
Syndiotactic Poly(4-methyl-1-pentene)-Based Stereoregular Diblock Copolymers: Synthesis and Self-Assembly Studies


Abstract
:
1. Introduction
2. Experimental Section
2.1. General Procedure
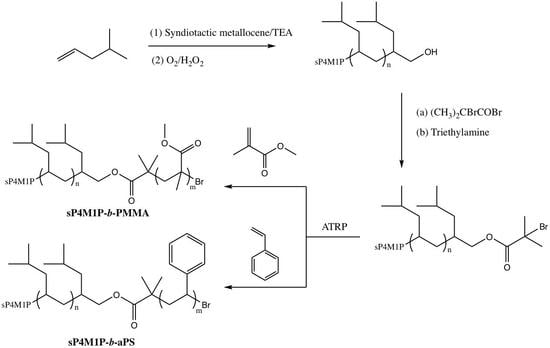
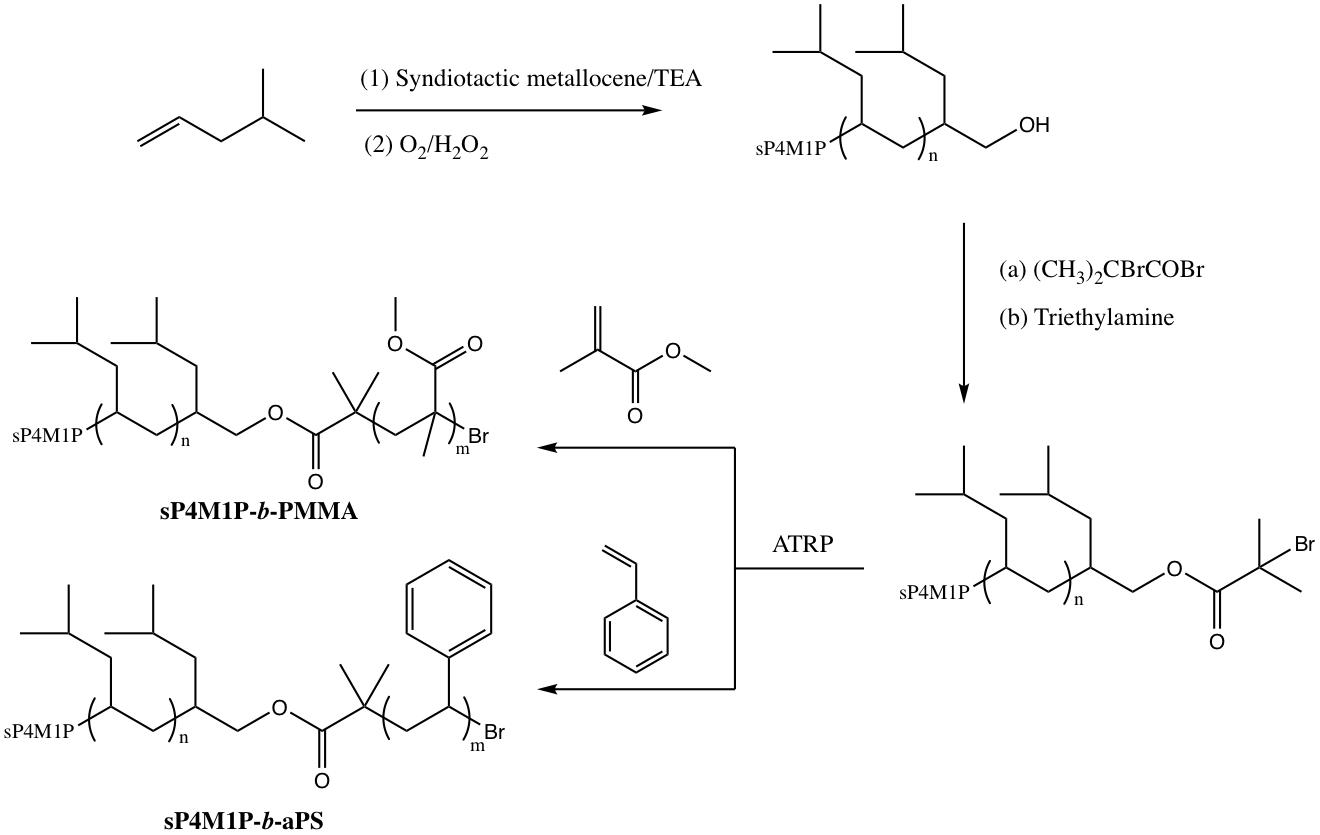
2.2. Preparation of OH-Capped sP4M1P
2.3. Fractionation of OH-Capped sP4M1P
2.4. Preparation of α-Bromoisobutylester-Capped sP4M1P
2.5. Preparation of sP4M1P-b-PMMA
2.6. Preparation of sP4M1P-b-aPS
2.7. Polymer Analysis
2.8. Bulk Sample Preparation
2.9. Small-Angle X-ray Scattering Measurement
3. Results and Discussion
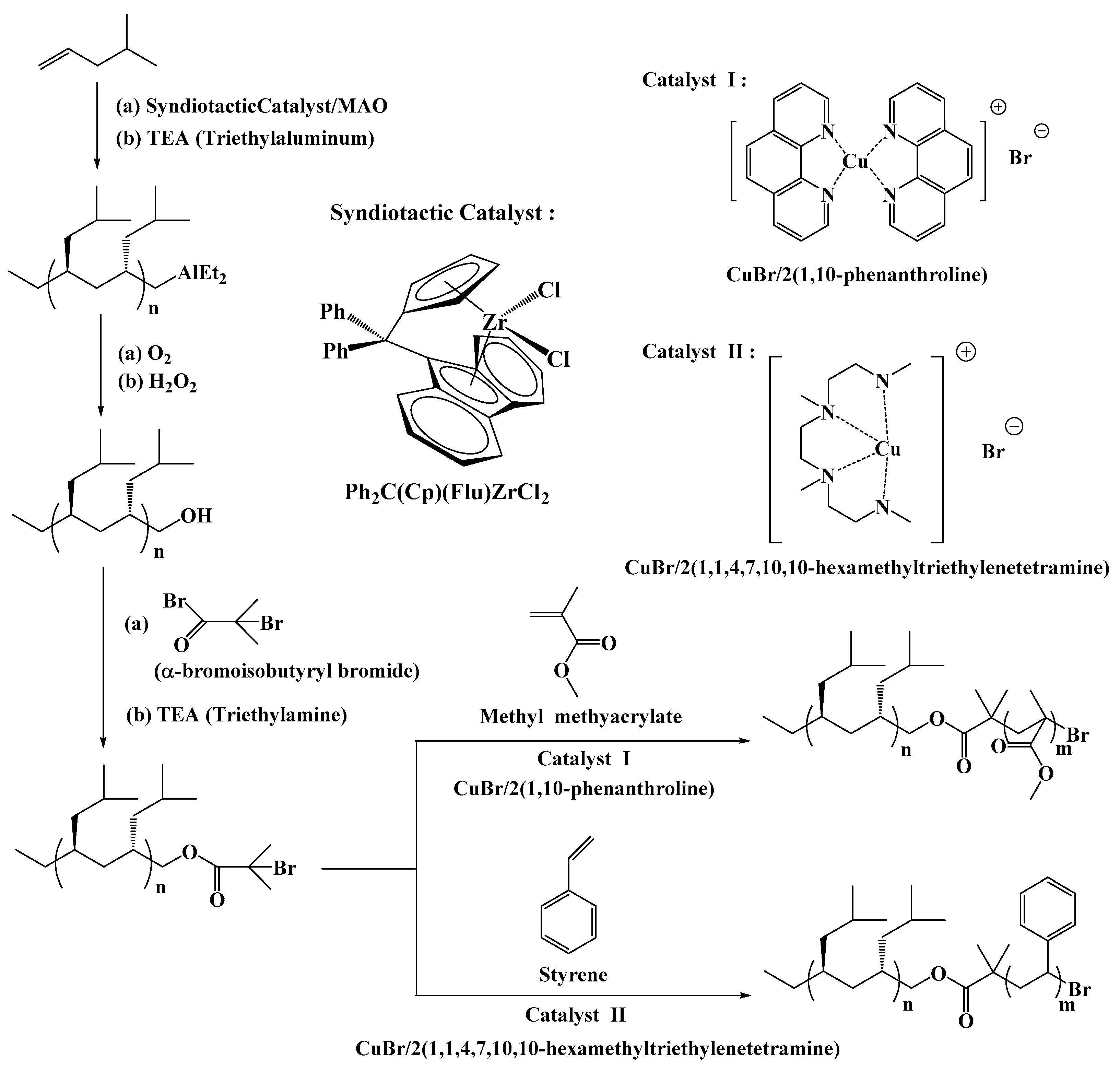
3.1. Preparation of Hydroxyl-Capped sP4M1P by Selective Chain Transfer to Alkylaluminum
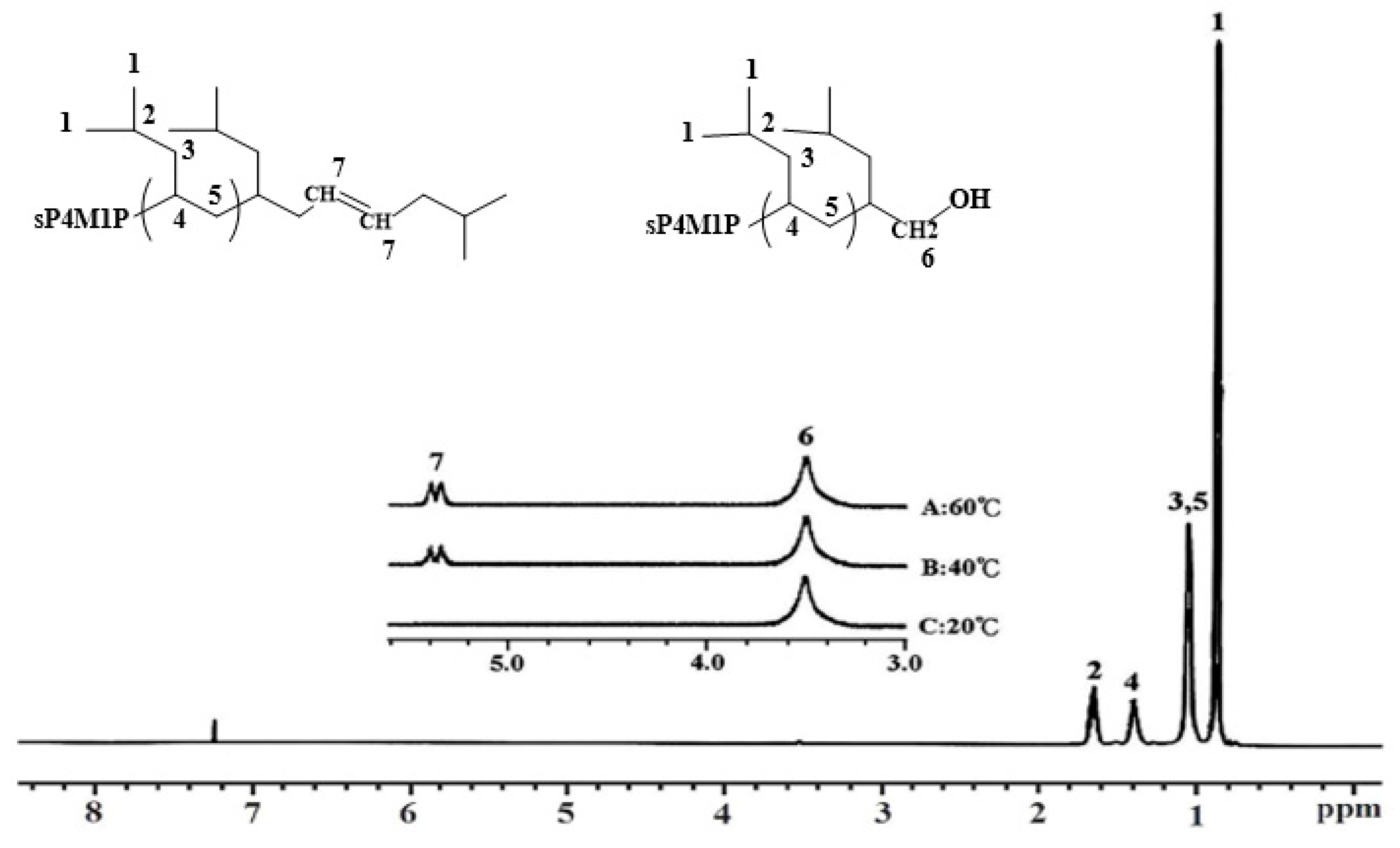
3.2. Structural Characterization of OH-Capped sP4M1P
3.3. Preparation of Syndiotactic 4-Methyl-1-pentene-Based Stereoregular Diblock Copolymers
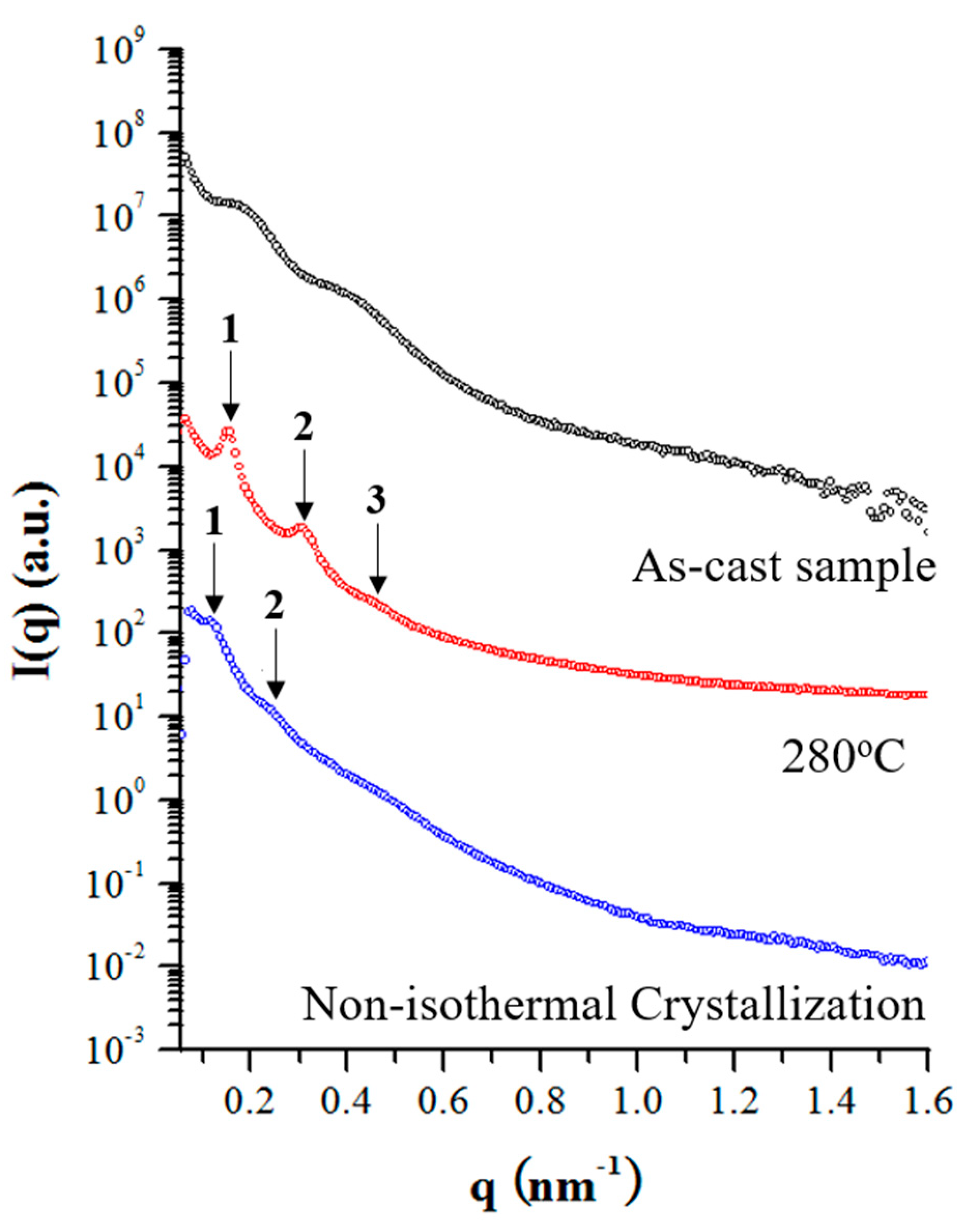
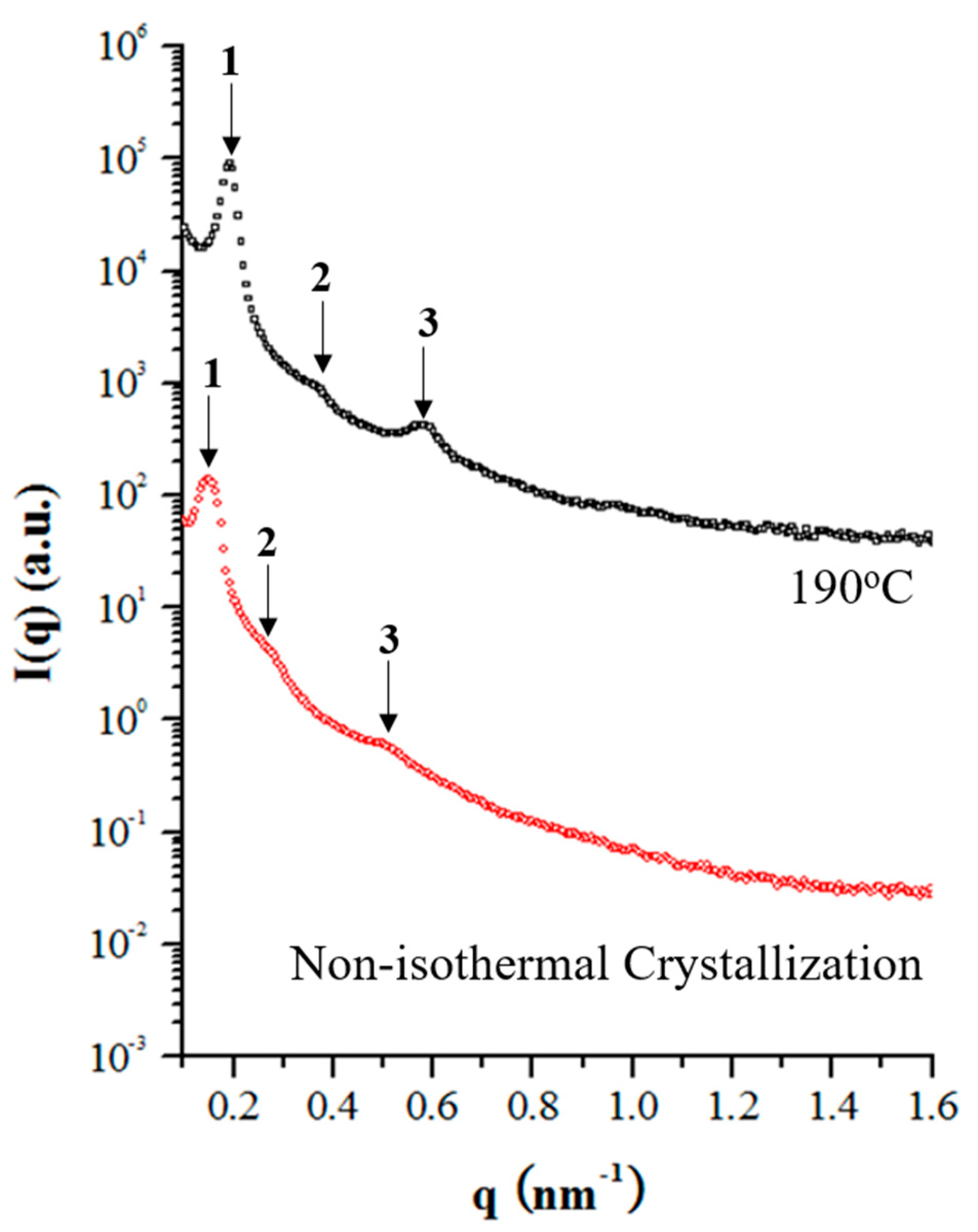
3.4. Self-Assembly of sP4MP-Based Stereoregular Block Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alexandridis, P.; Lindman, B. Amphiphilic Block Copolymers: Self-Assembly and Applications; Elsevier Science BV: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Calleja, F.B.; Roslaniec, Z. Block Copolymers; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Hadjichristidis, N.; Pispas, S.; Floudas, G. Block Copolymers: Synthetic Strategies, Physical Properties, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2003. [Google Scholar]
