1. Introduction
In recent years, electrospinning technology has gained more and more attention along with the development of nanotechnologies, which have been used by scientists and technicians in a wide variety of scientific and technological fields. It is important to mention that healthcare has been one of the pioneers in the development of nanotechnologies, with applications in such areas as fluorescent biological labeling, drug and gene delivery, bio-detection of pathogens, detection of proteins, probing of DNA structure, tissue engineering, tumor detection, separation and purification of biological molecules and cells, MRI contrast enhancement and pharmacokinetic studies [
1]. Nano-sized drug capsules also have gained increasing attention due to the fact that they are believed to have outstanding advantages over conventional sized capsules. Given the same drug mass, nano-sized capsules have a significantly higher total surface area as well as a higher drug decomposition and absorption rates. Another nanomaterial that exhibits outstanding advantages is electrospun fibers, which are used as a drug carriers. Due to their size, nano-sized capsules allow otherwise difficult-to-absorb drugs to be absorbed and slowly distributed into the body, thus enhancing their absorption and providing better therapeutic effect. In addition, biodegradable materials used as drug carriers can be broken down into small molecules that are absorbed or excreted from the body; during this process, the drug can be easily released and assimilated, improving its therapeutic function [
2]. The fabrication of nanofibers by the electrospinning technique follows the fundamental principle of fabricating nanofibers from molten polymers or polymer solutions under the effect of high-voltage current. In that process, the polymer solution is streamed under the action of a high-voltage electric field; while being ejected from the needle tip, the solution adopts a Taylor cone shape, then changes into a liquid jet that solidifies into nanofibers over the collector’s surface [
3,
4].
Chitosan is a linear polysaccharide composed of randomly distributed deacetylated units (β-(1,4)-D-glucosamine) and acetylated units (N-acetyl-D-glucosamine) (
Figure 1). Chitosan is obtained by deacetylation from chitin, which is the second most common polysaccharide in the world, only after cellulose. Usually, chitin with more than 60% degree of deacetylation [
5,
6] is considered as chitosan; the degree of deacetylation of chitosan is in the range of 60–98% [
7]. Known as an attractive material for medical use, chitosan possesses excellent biological properties such as biodegradability, lack of toxicity, antifungal effects, and the ability to accelerate wound healing and stimulate the immune system [
8,
9]. Due to its ability to function in a variety of forms, chitosan has attracted much attention in the fields of orthopedics and periodontitis therapy [
10,
11], tissue engineering [
10,
11,
12], wound healing [
10,
12], and drug transport [
10]. In biomedical applications, it has been used for artificial skin, surgical sutures, artificial blood vessels, controlled drug release, contact lenses, eye moisturizers, bandages, sponges, cholesterol control, anti-inflammatory treatment, tumor inhibition, antiviral drugs, inhibition of plaque, bone healing, accelerated wound healing; hemostatic, antibacterial, and antifungal applications; and weight loss effects [
10]. The reason for our preference for chitosan as a nanofiber-forming material was the distribution of amino groups along the molecule, as well as its polysaccharide structure. The amino groups can be protonated, providing the chitosan with different properties such as passing through characteristic neutralization reactions of alkaline compounds [
8] and ensuring solubility in dilute acidic aqueous solutions (pH < 6) [
5,
13]. It is known that pathological microenvironments are mostly acidic: for example, inflammation-associated acidic pH of 7.2–6.5 for the extracellular pH in tumors; pH 5.4 in inflamed tissue; pH 4.7 in fracture-related hematomas; and pH 5.7 in cardiac ischemia [
14,
15]. Thus, the base structure of chitosan will help the formed nanofibers to be highly targeted to pathological cells and tissues. As a polycationic polymer with a high density of positive charges, it can adhere to both hard and soft tissues such as epithelial and mucous tissues through hydration, hydrogen bonding and ionic interactions, and has been extensively investigated as a drug carrier for targeted drug delivery purposes [
16]. Cancerous cells are characterized by an abnormal glucose metabolism pathway that leads to the need for high glucose to rapidly generate energy for their survival [
17,
18,
19]. Chitosan has been shown to be suitable as a material to target cancerous cells due to its polysaccharide structure. In addition, as a polysaccharide that contains degradable glycosidic bonds, chitosan is biodegradable by several protease enzymes, and mainly by lysozyme [
5,
13]. Chitosan is a biopolymer that behaves like a hydrogel due to its three-dimensional structure, which can absorb and retain large amounts of water, allowing it to swell without the need for complete dissolution [
7]. In solution, chitosan macromolecules tend to entangle and form physical networks due to the abundant intermolecular hydrogen bonding, even at low concentrations. This causes some difficulties in the fabrication of nanofibers with only chitosan, requiring that it be combined with another polymer that has more favorable properties for nanofiber fabrication; PVA was chosen for our investigation.
