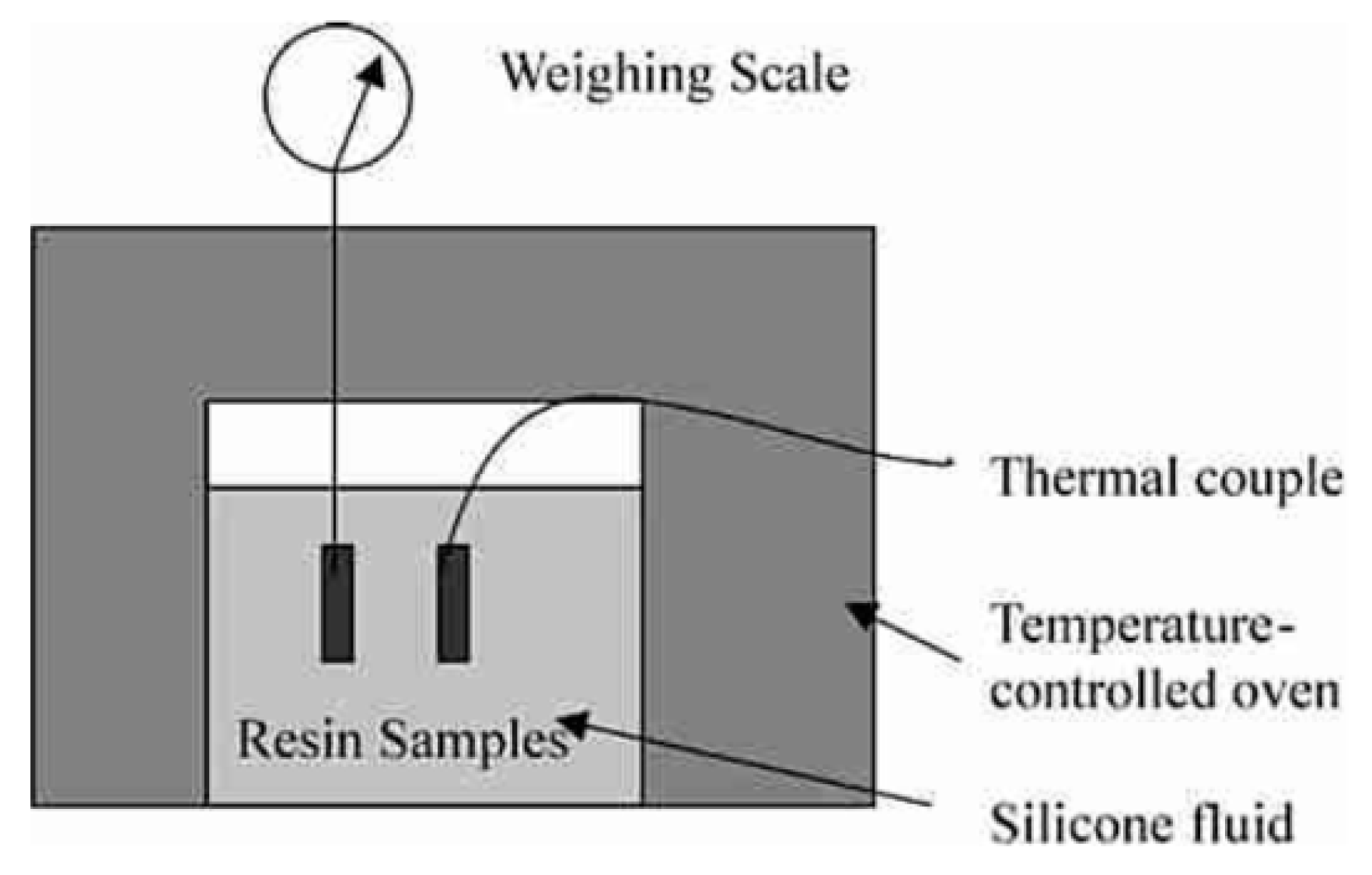
2.1. Measurement of Volume or Density Changes
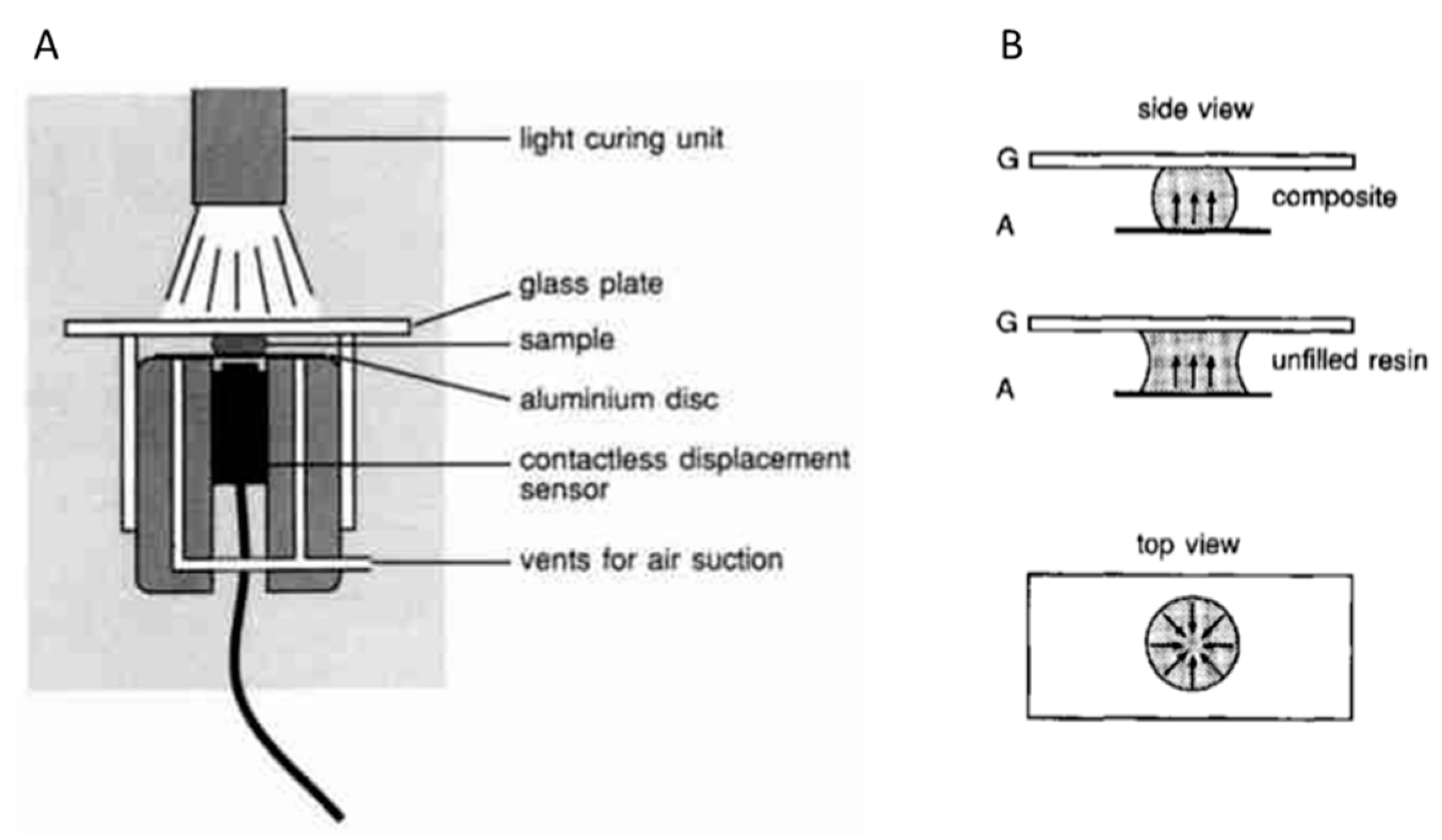
2.2. Measurement of Linear Shrinkage
2.3. Imaging Methods
3. Expanding Cycloalkanes and Cycloalkenes
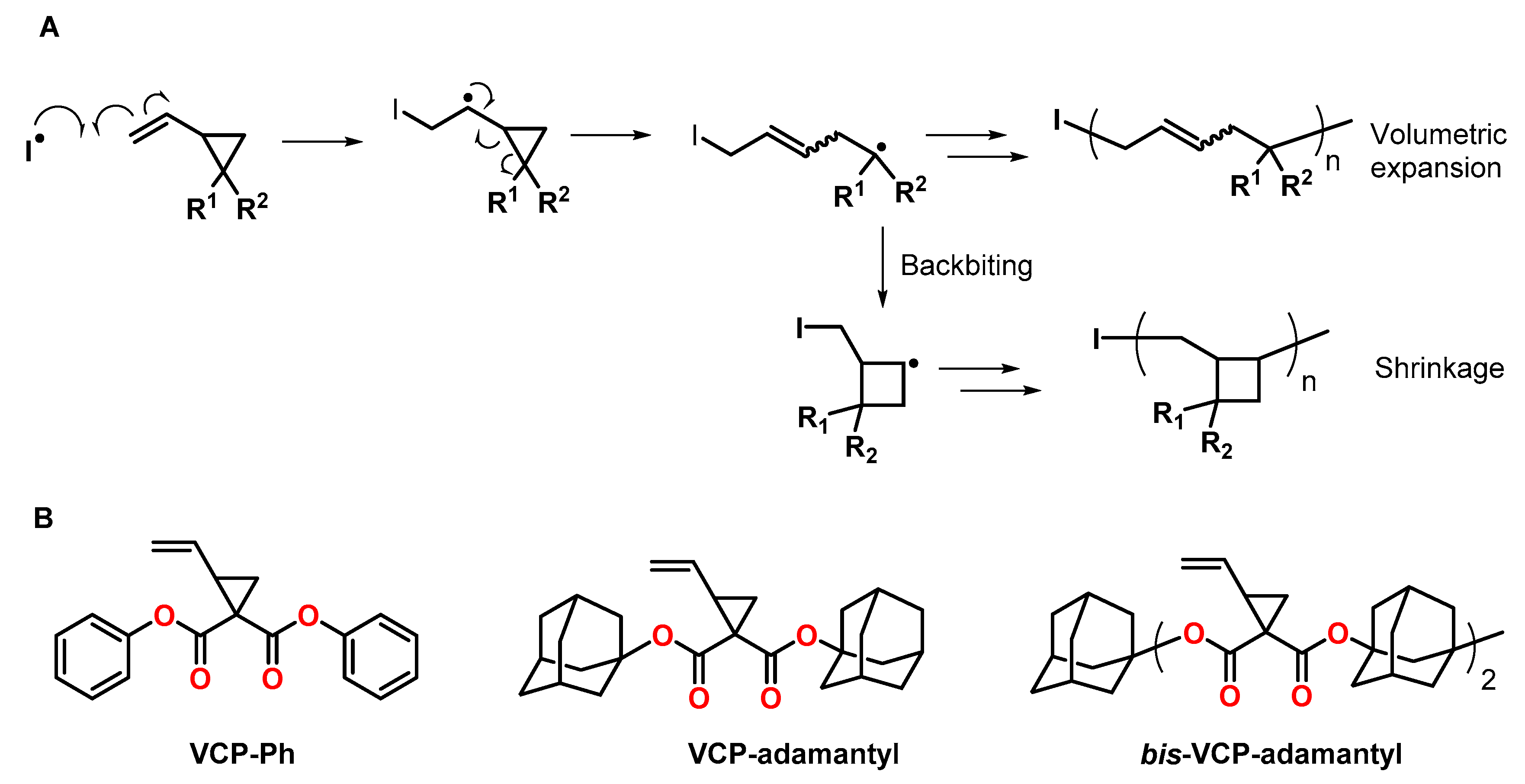
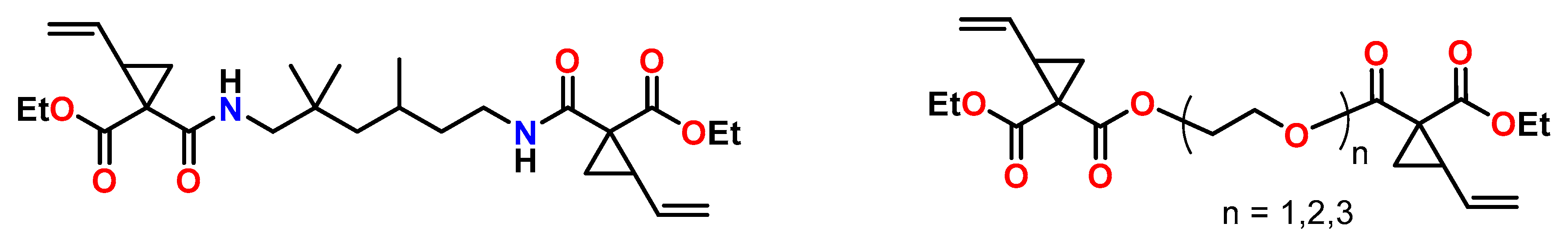
3.1. Vinylcyclopropanes
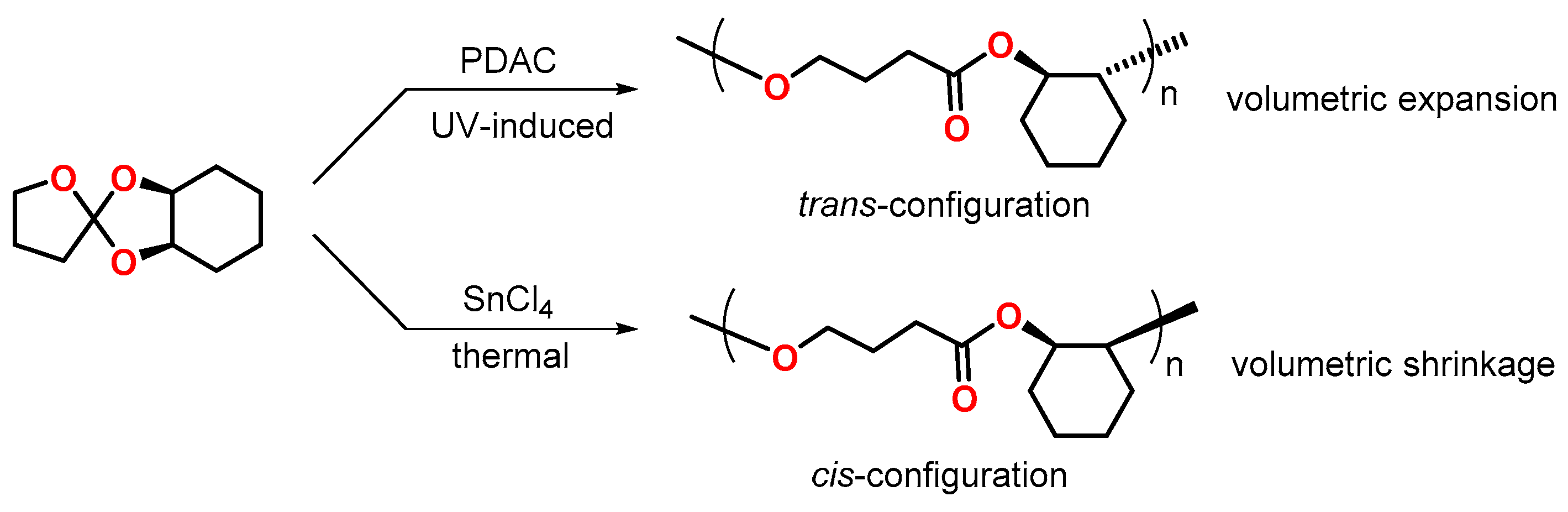
3.2. Norbornenes
4. Expanding Oxacycles
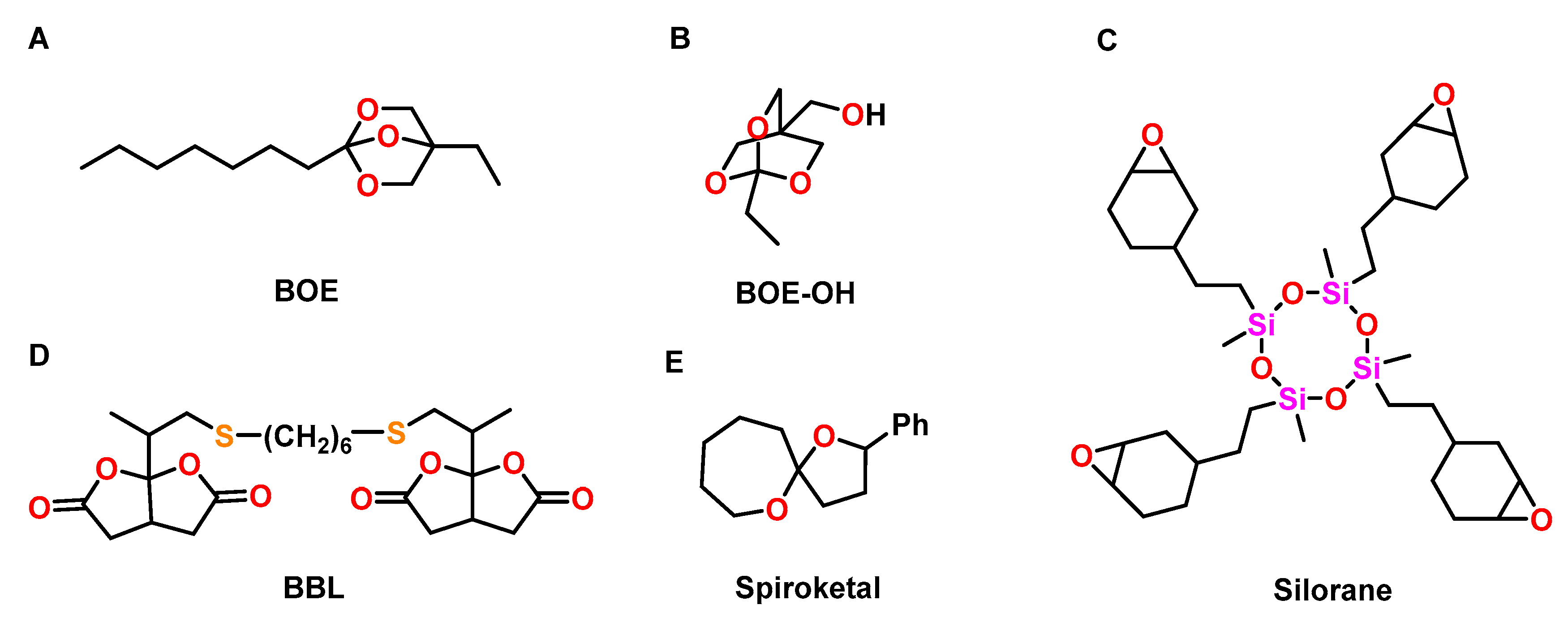
4.1. Oxetanes
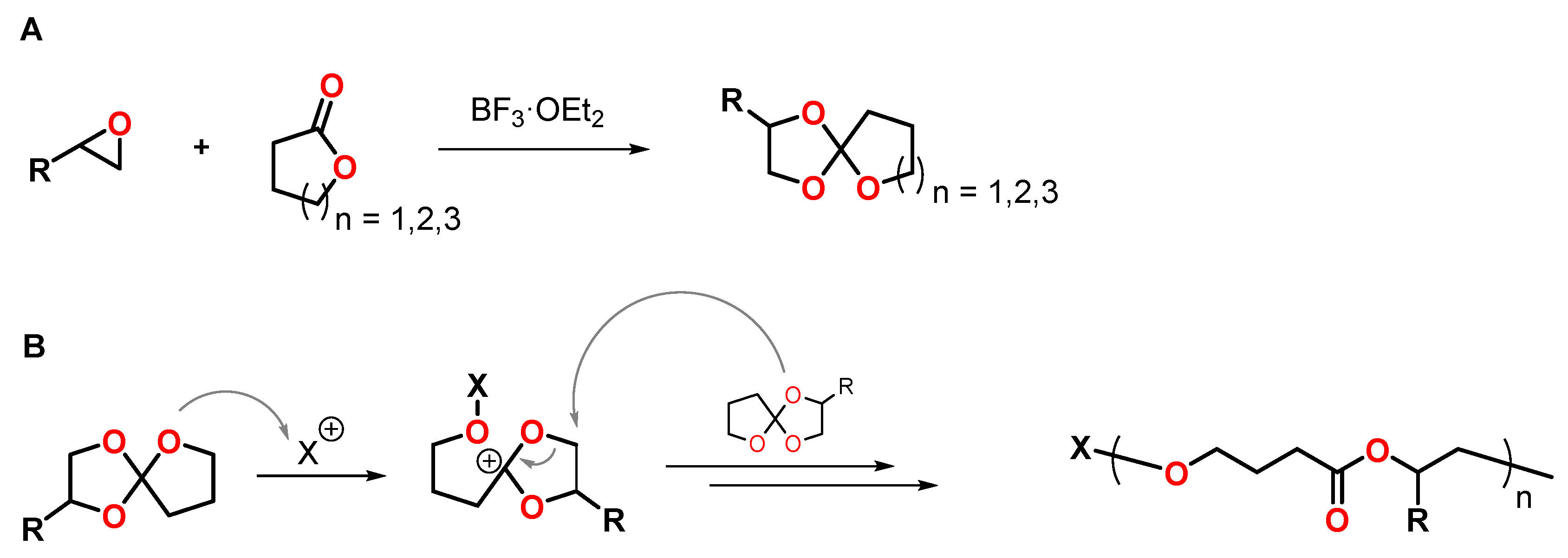
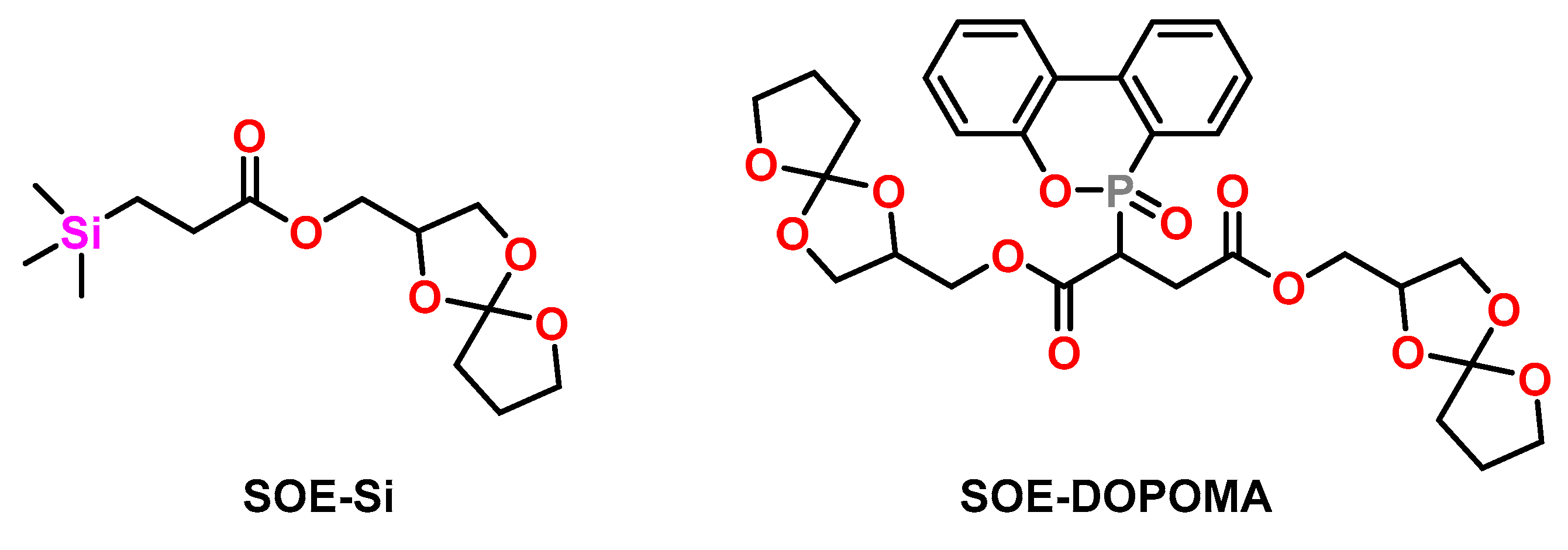
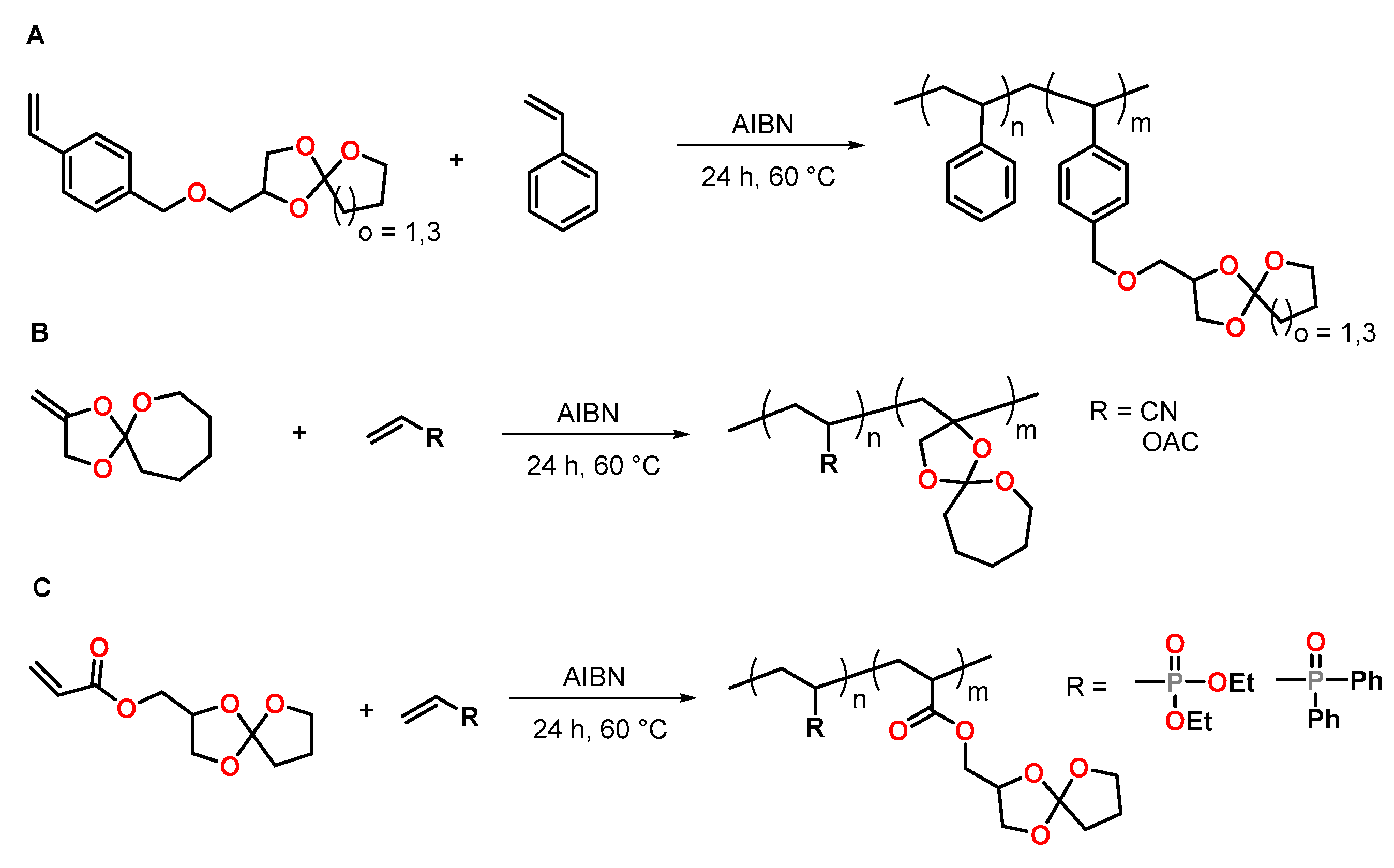
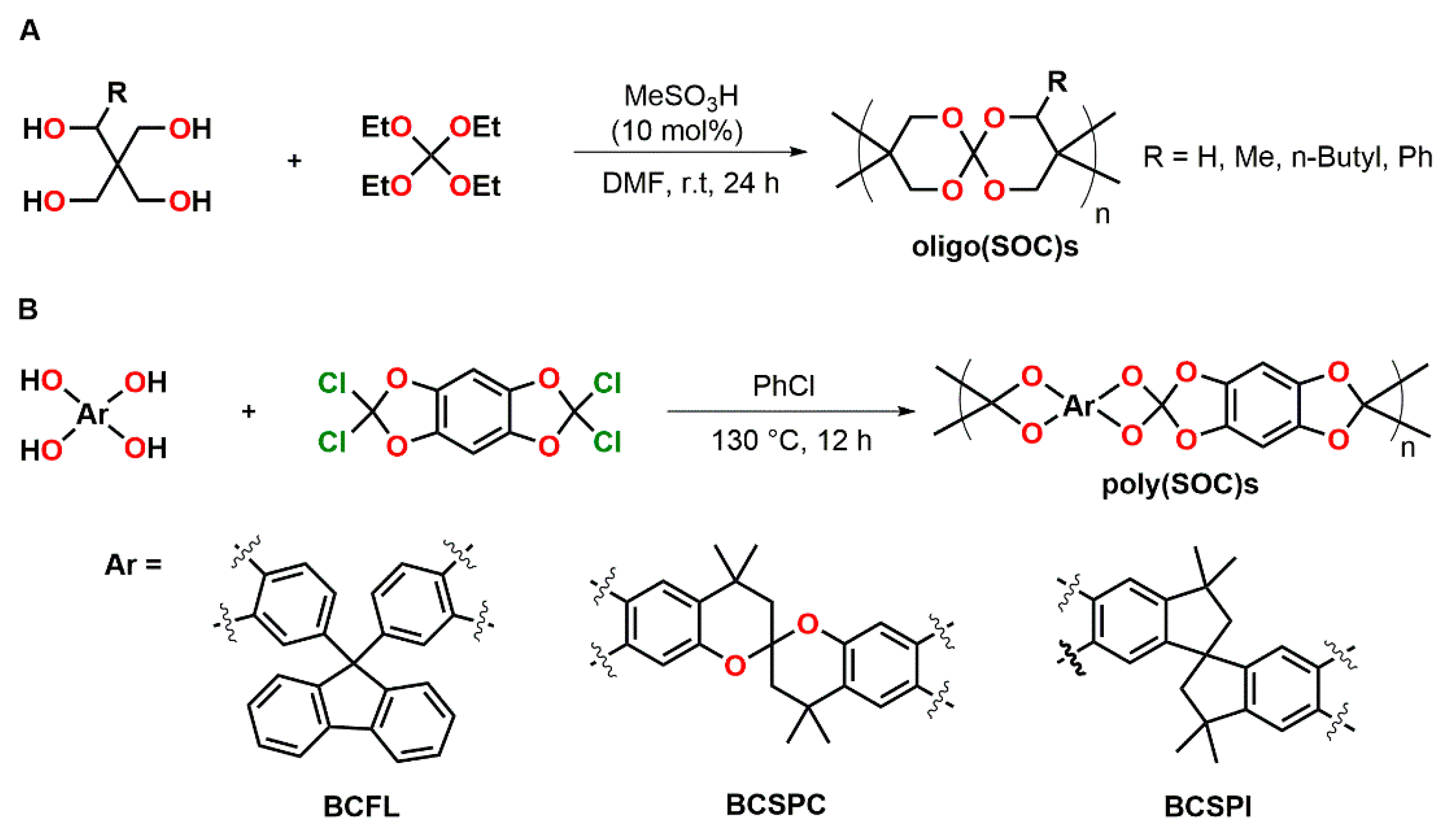
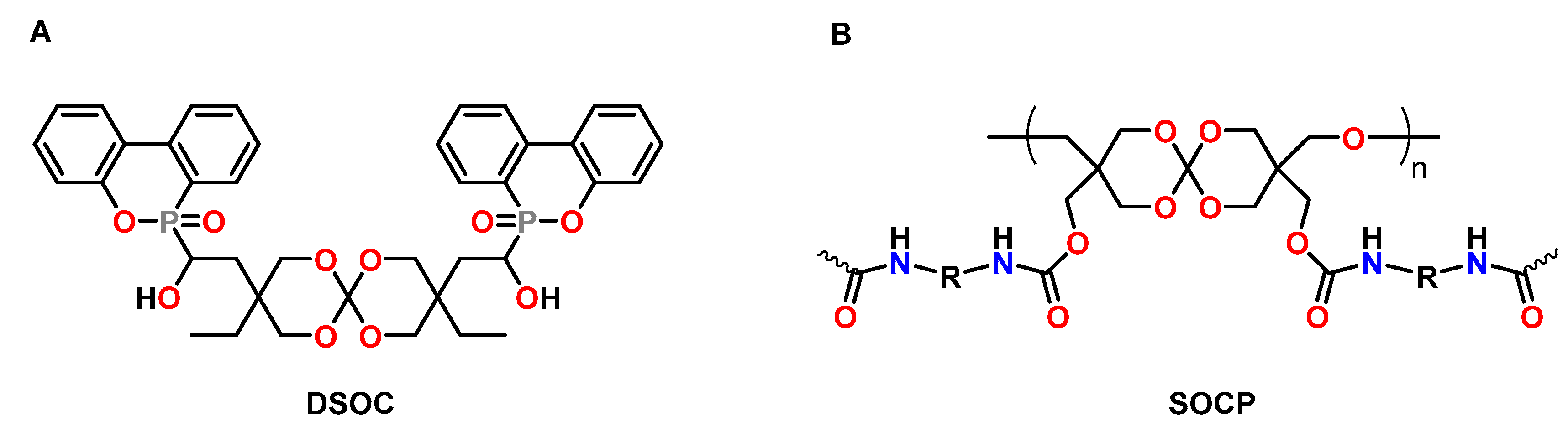
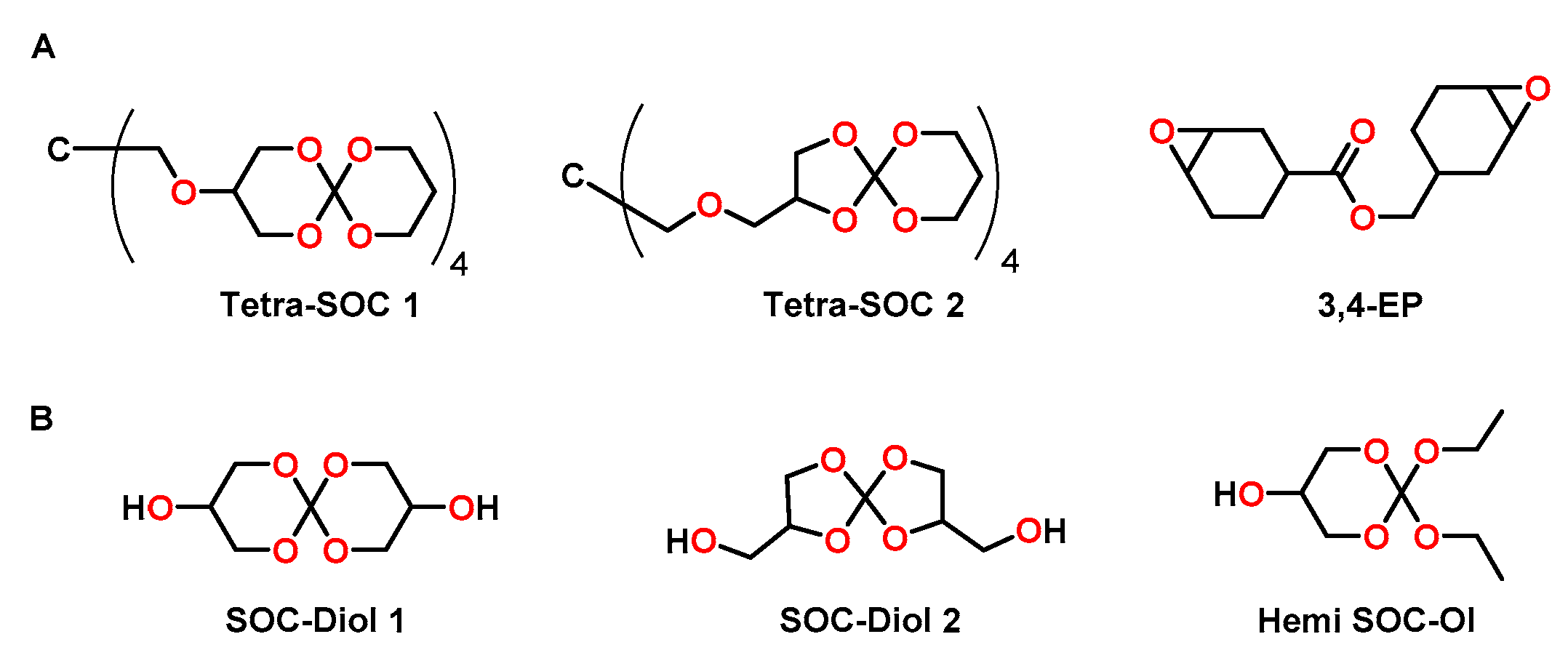
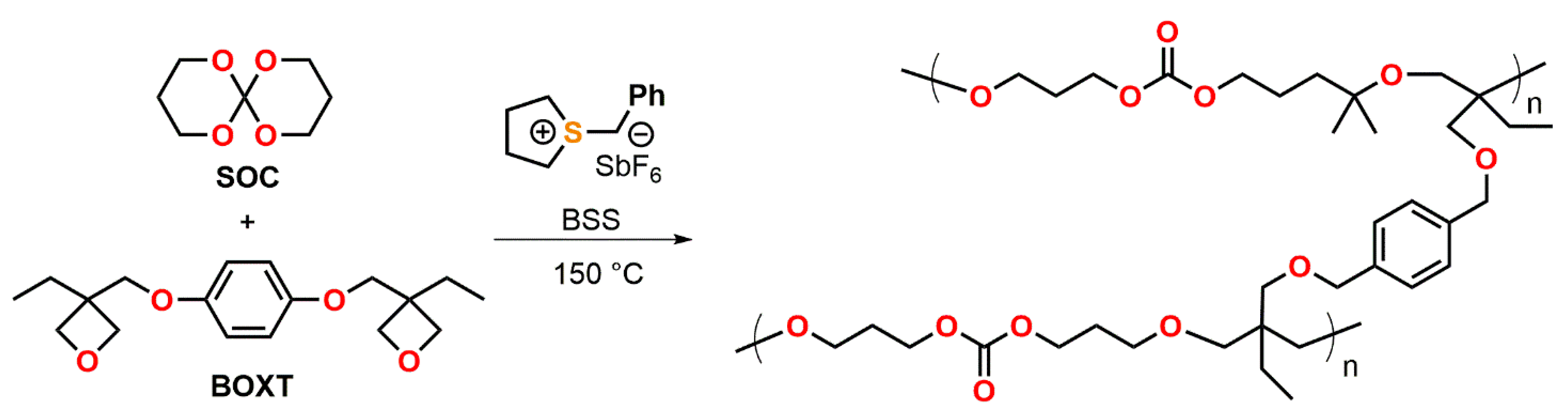
4.2. Spiroorthoesters
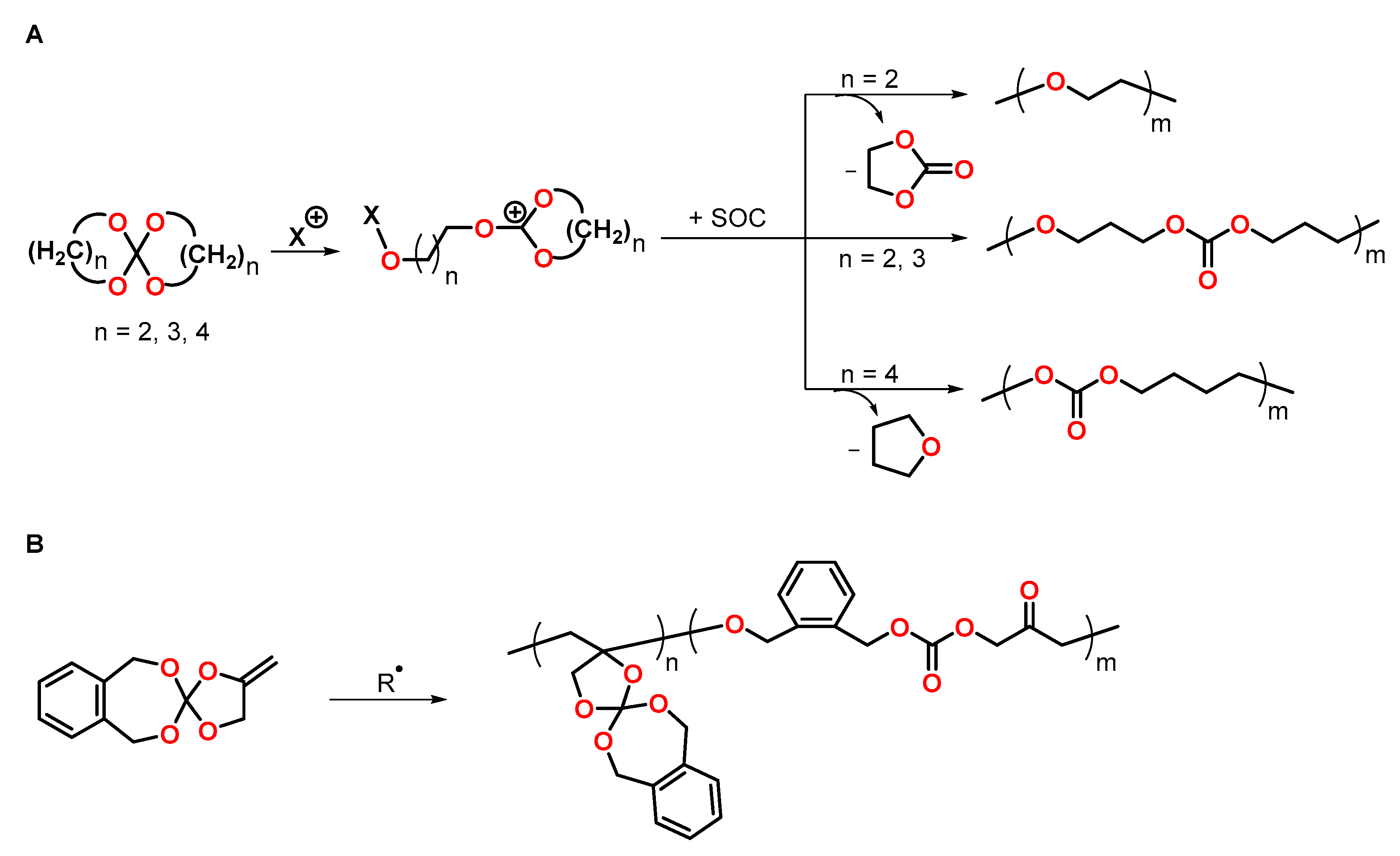
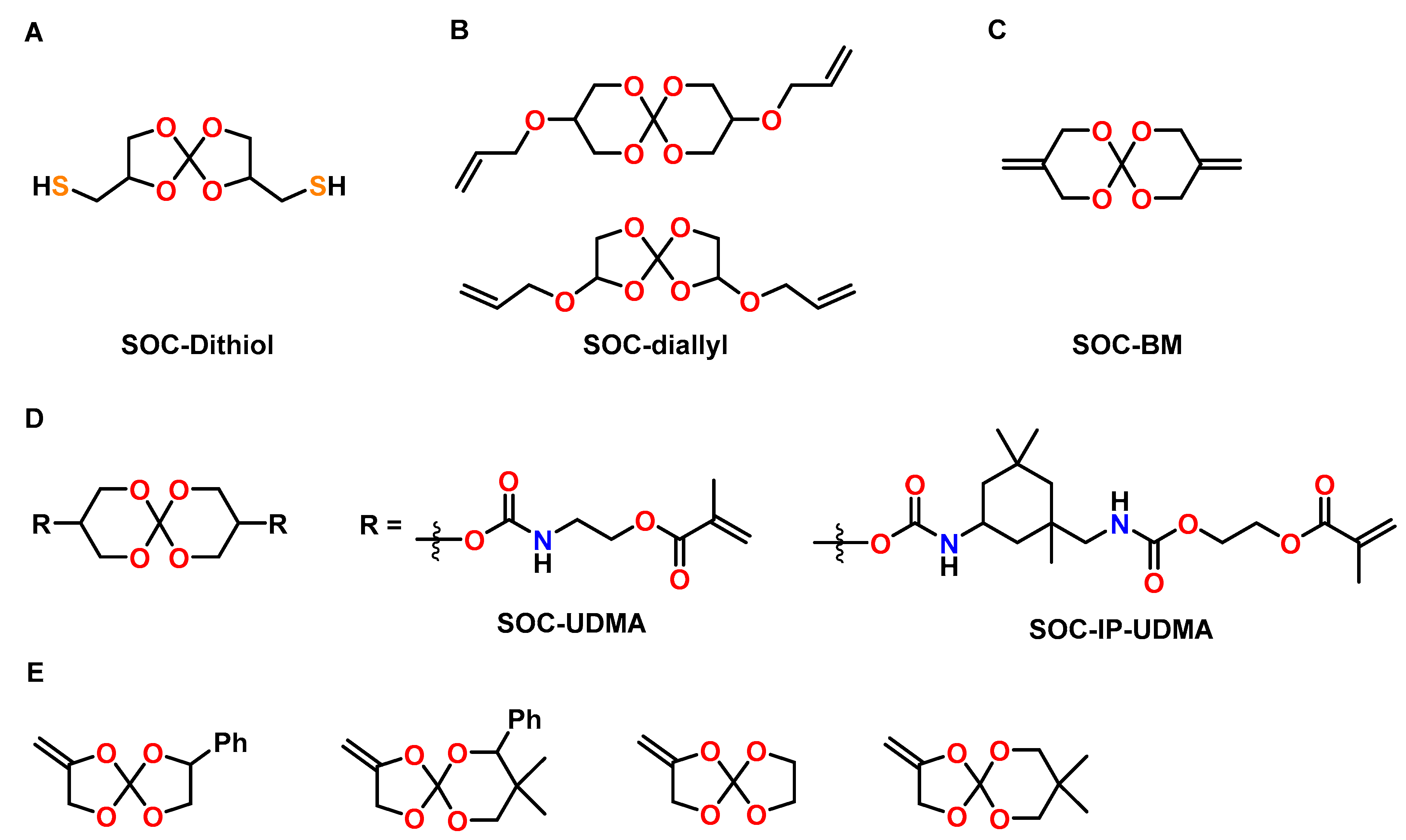
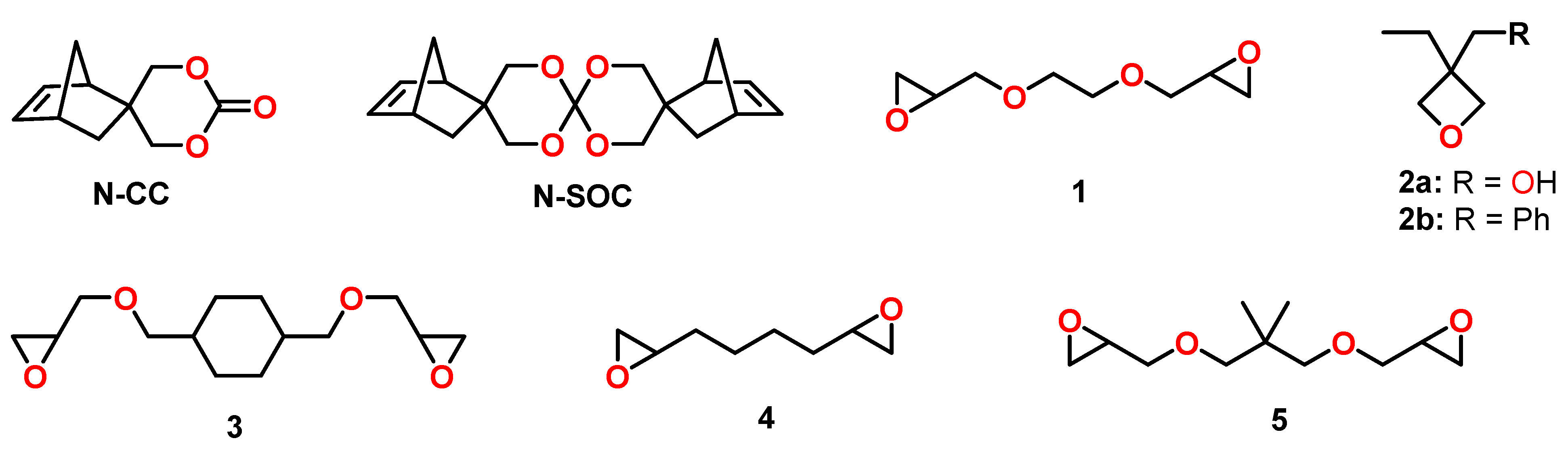
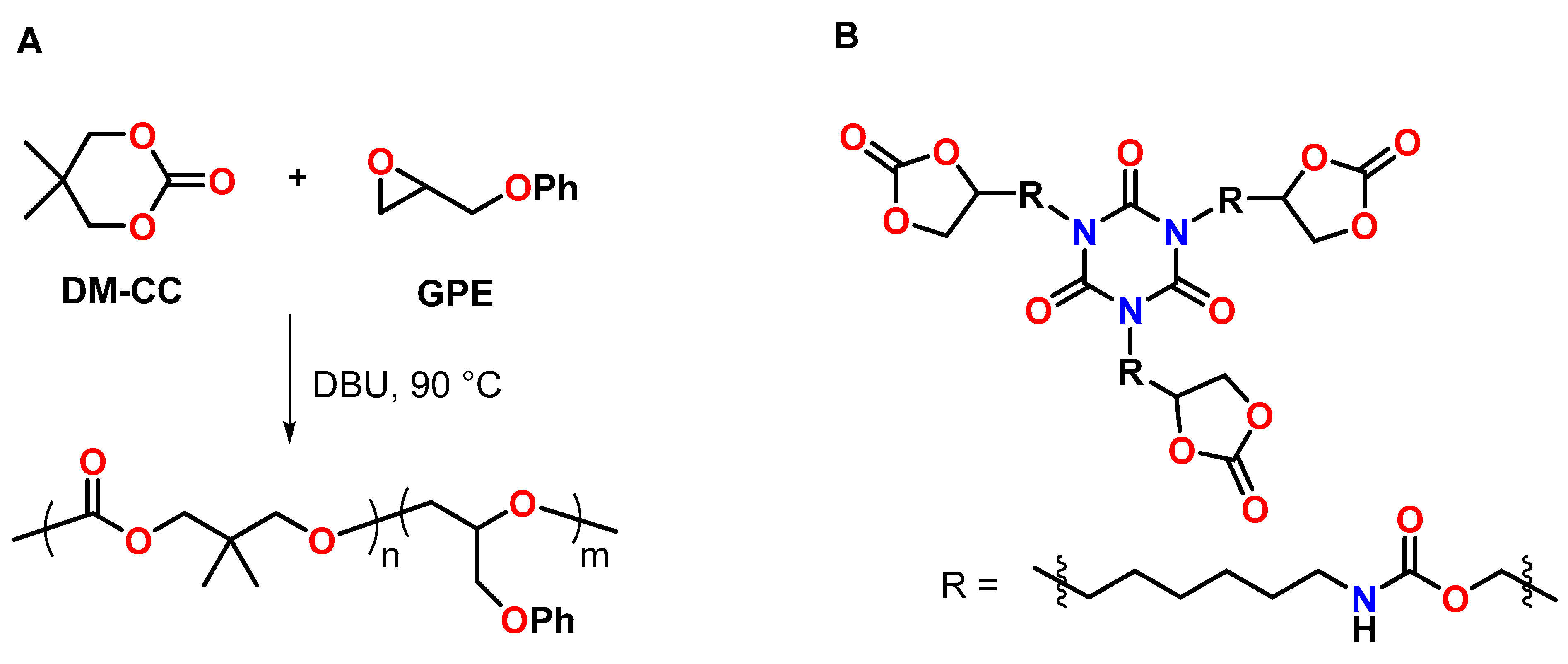
4.3. Spiroorthocarbonates
4.4. Cyclic Carbonates