- Murataj, I.; Cara, E.; Baglieri, N.; Pirri, C.F.; De Leo, N.; Ferrarese Lupi, F. Liquid Phase Infiltration of Block Copolymers. Polymers 2022, 14, 4317. [Google Scholar] [CrossRef]
- Siddique, S.K.; Sadek, H.; Lee, T.-L.; Tsai, C.-Y.; Chang, S.-Y.; Tsai, H.-H.; Lin, T.-S.; Manesi, G.-M.; Avgeropoulos, A.; Ho, R.-M. Block Copolymer Modified Nanonetwork Epoxy Resin for Superior Energy Dissipation. Polymers 2022, 14, 1891. [Google Scholar] [CrossRef]
- Lo, T.-Y.; Krishnan, M.R.; Lu, K.-Y.; Ho, R.-M. Silicon-containing block copolymers for lithographic applications. Prog. Polym. Sci. 2018, 77, 19–68. [Google Scholar] [CrossRef]
- Kannaiyan, D.; Kochuveedu, S.T.; Jang, Y.H.; Jang, Y.J.; Lee, J.Y.; Lee, J.; Lee, J.; Kim, J.; Kim, D.H. Enhanced photophysical properties of nanopatterned titania nanodots/nanowires upon hybridization with silica via block copolymer templated sol-gel process. Polymers 2010, 2, 490. [Google Scholar] [CrossRef] [Green Version]