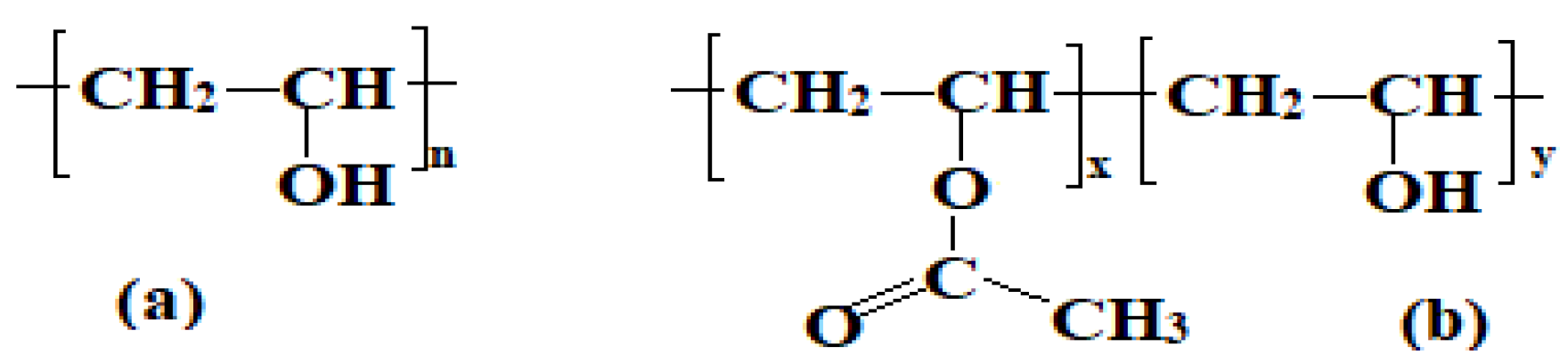
Poly (vinyl alcohol) (PVA) (
Figure 2) has attracted considerable research interest and is recognized as one of the most extensively produced synthetic polymers worldwide [
20]. PVA is a widely used thermoplastic polymer that is safe for living tissues, harmless and non-toxic. Orally administered poly (vinyl alcohol) has an LD
50 ≈ 15–20 g/kg [
21,
22]. PVA is a semicrystalline synthetic polymer, which is soluble in water, slightly soluble in ethanol and insoluble in other organic solvents [
22,
23,
24,
25]. It is also a biodegradable polymer, and its degradability is enhanced by hydrolysis due to the presence of hydroxyl groups. Under the action of the microbial community or some enzymes such as
ß-diketone hydrolase and
secondary alcohol oxidase, the PVA molecular chain can be partially cleaved at C–C bonds to form ketone and carboxylic compounds [
26,
27,
28]. The final breakdown product during PVA degradation is acetic acid, which is transferred into the central metabolic pathway. Acetic acid is readily metabolized by most human and animal tissues. It is also involved in the formation of phospholipids, neutral lipids, sterols, and saturated and unsaturated fatty acids in many human and animal tissues [
29,
30].