4.5. Other Oxygen-Containing Oligocyclic Monomers
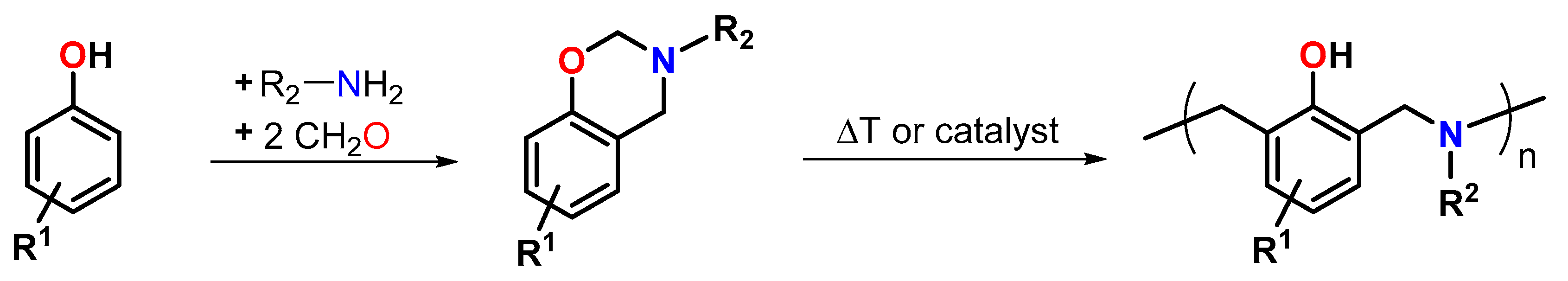
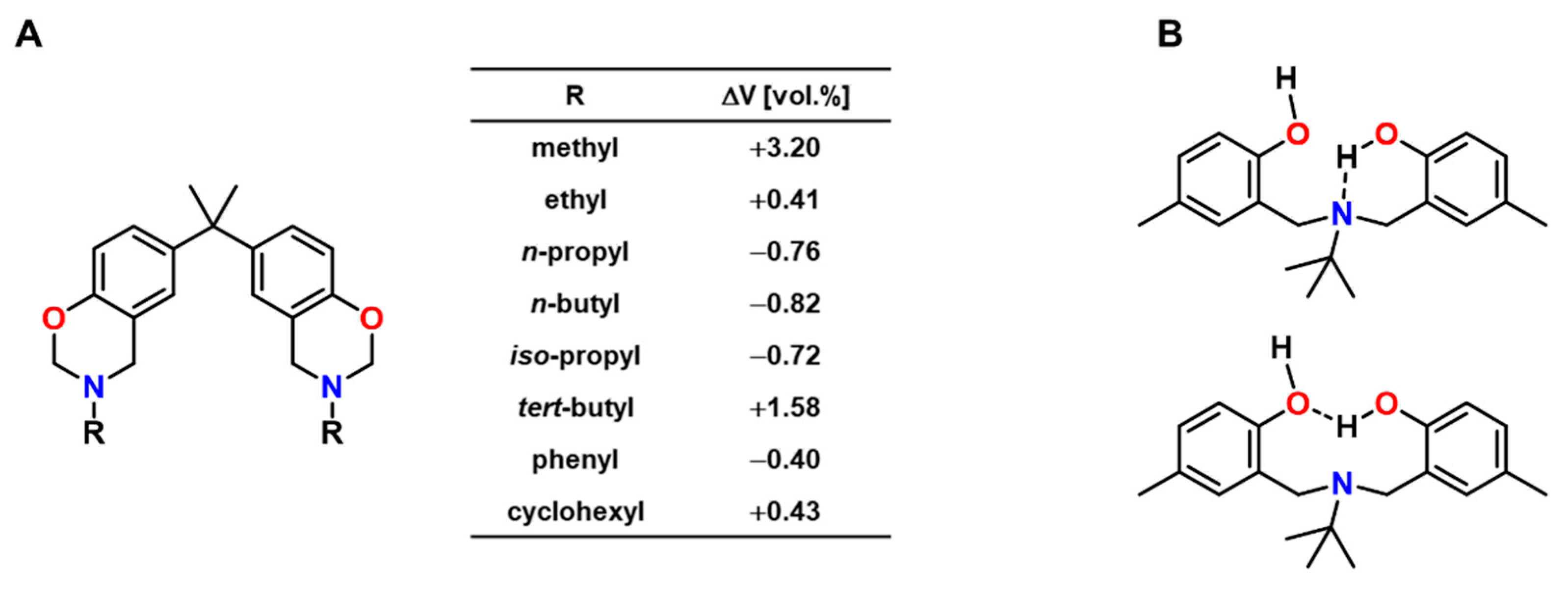
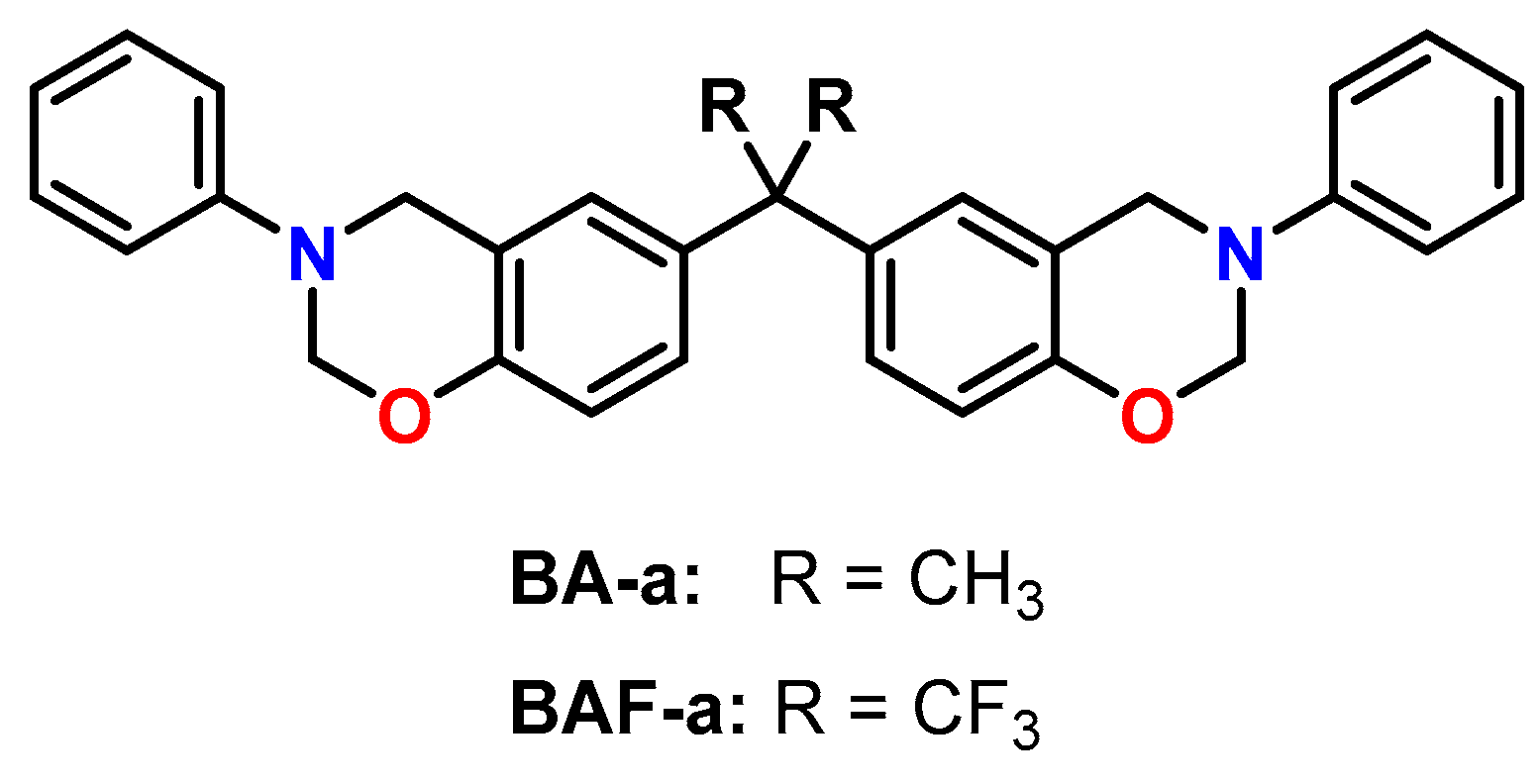
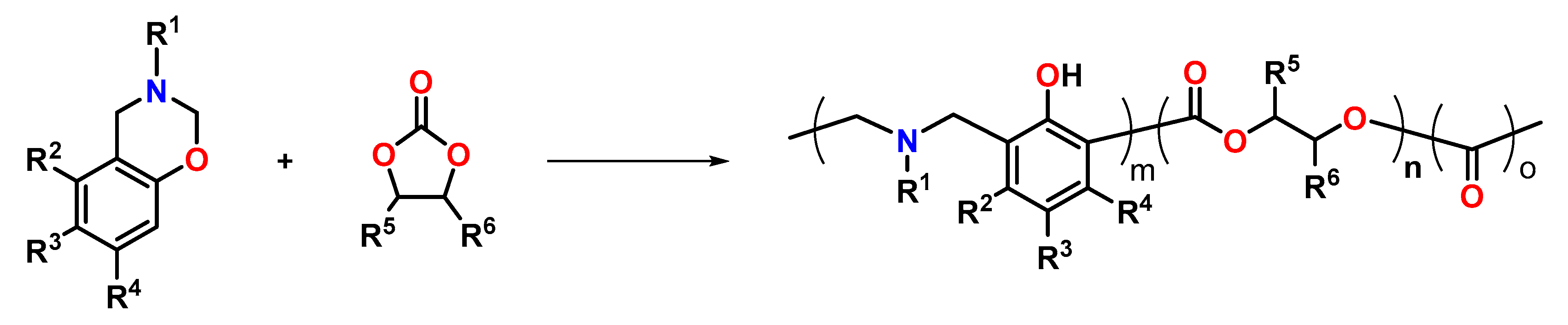
5. Expanding Benzoxazines
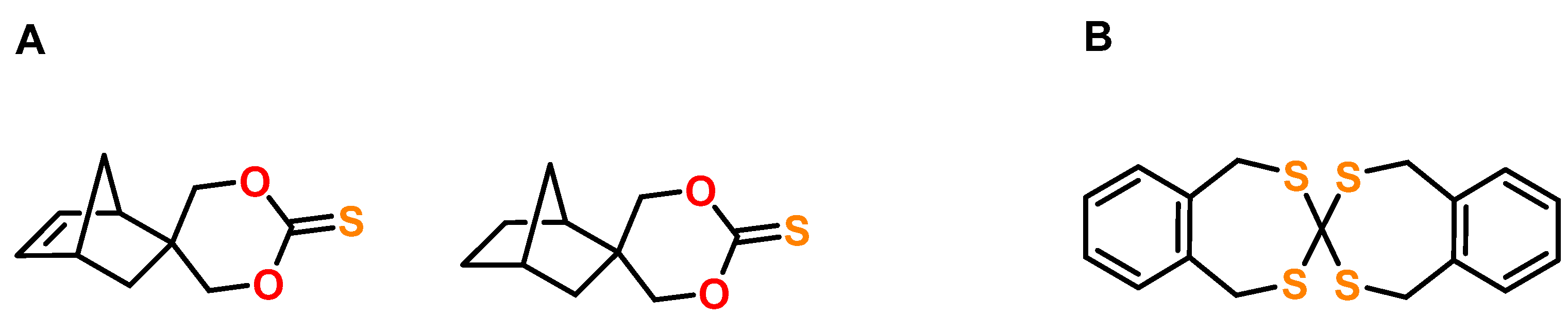
6. Expanding Thiocycles
7. Applicability of Expanding Monomers in Novel Products and Materials
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 1H-NMR | proton nuclear magnetic resonance |
| AIBN | azobisisobutyronitrile |
| BF3·NEt3 | boron trifluoride triethylamine |
| BF3·OEt2 | boron trifluoride diethyl etherate |
| bis-GMA | bisphenol A glyceroate dimethacrylate |
| BOE | bicyclic orthoester |
| CAD | computer-aided design |
| CC | cyclic carbonate |
| CH2Cl2 | dichloromethane |
| CROP | cationic ring-opening polymerization |
| Đ | dispersity index |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-en |
| DFT | density-functional theory |
| DGEBA | bisphenol A diglycidyl ether |
| DIC | digital image correlation |