- Saleem, S.; Rangou, S.; Abetz, C.; Lademann, B.; Filiz, V.; Abetz, V. Block copolymer membranes from polystyrene-b-poly (solketal methacrylate)(PS-b-PSMA) and amphiphilic polystyrene-b-poly (glyceryl methacrylate)(PS-b-PGMA). Polymers 2017, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, Y.; Konno, R.; Ishizone, T.; Goseki, R.; Hirao, A. Precise synthesis of block polymers composed of three or more blocks by specially designed linking methodologies in conjunction with living anionic polymerization system. Polymers 2013, 5, 1012–1040. [Google Scholar] [CrossRef]
- Yagci, Y.; Tasdelen, M.A. Mechanistic transformations involving living and controlled/living polymerization methods. Prog. Polym. Sci. 2006, 31, 1133–1170. [Google Scholar] [CrossRef]
- Domski, G.J.; Rose, J.M.; Coates, G.W.; Bolig, A.D.; Brookhart, M. Living alkene polymerization: New methods for the precision synthesis of polyolefins. Prog. Polym. Sci. 2007, 32, 30–92. [Google Scholar] [CrossRef]
- Capacchione, C.; Saviello, D.; Ricciardi, R.; Proto, A. Living, isoselective polymerization of 4-methyl-1, 3-pentadiene and styrenic monomers and synthesis of highly stereoregular block copolymers via sequential monomer addition. Macromolecules 2011, 44, 7940–7947. [Google Scholar] [CrossRef]
- Lin, C.-H.; Higuchi, T.; Chen, H.-L.; Tsai, J.-C.; Jinnai, H.; Hashimoto, T. Stabilizing the ordered bicontinuous double diamond structure of diblock copolymer by configurational regularity. Macromolecules 2018, 51, 4049–4058. [Google Scholar] [CrossRef]