PVA is commercially produced through the hydrolysis of poly (vinyl acetate) in a two-step process consisting of free radical polymerization of vinyl acetate followed by its hydrolysis [
20,
31]. Therefore, the structural properties of PVA are mainly dependent on the molecular weight of the polymer and the degree of hydrolysis, i.e., the percentage of vinyl alcohol in the polymer [
31,
32]. As the degree of vinyl acetate hydrolysis into vinyl alcohol increases, the polymer structure becomes more crystalline, which results in a highly durable PVA structure, which becomes chemically inert [
31]. The degree of crystallinity plays a major role in controlling the diffusion of drugs from PVA hydrogels [
33], which can be designed either as matrix or reservoir for drug delivery platforms [
34]. In general, due to its biocompatibility, drug compatibility, water solubility, film formation, mechanical properties and good swelling, PVA has been studied as a material for ocular inserts, ocular films, nanoparticles, microspheres, floating microspheres, mucoadhesives, transdermal patches, and intramuscular drug delivery systems, as well as targeted drug delivery for colon, rectal, buccal, transdermal, pH- and temperature-sensitive drug delivery systems and swelling-controlled drug delivery systems [
20,
31,
35]. Due to its susceptibility to hydrogen bonding and excessive crystallization, PVA is very sensitive to moisture; PVA hydrogels generally have low mechanical properties and offer very low swelling capacity, making them desirable only for specific biomedical and pharmaceutical applications [
31,
36,
37]. In order to improve its mechanical properties and stability, polyvinyl alcohol is often mixed with other biopolymers. It has been reported that the inclusion of chitosan into a polyvinyl alcohol matrix can improve its biocompatibility and mechanical properties [
24,
38,
39].
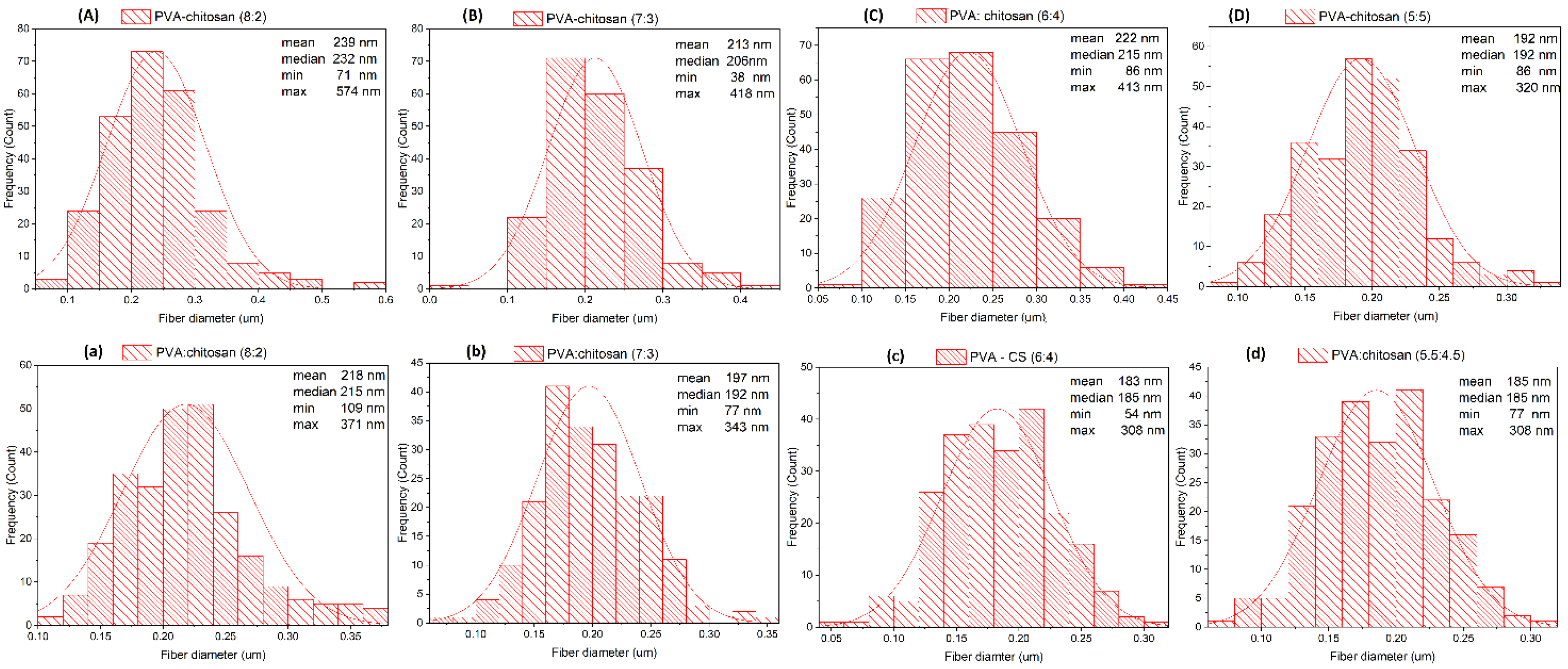
To date, there have been many reports referring to the fabrication of electrospun PVA–chitosan nanofibers, from which the smallest fibers are reported to have an average diameter of 20 to 100 nm [
39]. However, such fibers were interspersed with enlarged spindle-like sections of about 500 ± 100 nm in width. The addition of PVA facilitated the formation of chitosan nanofibers, but only when the chitosan content was equal or less than 25% [
39]. In another report on a morphological study of PVA–CS, nanofibers were prepared by electrospinning and collected on reticulated vitreous carbon; nanofibers with diameters from 132 to 212 nm were obtained from a mixture of 8% PVA and 2% chitosan in 2% acetic acid solution [