| DMF | N,N-dimethylformamide |
| EPOX | 3-ethyl-3-phenoxymethyl oxetane |
| Et3OBF4 | triethyloxonium tetrafluoroborate |
| FT-IR | Fourier-transformed infrared |
| GPE | phenyl glycidyl ether |
| La(OTf)3 | lanthanide triflate |
| LOI | limiting oxygen index |
| LS | linear shrinkage |
| MEK | methyl ethyl ketone |
| Mn | number-average molecular weight |
| Mw | weight-average molecular weight |
| PDAC | 9-phenyl-9,10-dihydroanthracen-10-ylium cation |
| PPh3 | triphenylphosphine |
| PSOE | 2-phenoxymethyl-1,4,6-trioxa-spiro[4.6]undecane |
| ROMP | ring-opening metathesis polymerization |
| ROP | ring-opening polymerization |
| RROP | radical ring-opening polymerization |
| Sc(OTf)3 | scandium triflate |
| SOC | spiroorthocarbonate |
| SOE | spiroorthoester |
| TEGDMA | tri(ethylene glycol) dimethacrylate |
| Tetra-SOCs | tetrafunctional SOCs |
| TfOH | trifluoromethanesulfonic acid |
| TfOMe | methyl triflate |
| Tg | glass-transition temperature |
| THF | tetrahydrofurane |
| TMA | thermomechanical analysis |