- Chiu, P.-T.; Sung, Y.-C.; Yang, K.-C.; Tsai, J.-C.; Wang, H.-F.; Ho, R.-M. Curving and twisting in self-assembly of triblock terpolymers driven by a chiral end block. Macromolecules 2022, 55, 1185–1195. [Google Scholar] [CrossRef]
- Wang, H.-F.; Chiu, P.-T.; Yang, C.-Y.; Xie, Z.-H.; Hung, Y.-C.; Lee, J.-Y.; Tsai, J.-C.; Prasad, I.; Jinnai, H.; Thomas, E.L. Networks with controlled chirality via self-assembly of chiral triblock terpolymers. Sci. Adv. 2020, 6, eabc3644. [Google Scholar] [CrossRef]
- Gao, H.; Liu, X.; Tang, Y.; Pan, J.; Wu, Q. Living/controlled polymerization of 4-methyl-1-pentene with α-diimine nickel-diethylaluminium chloride: Effect of alkylaluminium cocatalysts. Polym. Chem. 2011, 2, 1398–1403. [Google Scholar] [CrossRef]
- Leone, G.; Losio, S.; Piovani, D.; Sommazzi, A.; Ricci, G. Living copolymerization of ethylene with 4-methyl-1-pentene by an α-diimine Ni(II)/Et 2AlCl catalyst: Synthesis of diblock copolymers via sequential monomer addition. Polym. Chem. 2012, 3, 1987–1990. [Google Scholar] [CrossRef]
- Wentz, C.M.; Fischbach, D.M.; Sita, L.R. Stereomodulation of Poly (4-methyl-1-pentene): Adoption of a Neglected and Misunderstood Commercial Polyolefin. Angew. Chem., Int. Ed. 2022, 61, e202211992. [Google Scholar] [CrossRef]