| SnCl4 | tin(IV) chloride |
| UDMA | urethane dimethacrylate |
| UV | ultraviolet |
| VCP-adamantyl | 1,1-bis[(1-adamantyloxy)carbonyl]-2-vinylcyclopropane |
| VCP-Ph | 1,1-bis(phenoxycarbonyl)-2-vinylcyclopropane |
| VS | volumetric shrinkage |
| Yb(OTf)3 | ytterbium triflate |
| ZrO2 | zirconia |
References
- Charton, C.; Colon, P.; Pla, F. Shrinkage stress in light-cured composite resins: Influence of material and photoactivation mode. Dent. Mater. 2007, 23, 911–920. [Google Scholar] [CrossRef]
- Rodriguez, E.L. Microdelamination due to resin shrinkage in filament-wound fibreglass composites. J. Mater Sci. Lett. 1989, 8, 116–118. [Google Scholar] [CrossRef]
- Tsukrov, I.; Bayraktar, H.; Giovinazzo, M.; Goering, J.; Gross, T.; Fruscello, M.; Martinsson, L. Finite Element Modeling to Predict Cure-Induced Microcracking in Three-Dimensional Woven Composites. Int. J. Fract. 2011, 172, 209–216. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, Y.; Wang, X.; Zhang, Z. Study on the Effect of Curing Shrinkage Marks on Electrical Properties of Epoxy Composites. In Proceedings of the 21st International Symposium on High Voltage Engineering; Németh, B., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 150–156, ISBN 978-3-030-31679-2. [Google Scholar]
- Davidson, C.L.; Feilzer, A.J. Polymerization shrinkage and polymerization shrinkage stress in polymer-based restoratives. J. Dent. 1997, 25, 435–440. [Google Scholar] [CrossRef]
- Miletic, V.; Jakovljevic, N.; Manojlovic, D.; Marjanovic, J.; Rosic, A.A.; Dramićanin, M.D. Refractive indices of unfilled resin mixtures and cured composites related to color and translucency of conventional and low-shrinkage composites. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Tjong, S.C. Structural and mechanical properties of polymer nanocomposites. Mater. Sci. Eng. R Rep. 2006, 53, 73–197. [Google Scholar] [CrossRef]
- 1,3-Butadiene (Butadiene). In Handbook of Compressed Gases; Springer: Boston, MA, USA, 1990; pp. 276–283. ISBN 978-1-4612-8020-0.
- Willstätter, R.; Bruce, J. Zur Kenntnis der Cyclobutanreihe. Ber. Dtsch. Chem. Ges. 1907, 40, 3979–3999. [Google Scholar] [CrossRef] [Green Version]
- Willstätter, R.; Veraguth, H. Zur Kenntnis der Cyclooctanreihe. Ber. Dtsch. Chem. Ges. 1907, 40, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Scifinder Scholar, Scifinder-n; Chemical Abstracts Service: Columbus, OH, USA, 2021; RN 107-21-1 (accessed on 11 February 2021); Calculated Using ACD/Labs Software, Version 11.02; ACD/Labs.
- Dolgoplosk, B.A.; Belonovskaja, G.P.; Boldyreva, I.I.; Kropacheva, E.N.; Nelson, K.V.; Rosinoer, J.M.; Chernova, J.D. Investigation of the factors causing chain structure change in ionic diene polymerization. J. Polym. Sci. 1961, 53, 209–216. [Google Scholar] [CrossRef]
- Takata, T.; Endo, T. Recent advances in the development of expanding monomers: Synthesis, polymerization and volume change. Prog. Polym. Sci. 1993, 18, 839–870. [Google Scholar] [CrossRef]
- Rokicki, G. Aliphatic cyclic carbonates and spiroorthocarbonates as monomers. Prog. Polym. Sci. 2000, 25, 259–342. [Google Scholar] [CrossRef]
- Sadhir, R.K.; Luck, R.M. Expanding Monomers: Synthesis, Characterization, and Applications; CRC Press: Boca Raton, FL, USA, 1992; ISBN 1000098540. [Google Scholar]
- Moszner, N.; Salz, U. New developments of polymeric dental composites. Prog. Polym. Sci. 2001, 26, 535–576. [Google Scholar] [CrossRef]
- Acosta Ortiz, R.; Savage Gomez, A.G.; García Valdez, A.E.; Aguirre Flores, R.; Sangermano, M. Development of Low-Shrinkage Polymers by Using Expanding Monomers. Macromol. Symp. 2017, 374, 1600092. [Google Scholar] [CrossRef]
- Ghavami-Lahiji, M.; Hooshmand, T. Analytical methods for the measurement of polymerization kinetics and stresses of dental resin-based composites: A review. Dent. Res. J. (Isfahan) 2017, 14, 225–240. [Google Scholar]
- He, Y.; Yao, M.; Nie, J. Shrinkage in UV-Curable Coatings. In Protective Coatings: Film Formation and Properties; Wen, M., Dušek, K., Eds.; Springer: Cham, Switzerland, 2017; pp. 195–223. ISBN 978-3-319-51625-7. [Google Scholar]
- Nawab, Y.; Shahid, S.; Boyard, N.; Jacquemin, F. Chemical shrinkage characterization techniques for thermoset resins and associated composites. J. Mater. Sci. 2013, 48, 5387–5409. [Google Scholar] [CrossRef] [Green Version]
- Jongsma, L.A.; Kleverlaan, C.J. Influence of temperature on volumetric shrinkage and contraction stress of dental composites. Dent. Mater. 2015, 31, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Miyazaki, M.; Rikuta, A.; Kurokawa, H.; Takamizawa, T. Comparison of two methods for measuring the polymerization characteristics of flowable resin composites. Dent. Mater. 2007, 23, 792–798. [Google Scholar] [CrossRef]
- Oberholzer, T.G.; Grobler, S.R.; Pameijer, C.H.; Rossouw, R.J. A modified dilatometer for determining volumetric polymerization shrinkage of dental materials. Meas. Sci. Technol. 2002, 13, 78–83. [Google Scholar] [CrossRef]
- Amore, R.; Pagani, C.; Youssef, M.N.; Anauate Netto, C.; Lewgoy, H.R. Polymerization shrinkage evaluation of three packable composite resins using a gas pycnometer. Pesqui. Odontol. Bras. 2003, 17, 273–277. [Google Scholar] [CrossRef]
- Cook, W.D.; Forrest, M.; Goodwin, A.A. A simple method for the measurement of polymerization shrinkage in dental composites. Dent. Mater. 1999, 15, 447–449. [Google Scholar] [CrossRef]
- Shah, D.U.; Schubel, P.J. Evaluation of cure shrinkage measurement techniques for thermosetting resins. Polym. Test. 2010, 29, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Potter, K.; Wisnom, M.R.; Stringer, G. In-situ measurement of chemical shrinkage of MY750 epoxy resin by a novel gravimetric method. Compos. Sci. Technol. 2004, 64, 55–64. [Google Scholar] [CrossRef]
- de Gee, A.J.; Feilzer, A.J.; Davidson, C.L. True linear polymerization shrinkage of unfilled resins and composites determined with a linometer. Dent. Mater. 1993, 9, 11–14. [Google Scholar] [CrossRef]
- Watts, D.C.; Cash, A.J. Determination of polymerization shrinkage kinetics in visible-light-cured materials: Methods development. Dent. Mater. 1991, 7, 281–287. [Google Scholar] [CrossRef]
- Lee, I.-B.; Cho, B.-H.; Son, H.-H.; Um, C.-M.; Lim, B.-S. The effect of consistency, specimen geometry and adhesion on the axial polymerization shrinkage measurement of light cured composites. Dent. Mater. 2006, 22, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Fogleman, E.A.; Kelly, M.T.; Grubbs, W.T. Laser interferometric method for measuring linear polymerization shrinkage in light cured dental restoratives. Dent. Mater. 2002, 18, 324–330. [Google Scholar] [CrossRef]
- Fano, V.; Ortalli, I.; Pizzi, S.; Bonanini, M. Polymerization shrinkage of microfilled composites determined by laser beam scanning. Biomaterials 1997, 18, 467–470. [Google Scholar] [CrossRef]
- Jian, Y.; He, Y.; Jiang, T.; Li, C.; Yang, W.; Nie, J. Volume shrinkage of UV-curable coating formulation investigated by real-time laser reflection method. J. Coat. Technol. Res. 2013, 10, 231–237. [Google Scholar] [CrossRef]
- Zhou, W.X.; Chan-Park, M.B. Effect of oligomer length on the buckling of long and high aspect ratio microwalls UV embossed from oligomer/monomer mixtures. Sens. Actuators B Chem. 2007, 128, 12–22. [Google Scholar] [CrossRef]
- Khoun, L.; Centea, T.; Hubert, P. Characterization Methodology of Thermoset Resins for the Processing of Composite Materials—Case Study: CYCOM 890RTM Epoxy Resin. J. Compos. Mater. 2010, 44, 1397–1415. [Google Scholar] [CrossRef]
- Schmidt, C.; Scherzer, T. Monitoring of the shrinkage during the photopolymerization of acrylates using hyphenated photorheometry/near-infrared spectroscopy. J. Polym. Sci. B Polym. Phys. 2015, 53, 729–739. [Google Scholar] [CrossRef]
- Brauner, C.; Block, T.B.; Purol, H.; Herrmann, A.S. Microlevel manufacturing process simulation of carbon fiber/epoxy composites to analyze the effect of chemical and thermal induced residual stresses. J. Compos. Mater. 2012, 46, 2123–2143. [Google Scholar] [CrossRef]
- Exner, W.; Kühn, A.; Szewieczek, A.; Opitz, M.; Mahrholz, T.; Sinapius, M.; Wierach, P. Determination of volumetric shrinkage of thermally cured thermosets using video-imaging. Polym. Test. 2016, 49, 100–106. [Google Scholar] [CrossRef]
- Sharp, L.J.; Choi, I.B.; Lee, T.E.; Sy, A.; Suh, B.I. Volumetric shrinkage of composites using video-imaging. J. Dent. 2003, 31, 97–103. [Google Scholar] [CrossRef]
- Lau, A.; Li, J.; Heo, Y.C.; Fok, A. A study of polymerization shrinkage kinetics using digital image correlation. Dent. Mater. 2015, 31, 391–398. [Google Scholar] [CrossRef]
- Li, J.; Thakur, P.; Fok, A.S. Shrinkage of dental composite in simulated cavity measured with digital image correlation. J. Vis. Exp. Jove 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, Y.-C.; Rösch, P.; Dabanoglu, A.; Lin, C.-P.; Hickel, R.; Kunzelmann, K.-H. Polymerization composite shrinkage evaluation with 3D deformation analysis from microCT images. Dent. Mater. 2010, 26, 223–231. [Google Scholar] [CrossRef]
- de Melo Monteiro, G.Q.; Montes, M.A.J.R.; Rolim, T.V.; de Oliveira Mota, C.C.B.; de Barros Correia Kyotoku, B.; Gomes, A.S.L.; de Freitas, A.Z. Alternative methods for determining shrinkage in restorative resin composites. Dent. Mater. 2011, 27, e176–e185. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Eidelman, N.; Lin-Gibson, S. 3D mapping of polymerization shrinkage using X-ray micro-computed tomography to predict microleakage. Dent. Mater. 2009, 25, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Moszner, N.; Zeuner, F.; Völkel, T.; Rheinberger, V. Synthesis and polymerization of vinylcyclopropanes. Macromol. Chem. Phys. 1999, 200, 2173–2187. [Google Scholar] [CrossRef]
- Endo, T.; Sudo, A. Radical Ring-Opening Polymerization: Molecular Designs, Polymerization Mechanisms, and Living/Controlled Systems. In Controlled Radical Polymerization; Matyjaszewski, K., Sumerlin, B.S., Tsarevsky, N.V., Chiefari, J., Eds.; American Chemical Society: Washington, DC, USA, 2015; pp. 19–50. ISBN 0-8412-3048-X. [Google Scholar]
- Sugiyama, J.; Ohashi, K.; Ueda, M. Free Radical Polymerization of Volume Expandable Vinylcyclopropane. Macromolecules 1994, 27, 5543–5546. [Google Scholar] [CrossRef]
- Sugiyama, J.-i.; Kayamori, N.; Shimada, S.-i. Free Radical Ring-Opening Polymerization of 1,1-Bis[(1-adamantyloxy)carbonyl]-2-vinylcyclopropane. Macromolecules 1996, 29, 1943–1950. [Google Scholar] [CrossRef]
- Chiba, H.; Kitazume, K.; Yamada, S.; Endo, T. Synthesis and radical ring-opening polymerization of adamantane-containing bifunctional vinylcyclopropane undergoing volume expansion on polymerization. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 39–43. [Google Scholar] [CrossRef]
- Contreras, P.P.; Tyagi, P.; Agarwal, S. Low volume shrinkage of polymers by photopolymerization of 1,1-bis(ethoxycarbonyl)-2-vinylcyclopropanes. Polym. Chem. 2015, 6, 2297–2304. [Google Scholar] [CrossRef] [Green Version]
- Schoerpf, S.; Catel, Y.; Moszner, N.; Gorsche, C.; Liska, R. Enhanced reduction of polymerization-induced shrinkage stress via combination of radical ring opening and addition fragmentation chain transfer. Polym. Chem. 2019, 10, 1357–1366. [Google Scholar] [CrossRef] [Green Version]
- Catel, Y.; Fässler, P.; Fischer, U.; Gorsche, C.; Schörpf, S.; Tauscher, S.; Liska, R.; Moszner, N. Evaluation of Difunctional Vinylcyclopropanes as Reactive Diluents for the Development of Low-Shrinkage Composites. Macromol. Mater. Eng. 2017, 302, 1700021. [Google Scholar] [CrossRef]
- Catel, Y.; Fässler, P.; Fischer, U.; Gorsche, C.; Liska, R.; Schörpf, S.; Tauscher, S.; Moszner, N. Synthesis and polymerization of vinylcyclopropanes bearing urethane groups for the development of low-shrinkage composites. Eur. Polym. J. 2018, 98, 439–447. [Google Scholar] [CrossRef]
- Watts, D. Photo-polymerization shrinkage-stress kinetics in resin-composites: Methods development. Dent. Mater. 2003, 19, 1–11. [Google Scholar] [CrossRef]