- Zhang, Y.; Keaton, R.J.; Sita, L.R. Degenerative transfer living Ziegler−Natta polymerization: Application to the synthesis of monomodal stereoblock polyolefins of narrow polydispersity and tunable block length. J. Am. Chem. Soc. 2003, 125, 9062–9069. [Google Scholar] [CrossRef]
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific olefin polymerization with chiral metallocene catalysts. Angew. Chem. Int. Ed. 1995, 34, 1143–1170. [Google Scholar] [CrossRef] [Green Version]
- Matyjaszewski, K.; Xia, J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. [Google Scholar] [CrossRef]
- Wen, T.; Lee, J.-Y.; Li, M.-C.; Tsai, J.-C.; Ho, R.-M. Competitive interactions of π–π junctions and their role on microphase separation of chiral block copolymers. Chem. Mater. 2017, 29, 4493–4501. [Google Scholar] [CrossRef]
- Huang, S.-H.; Huang, Y.-W.; Chiang, Y.-W.; Hsiao, T.-J.; Mao, Y.-C.; Chiang, C.-H.; Tsai, J.-C. Nanoporous Crystalline Templates from Double-Crystalline Block Copolymers by Control of Interactive Confinement. Macromolecules 2016, 49, 9048–9059. [Google Scholar] [CrossRef]
- Chiang, Y.-W.; Huang, Y.-W.; Huang, S.-H.; Huang, P.-S.; Mao, Y.-C.; Tsai, C.-K.; Kang, C.-S.; Tasi, J.-C.; Su, C.-J.; Jeng, U.-S. Control of nanostructural dimension by crystallization in a double-crystalline syndiotactic poly (4-methyl-1-pentene)-block-poly (l-lactide) block copolymer. J. Phys. Chem. C 2014, 118, 19402–19414. [Google Scholar] [CrossRef]
- Razavi, A.; Ferrara, J. Preparation and crystal structures of the complexes (η5-C5H4CMe2η5-C13H8) MCl2 (M = Zr, Hf) and their role in the catalytic formation of syndiotactic polypropylene. J. Organomet. Chem. 1992, 435, 299–310. [Google Scholar] [CrossRef]
- Resconi, L.; Piemontesi, F.; Camurati, I.; Sudmeijer, O.; Nifant’ev, I.E.; Ivchenko, P.V.; Kuz’mina, L.G. Highly regiospecific zirconocene catalysts for the isospecific polymerization of propene. J. Am. Chem. Soc. 1998, 120, 2308–2321. [Google Scholar] [CrossRef]
- Bochmann, M.; Lancaster, S.J. Base-free cationic zirconium benzyl complexes as highly active polymerization catalysts. Organometallics 1993, 12, 633–640. [Google Scholar] [CrossRef]
- Han, C.J.; Lee, M.S.; Byun, D.-J.; Kim, S.Y. Synthesis of hydroxy-terminated polyethylene via controlled chain transfer reaction and poly (ethylene-b-caprolactone) block copolymer. Macromolecules 2002, 35, 8923–8925. [Google Scholar] [CrossRef]
- Byun, D.-J.; Kim, S.Y. Selective chain transfer reactions in metallocene catalyzed copolymerization of ethylene with allylbenzene. Macromolecules 2000, 33, 1921–1923. [Google Scholar] [CrossRef]
- Shiono, T.; Kang, K.K.; Hagihara, H.; Ikeda, T. Novelty of vinylidene-terminated polypropylene prepared by a MgCl2-supported TiCl4 catalyst combined with AlEt3 as cocatalyst. Macromolecules 1997, 30, 5997–6000. [Google Scholar] [CrossRef]