- Hino, T.; Inoue, N.; Endo, T. Unexpected Volume Expanding Behavior on Ring-Opening Metathesis Polymerization of Norbornene Bearing a Five- or Six-Membered-Ring Cyclic Carbonate Followed by a Cationic Ring-Opening Cross-Linking Reaction. Macromolecules 2004, 37, 9660–9663. [Google Scholar] [CrossRef]
- Hino, T.; Inoue, N.; Endo, T. Detailed study of the ring-opening metathesis polymerization of norbornene bearing a five- or six-membered ring cyclic carbonate along with volume expansion. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 395–405. [Google Scholar] [CrossRef]
- Hino, T.; Inoue, N.; Endo, T. Ring-opening metathesis copolymerization behaviors of cyclooctene and norbornene bearing a five- or six-membered ring cyclic carbonate. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 6599–6604. [Google Scholar] [CrossRef]
- Nuyken, O.; Böhner, R.; Erdmann, C. Oxetane photopolymerization—A system with low volume shrinkage. Macromol. Symp. 1996, 107, 125–138. [Google Scholar] [CrossRef]
- Verstegen, E.J.K.; Kloosterboer, J.G.; Lub, J. Synthesis and photopolymerization of oxetanes derived from bisphenol A. J. Appl. Polym. Sci. 2005, 98, 1697–1707. [Google Scholar] [CrossRef]
- Lub, J.; Recaj, V.; Puig, L.; Forcén, P.; Luengo, C. Synthesis, properties and photopolymerization of liquid crystalline dioxetanes. Liq. Cryst. 2004, 31, 1627–1637. [Google Scholar] [CrossRef]
- Trathnigg, B.; Hippmann, G.; Junek, H. Polymerization of spiro-orthoesters. III. Syntheses of monomers. Die Angew. Makromol. Chem. 1982, 105, 1–7. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanda, F.; Endo, T. Synthesis and cationic ring-opening polymerization of mono- and bifunctional spiro orthoesters containing ester groups and depolymerization of the obtained polymers: An approach to chemical recycling for polyesters as a model system. J. Polym. Sci. A Polym. Chem. 1999, 37, 2551–2558. [Google Scholar] [CrossRef]
- Jung, H.-J.; Chang, C.; Yu, I.; Aluthge, D.C.; Ebrahimi, T.; Mehrkhodavandi, P. Coupling of Epoxides and Lactones by Cationic Indium Catalysts To Form Functionalized Spiro-Orthoesters. ChemCatChem 2018, 10, 3219–3222. [Google Scholar] [CrossRef]
- Nishida, H.; Morikawa, H.; Nakahara, T.; Ogata, T.; Kusumoto, K.; Endo, T. Catalytic double ring-opening polyaddition of spiro orthoester with acid chloride for shrinkage-controlled molding. Polymer 2005, 46, 2531–2540. [Google Scholar] [CrossRef]
- Hsu, Y.-G.; Wan, Y.-S.; Lin, W.-Y.; Hsieh, W.-L. Cationic Polymerization of cis -2,3-Tetramethylene-1,4,6-trioxaspiro[4,4]nonane Photosensitized by Anthracene. Macromolecules 2010, 43, 8430–8435. [Google Scholar] [CrossRef]
- Hsu, Y.-G.; Lin, W.-Y.; Hsieh, W.-L.; Tsai, S.-Y. Regiospecific cationic polymerization of spiroorthoesters with different cyclic ether ring sizes. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 720–728. [Google Scholar] [CrossRef]
- Canadell, J.; Mantecón, A.; Cádiz, V. Microwave-Accelerated Polymerization of 2-Phenoxymethyl-1,4,6-trioxaspiro[4,4]nonane with Diglycidyl Ether of Bisphenol A. Macromol. Chem. Phys. 2007, 208, 2018–2025. [Google Scholar] [CrossRef]
- Fernández-Francos, X.; Salla, J.M.; Pérez, G.; Mantecón, A.; Serra, À.; Ramis, X. New Thermosets Obtained by Thermal and UV-Induced Cationic Copolymerization of DGEBA with 4-Phenyl- γ -butyrolactone. Macromol. Chem. Phys. 2009, 210, 1450–1460. [Google Scholar] [CrossRef]
- Canadell, J.; Mantecón, A.; Cádiz, V. Novel silicon-containing spiroorthoester to confer combined flame retardancy and low shrinkage properties to epoxy resins. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4211–4224. [Google Scholar] [CrossRef]
- Canadell, J.; Mantecón, A.; Cádiz, V. Synthesis of a novel bis-spiroorthoester containing 9,10-dihydro-9-oxa-10-phosphaphenantrene-10-oxide as a substituent: Homopolymerization and copolymerization with diglycidyl ether of bisphenol A. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1980–1992. [Google Scholar] [CrossRef]
- Canadell, J.; Mantecón, A.; Cádiz, V. Copolymerization of a silicon-containing spiroorthoester with a phosphorus-containing diglycidyl compound: Influence on flame retardancy and shrinkage. Polym. Degrad. Stab. 2007, 92, 1934–1941. [Google Scholar] [CrossRef]
- Marx, P.; Romano, A.; Fischer, R.; Roppolo, I.; Sangermano, M.; Wiesbrock, F. Dual-Cure Coatings: Spiroorthoesters as Volume-Controlling Additives in Thiol–Ene Reactions. Macromol. Mater. Eng. 2019, 304, 1800627. [Google Scholar] [CrossRef]
- Marx, P.; Romano, A.; Roppolo, I.; Chemelli, A.; Mühlbacher, I.; Kern, W.; Chaudhary, S.; Andritsch, T.; Sangermano, M.; Wiesbrock, F. 3D-Printing of High-κ Thiol-Ene Resins with Spiro-Orthoesters as Anti-Shrinkage Additive. Macromol. Mater. Eng. 2019, 304, 1900515. [Google Scholar] [CrossRef]
- Li, Z.; Cho, Y.H.; Kawakami, Y. Fabrication of transmission holographic gratings with spiroorthoester and trimethylolpropane triacrylate. Polym. Int. 2007, 56, 666–673. [Google Scholar] [CrossRef]
- Fu, J.; Liu, W.; Liu, X.; Tuladhar, S.L.; Wan, Q.; Wang, H. Properties of a new dental photocurable resin based on the expanding monomer and three-component photoinitiator system. J. Wuhan Univ. Technol. Mat. Sci. Ed. 2014, 29, 384–390. [Google Scholar] [CrossRef]
- Nagasawa, T.; Ochiai, B.; Endo, T. Infrared thermographic analysis on copolymerization of spiroorthoester with oxetane. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1388–1393. [Google Scholar] [CrossRef]
- Ochiai, B.; Nagasawa, T.; Asano, Y.; Nagai, D.; Sudo, A.; Endo, T. Novel analytical method for the crosslinking process: Infrared thermographic analysis of the thermally latent cationic polymerization of a spiroorthoester and a bifunctional oxetane for the construction of a low-shrinkage curing system. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 2820–2826. [Google Scholar] [CrossRef]
- Miyata, T.; Matsumoto, K.; Yonemori, S.; Endo, T. Synthesis and polymerization of styrene monomers bearing spiroorthoester structure and volume change during crosslinking by double ring-opening of the pendant spiroorthoesters of the obtained polymers. J. Polym. Sci. A Polym. Chem. 2014, 52, 1790–1795. [Google Scholar] [CrossRef]
- Kume, M.; Hirano, A.; Ochiai, B.; Endo, T. Copolymers containing a spiro orthoester moiety that undergo no shrinkage during cationic crosslinking. J. Polym. Sci. A Polym. Chem. 2006, 44, 3666–3673. [Google Scholar] [CrossRef]
- Canadell, J.; Hunt, B.J.; Cook, A.G.; Mantecón, A.; Cádiz, V. Phosphorylated copolymers containing pendant, crosslinkable spiro orthoester moieties. J. Polym. Sci. A Polym. Chem. 2006, 44, 6728–6737. [Google Scholar] [CrossRef]
- Canadell, J.; Mantecón, A.; Cádiz, V. Crosslinking of a polyacrylate bearing a spiroorthoester pendant group with mixtures of diglycidyl ether of bisphenol A and phosphorus-containing glycidyl derivatives. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1920–1930. [Google Scholar] [CrossRef]
- Takata, T.; Endo, T. New aspect of cationic ring-opening polymerization of seven-membered spiroorthocarbonates: Synthesis and polymerization of substituted 1,6,8,13-tetraoxaspiro[6.6]tridecanes. Macromolecules 1988, 21, 900–904. [Google Scholar] [CrossRef]
- Moszner, N.; Zeuner, F.; Rheinberger, V. Polymerization of cyclic monomers, 1. Radical polymerization of unsaturated spiro orthocarbonates. Macromol. Rapid Commun. 1995, 16, 667–672. [Google Scholar] [CrossRef]
- Sanda, F.; Takata, T.; Endo, T. Molecular orbital study on the mechanism of radical polymerization of spiro-orthocarbonates bearing exo-methylene groups. Macromol. Theory Simul. 1995, 4, 221–231. [Google Scholar] [CrossRef]
- Mori, Y.; Sudo, A.; Endo, T. Efficient synthesis and properties of soluble aliphatic oligo(spiroorthocarbonate)s from pentaerythritol derivatives. J. Polym. Sci. Part A Polym. Chem. 2019. [Google Scholar] [CrossRef]
- Moritsugu, M.; Seto, R.; Matsumoto, K.; Endo, T. Synthesis of thermally stable aromatic poly(spiroorthocarbonate)s having a Cardo or bent structure. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 1409–1416. [Google Scholar] [CrossRef]
- Mori, Y.; Sudo, A.; Endo, T. Effect of oligo(spiroorthocarbonate)s on the volume shrinkage of epoxides during crosslinking by sulfonium salt-initiated cationic polymerization of epoxides. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 1564–1568. [Google Scholar] [CrossRef]
- Xu, X.; Wu, Y.; Wang, C. Synthesis of DOPO-based spiroorthocarbonate and its application in epoxy resin. Des. Monomers Polym. 2015, 18, 690–697. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhou, L.; Liang, B.; Wu, Y.; Wang, C. Synthesis of Copolymers Containing Double Spiro Orthocarbonate and Used as Anti-shrinkage Additives in Epoxy Resin Composite. Polym. Plast. Technol. Eng. 2014, 53, 753–759. [Google Scholar] [CrossRef]
- Acosta Ortiz, R.; Garcia Valdez, A.E.; Aguirre Flores, R.; Lozano Palacios, R.I.; Berlanga Duarte, M.L. Synthesis of a novel highly hindered spiroorthocarbonate and the study of its efficiency to eliminate the shrinkage in the photopolymerization of an epoxycycloaliphatic resin. J. Polym. Res. 2015, 22. [Google Scholar] [CrossRef]
- Acosta Ortiz, R.; Berlanga Duarte, M.L.; Robles Olivares, J.L.; Sangermano, M. Synthesis of the fluorene spiroorthocarbonate and the evaluation of its antishrinking activity in the cationic photopolymerization of an epoxy resin. Des. Monomers Polym. 2013, 16, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Min, H.; Zheng, N.; Fan, Z.; Jiang, Y.; Cheng, X. UV-curable nanoimprint resist with liquid volume-expanding monomers. Microelectron. Eng. 2019, 205, 32–36. [Google Scholar] [CrossRef]
- Ortiz, R.A.; Berlanga Duarte, M.L.; Gómez, A.G.S.; Sangermano, M.; García Valdez, A.E.; Ramírez, M.P. Novel Tetraspiroorthocarbonates as Successful Anti-shrinking Agents for the Photopolymerization of Epoxy Monomers. J. Macromol. Sci. Part A 2012, 49, 361–368. [Google Scholar] [CrossRef]
- Acosta Ortiz, R.; Duarte, M.L.B.; Gómez, A.G.S.; Sangermano, M.; García Valdez, A.E. Novel diol spiro orthocarbonates derived from glycerol as anti-shrinkage additives for the cationic photopolymerization of epoxy monomers. Polym. Int. 2010, 59, 680–685. [Google Scholar] [CrossRef]