- Lieber, S.; Brintzinger, H.-H. Propene polymerization with catalyst mixtures containing different ansa-zirconocenes: Chain transfer to alkylaluminum cocatalysts and formation of stereoblock polymers. Macromolecules 2000, 33, 9192–9199. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | AlR3 b (mmol) | Temp. (°C) | Activity c | Mnd | PDI d | Tm e (°C) | End Group Ratio (%) f | ||
|---|---|---|---|---|---|---|---|---|---|
| Hydroxy | Vinylene | ||||||||
| 1 | TMA | 2 | 40 | 177.21 | 8730 | 1.74 | 166.5 | 75.8 | 24.2 |
| 2 | TMA | 2 | 20 | 130.62 | 19,730 | 1.69 | 171.7 | 84.3 | 15.7 |
| 3 | TMA | 4 | 20 | 121.12 | 18,870 | 1.69 | 172.42 | 86.4 | 13.6 |
| 4 | TMA | 6 | 20 | 115.97 | 17,160 | 1.65 | 175.2 | 88.1 | 11.9 |
| 5 | TMA | 8 | 20 | 104.92 | 16,570 | 1.64 | 172.5 | 90.2 | 9.8 |
| 6 | TEA | 4 | 40 | 29.18 | 3600 | 1.58 | 163.6 | 82.4 | 17.6 |
| 7 | TEA | 4 | 60 | 58.60 | 1290 | 1.52 | - | 66.5 | 33.5 |
| 8 | TEA | 2 | 20 | 27.45 | 9890 | 1.65 | 174.0 | >99.0 | - |
| 9 | TEA | 4 | 20 | 18.83 | 5740 | 1.63 | 174.3 | >99.0 | - |
| 10 | TEA | 6 | 20 | 14.43 | 3880 | 1.62 | 170.8 | > 99.0 | - |
| 11 | TEA | 8 | 20 | 5.55 | 3290 | 1.56 | 164.2 | >99.0 | - |
| 12 | TEA | 4 | 0 | 11.43 | 6610 | 1.72 | 191.9 | >99.0 | - |
| Sample | Mn,exp a | PDI a | Volume Fraction of Polyolefin Block b | Morphologies of Microstructure c | |
|---|---|---|---|---|---|
| 1 | sP4M1P | 14,500 | 1.32 | 1.00 | |
| 2 | sP4M1P-b-PMMA | 32,000 | 1.20 | 0.46 | Lamella |
| 3 | sP4M1P | 10,500 | 1.40 | 1.00 | |
| 4 | sP4M1P-b-aPS | 19,000 | 1.34 | 0.39 | Lamella |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sung, Y.-C.; Huang, P.-S.; Huang, S.-H.; Chiang, Y.-W.; Tsai, J.-C. Syndiotactic Poly(4-methyl-1-pentene)-Based Stereoregular Diblock Copolymers: Synthesis and Self-Assembly Studies. Polymers 2022, 14, 4815. https://doi.org/10.3390/polym14224815
Sung Y-C, Huang P-S, Huang S-H, Chiang Y-W, Tsai J-C. Syndiotactic Poly(4-methyl-1-pentene)-Based Stereoregular Diblock Copolymers: Synthesis and Self-Assembly Studies. Polymers. 2022; 14(22):4815. https://doi.org/10.3390/polym14224815
Chicago/Turabian StyleSung, Yu-Chuan, Pei-Sun Huang, Shih-Hung Huang, Yeo-Wan Chiang, and Jing-Cherng Tsai. 2022. "Syndiotactic Poly(4-methyl-1-pentene)-Based Stereoregular Diblock Copolymers: Synthesis and Self-Assembly Studies" Polymers 14, no. 22: 4815. https://doi.org/10.3390/polym14224815
APA StyleSung, Y. -C., Huang, P. -S., Huang, S. -H., Chiang, Y. -W., & Tsai, J. -C. (2022). Syndiotactic Poly(4-methyl-1-pentene)-Based Stereoregular Diblock Copolymers: Synthesis and Self-Assembly Studies. Polymers, 14(22), 4815. https://doi.org/10.3390/polym14224815