- Sangermano, M.; Duarte, M.L.B.; Ortiz, R.A.; Gómez, A.G.S.; Valdez, A.E.G. Diol spiroorthocarbonates as antishrinkage additives for the cationic photopolymerization of bisphenol-A–diglycidyl ether. React. Funct. Polym. 2010, 70, 98–102. [Google Scholar] [CrossRef]
- Nagai, D.; Nishida, M.; Nagasawa, T.; Ochiai, B.; Miyazaki, K.; Endo, T. Non-Shrinking Networked Materials from the Cross-Linking Copolymerization of Spiroorthocarbonate with Bifunctional Oxetane. Macromol. Rapid Commun. 2006, 27, 921–925. [Google Scholar] [CrossRef]
- Sangermano, M.; Giannelli, S.; Ortiz, R.A.; Duarte, M.L.B.; Gonzalez, A.K.R.; Valdez, A.E.G. Synthesis of an oxetane-functionalized hemispiroorthocarbonate used as a low-shrinkage additive in the cationic ultraviolet curing of oxetane monomers. J. Appl. Polym. Sci. 2009, 112, 1780–1787. [Google Scholar] [CrossRef]
- Acosta Ortiz, R.; Savage Gomez, A.G.; Berlanga Duarte, M.L.; Garcia Valdez, A.E. The effect of a dithiol spiroorthocarbonate on mechanical properties and shrinkage of a dental resin. Des. Monomers Polym. 2015, 18, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Acosta Ortiz, R.; Reyna Medina, L.A.; Berlanga Duarte, M.L.; Ibarra Samaniego, L.; Garcia Valdez, A.E.; García Mendez, Z.L.; Mendez Gonzalez, L. Synthesis of glycerol-derived diallyl spiroorthocarbonates and the study of their antishrinking properties in acrylic dental resins. J. Mater. Sci. Mater. Med. 2013, 24, 2077–2084. [Google Scholar] [CrossRef]
- Fu, J.; Liu, W.; Hao, Z.; Wu, X.; Yin, J.; Panjiyar, A.; Liu, X.; Shen, J.; Wang, H. Characterization of a low shrinkage dental composite containing bismethylene spiroorthocarbonate expanding monomer. Int. J. Mol. Sci. 2014, 15, 2400–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlanga Duarte, M.L.; Reyna Medina, L.A.; Torres Reyes, P.; Esparza González, S.C.; Herrera González, A.M. Dental restorative composites containing methacrylic spiroorthocarbonate monomers as antishrinking matrixes. J. Appl. Polym. Sci. 2019, 136, 47114. [Google Scholar] [CrossRef]
- Yoo, S.H.; Park, K.; Kim, J.; Kim, C.K. Characteristics of dental restorative composites fabricated from Bis-GMA alternatives and spiro orthocarbonates. Macromol. Res. 2011, 19, 27–32. [Google Scholar] [CrossRef]
- Takata, T.; Sanda, F.; Ariga, T.; Nemoto, H.; Endo, T. Cyclic carbonates, novel expandable monomers on polymerization. Macromol. Rapid Commun. 1997, 18, 461–469. [Google Scholar] [CrossRef]
- Hino, T.; Inoue, N.; Endo, T. Cationic copolymerization behavior of cyclic ether monomers with norbornene-containing cyclic carbonate or spiro–orthoether structure. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5113–5120. [Google Scholar] [CrossRef]
- Morikawa, H.; Sudo, A.; Nishida, H.; Endo, T. Volume-expandable monomer 5,5-dimethyl-1,3-dioxolan-2-one: Its copolymerization behavior with epoxide and its applications to shrinkage-controlled epoxy-curing systems. J. Appl. Polym. Sci. 2005, 96, 372–378. [Google Scholar] [CrossRef]
- Rajagopal, R.V.; Yamanaka, K. Methods of Making Low Shrinkage and Expandable Compositions and Expandable Monomers. WO Patent 2014us19419 20140228, 28 February 2014. [Google Scholar]
- Sangermano, M.; Malucelli, G.; Delleani, G.; Priola, A. Bicyclo-orthoester as a low-shrinkage additive in cationic UV curing. Polym. Int. 2007, 56, 1224–1229. [Google Scholar] [CrossRef]
- Kume, M.; Endo, T. Cationic copolymerization behavior of a bicyclic orthoester having hydroxy group with glycidyl phenyl ether and volume change on their copolymerization. J. Appl. Polym. Sci. 2006, 101, 1356–1361. [Google Scholar] [CrossRef]
- Ohsawa, S.; Morino, K.; Sudo, A.; Endo, T. Synthesis of bicyclic bis(γ -butyrolactone) derivatives bearing sulfide moieties and their alternating copolymers with epoxide. J. Polym. Sci. A Polym. Chem. 2012, 50, 4666–4673. [Google Scholar] [CrossRef]
- Fukuchi, I.; Sanda, F.; Endo, T. Synthesis and Cationic Polymerization of 2-Phenyl-1,6-dioxaspiro[4.6]undecane. A Novel Expandable Monomer Undergoing Cationic Double Ring-Opening Polymerization. Macromolecules 2001, 34, 4296–4298. [Google Scholar] [CrossRef]
- Weinmann, W.; Thalacker, C.; Guggenberger, R. Siloranes in dental composites. Dent. Mater. 2005, 21, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Lee, J.-R. Evidence for the Volumetric Expansion of N-Benzylpyrazinium Hexafluoroantimonate and Epoxy Mixture during Curing Reaction. Polym. J. 1998, 30, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Kiskan, B. Adapting benzoxazine chemistry for unconventional applications. React. Funct. Polym. 2018, 129, 76–88. [Google Scholar] [CrossRef]
- Lligadas, G.; Tüzün, A.; Ronda, J.C.; Galià, M.; Cádiz, V. Polybenzoxazines: New players in the bio-based polymer arena. Polym. Chem. 2014, 5, 6636–6644. [Google Scholar] [CrossRef]
- Ishida, H.; Allen, D.J. Physical and mechanical characterization of near-zero shrinkage polybenzoxazines. J. Polym. Sci. B Polym. Phys. 1996, 34, 1019–1030. [Google Scholar] [CrossRef]
- Ishida, H.; Low, H.Y. A Study on the Volumetric Expansion of Benzoxazine-Based Phenolic Resin. Macromolecules 1997, 30, 1099–1106. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Ishida, H. Cationic ring-opening polymerization of benzoxazines. Polymer 1999, 40, 4563–4570. [Google Scholar] [CrossRef]
- Dunkers, J.; Ishida, H. Reaction of benzoxazine-based phenolic resins with strong and weak carboxylic acids and phenols as catalysts. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 1913–1921. [Google Scholar] [CrossRef]
- Kasapoglu, F.; Cianga, I.; Yagci, Y.; Takeichi, T. Photoinitiated cationic polymerization of monofunctional benzoxazine. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3320–3328. [Google Scholar] [CrossRef]
- Dunkers, J.; Zarate, E.A.; Ishida, H. Crystal Structure and Hydrogen-Bonding Characteristics of N,N-Bis(3,5-dimethyl-2-hydroxybenzyl)methylamine, A Benzoxazine Dimer. J. Phys. Chem. 1996, 100, 13514–13520. [Google Scholar] [CrossRef]
- Liu, X.; Gu, Y. Study on the volumetric expansion of benzoxazine curing with different catalysts. J. Appl. Polym. Sci. 2002, 84, 1107–1113. [Google Scholar] [CrossRef]
- Lotfi, L.; Javadpour, J.; Naimi-Jamal, M.R. Biological and nano-indentation properties of polybenzoxazine-based composites reinforced with zirconia particles as a novel biomaterial. Biomed. Mater. Eng. 2018, 29, 369–387. [Google Scholar] [CrossRef] [PubMed]
- Kanchanasopa, M.; Yanumet, N.; Hemvichian, K.; Ishida, H. The Effect of Polymerization Conditions on the Density and Tg of Bisphenol-A and Hexafluoroisopropylidene-Containing Polybenzoxazines. Polym. Polym. Compos. 2001, 9, 367–376. [Google Scholar] [CrossRef]
- Wiesbrock, F.; Windberger, M.S. Novel Expanding Copolymers. WO Patent 2019EP55862 20190308, 8 March 2019. [Google Scholar]
- Saiev, S.; Bonnaud, L.; Dubois, P.; Beljonne, D.; Lazzaroni, R. Modeling the formation and thermomechanical properties of polybenzoxazine thermosets. Polym. Chem. 2017, 8, 5988–5999. [Google Scholar] [CrossRef]
- Kakimoto, K.; Nemoto, N.; Sanda, F.; Endo, T. Anionic Ring-Opening Polymerization of Cyclic Thiocarbonates Containing Norbornene and Norbornane Groups Undergoing Volume Expansion on Polymerization. Chem. Lett. 2002, 31, 156–157. [Google Scholar] [CrossRef]
- Kameshima, H.; Nemoto, N.; Sanda, F.; Endo, T. Cationic Ring-Opening Polymerization of Five-Membered Cyclic Thiocarbonate Bearing an Adamantane Moiety via Selective Ring-Opening Direction. Macromolecules 2002, 35, 5769–5773. [Google Scholar] [CrossRef]
- Ruppitsch, L.A.; Peer, G.; Ehrmann, K.; Koch, T.; Liska, R. Photopolymerization of difunctional cyclopolymerizable monomers with low shrinkage behavior. J. Polym. Sci. 2021. [Google Scholar] [CrossRef]
- Peer, G.; Kury, M.; Gorsche, C.; Catel, Y.; Frühwirt, P.; Gescheidt, G.; Moszner, N.; Liska, R. Revival of Cyclopolymerizable Monomers as Low-Shrinkage Cross-Linkers. Macromolecules 2020, 53, 8374–8381. [Google Scholar] [CrossRef]




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
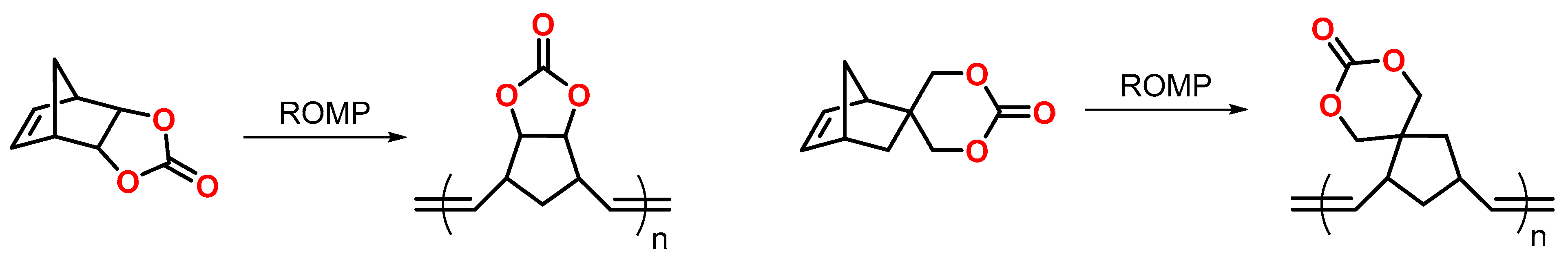{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Potential Shortcomings |
|---|---|
| Capillary Dilatometry | Sticking of the resin to the capillary walls and the challenges of temperature control in case of strongly exothermic polymerization reactions can affect the accuracy of the measurements. |
| Gas Pycnometry | For the quantification of volumetric changes during curing reactions, the volume of the sample needs to be measured before and after curing. |
| Buoyancy Method | While the method is independent of the size or geometry of the specimens, voids inside the material and air bubbles on the surface can strongly affect the results of the measurements. |
| Linometry | Three-dimensional volumetric expansion can be only estimated in case of isotropic expansion. |
| Bonded Disk Method | The obtained values for linear shrinkage depend on the dimension of the tested specimens |
| Laser-Based Methods | High equipment expenditure and high security demands. |
| Rheometry | Three-dimensional volumetric expansion can be only estimated in case of isotropic expansion. |
| Thermomechanical Analyses | Standard set-ups are exclusively applicable for solid samples. |
| Imaging Methods | High equipment expenditure (and eventually high security demands). |
| Cyclic Carbonate | Boiling Point (°C) | ρ (g/mL) | μ (Debye) |
|---|---|---|---|
 | 243–244 | 1.321 | 4.87 |
 | 242 | 1.189 | 4.94 |
 | 90 | 1.069 | _ |
 | 126 | 0.975 | 0.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marx, P.; Wiesbrock, F. Expanding Monomers as Anti-Shrinkage Additives. Polymers 2021, 13, 806. https://doi.org/10.3390/polym13050806
Marx P, Wiesbrock F. Expanding Monomers as Anti-Shrinkage Additives. Polymers. 2021; 13(5):806. https://doi.org/10.3390/polym13050806
Chicago/Turabian StyleMarx, Philipp, and Frank Wiesbrock. 2021. "Expanding Monomers as Anti-Shrinkage Additives" Polymers 13, no. 5: 806. https://doi.org/10.3390/polym13050806
APA StyleMarx, P., & Wiesbrock, F. (2021). Expanding Monomers as Anti-Shrinkage Additives. Polymers, 13(5), 806. https://doi.org/10.3390/polym13050806