40]. PVA–CS nanofibers produced by the electrospinning technique were also obtained from an acetic acid solution with up to 70% concentration and PVA: CS ratio of 70:30 [
41]. There are obviously major differences in PVA–CS nanofiber fabrication methods and results, so more detailed and specific studies are needed. In this study, we conducted our experiments step-by-step to estimate the correlation between the influence of the solution conditions and the electrospinning parameters on the formation of PVA–CS nanofibers.
4. Conclusions
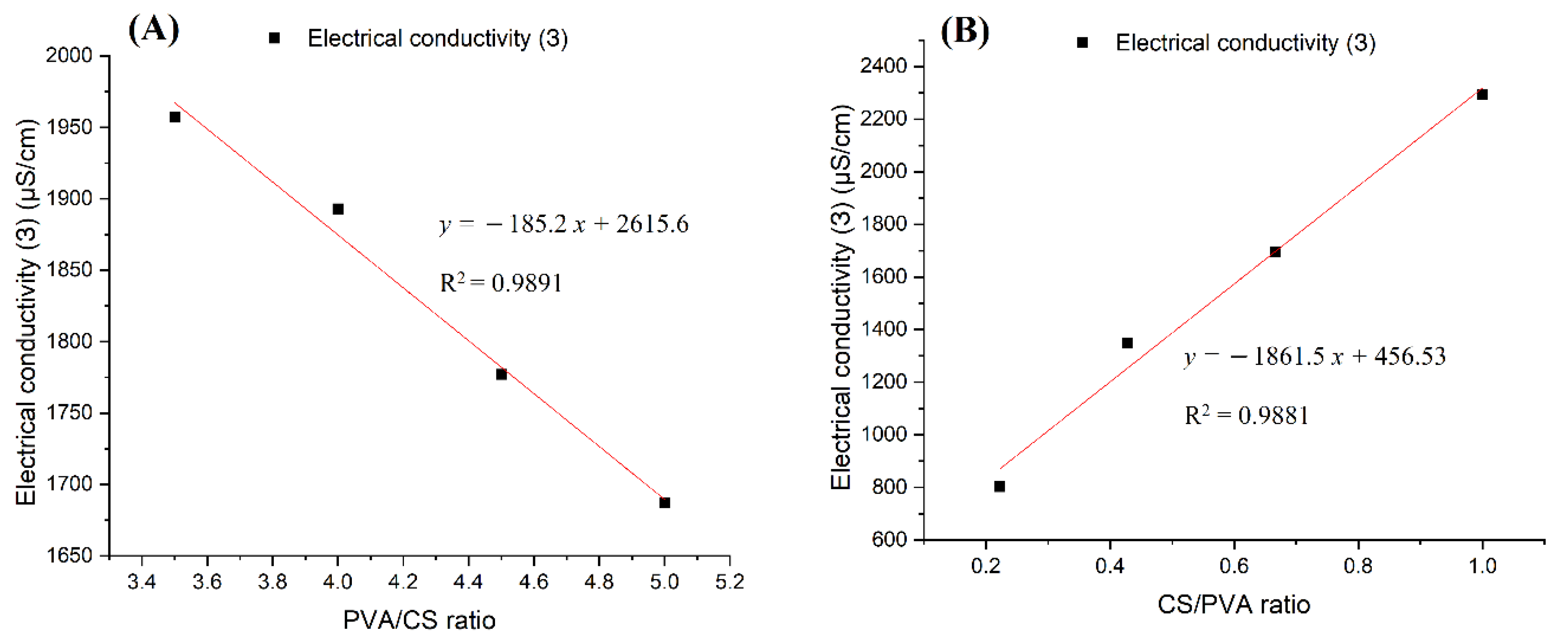
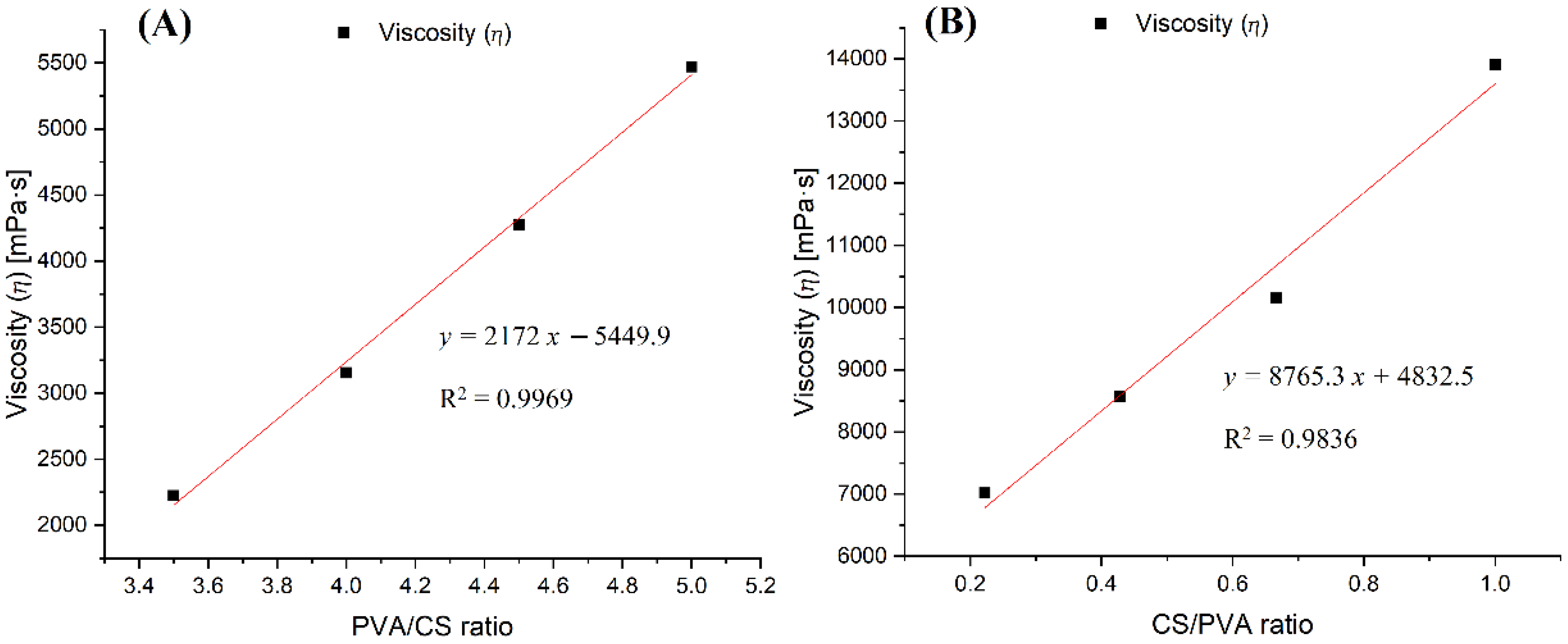
The investigation of nanofiber fabrication and rheological properties of the solutions containing PVA and CS polymers together with acetic acid as solvent showed that these factors were closely related to each other. Any increase in the concentration of either the acetic acid or the polymers led to an increase in the solution’s viscosity. While PVA and acetic acid reduced the conductivity of the solution, CS greatly increased the solution’s conductivity. For this reason, it is easier to obtain fibers from solutions with a high concentration of PVA; however, the fibers obtained from these solutions have larger sizes and more defects. In contrast, although requiring more precise electrospinning parameters, solutions with high CS content resulted in more nanoscale fibers.
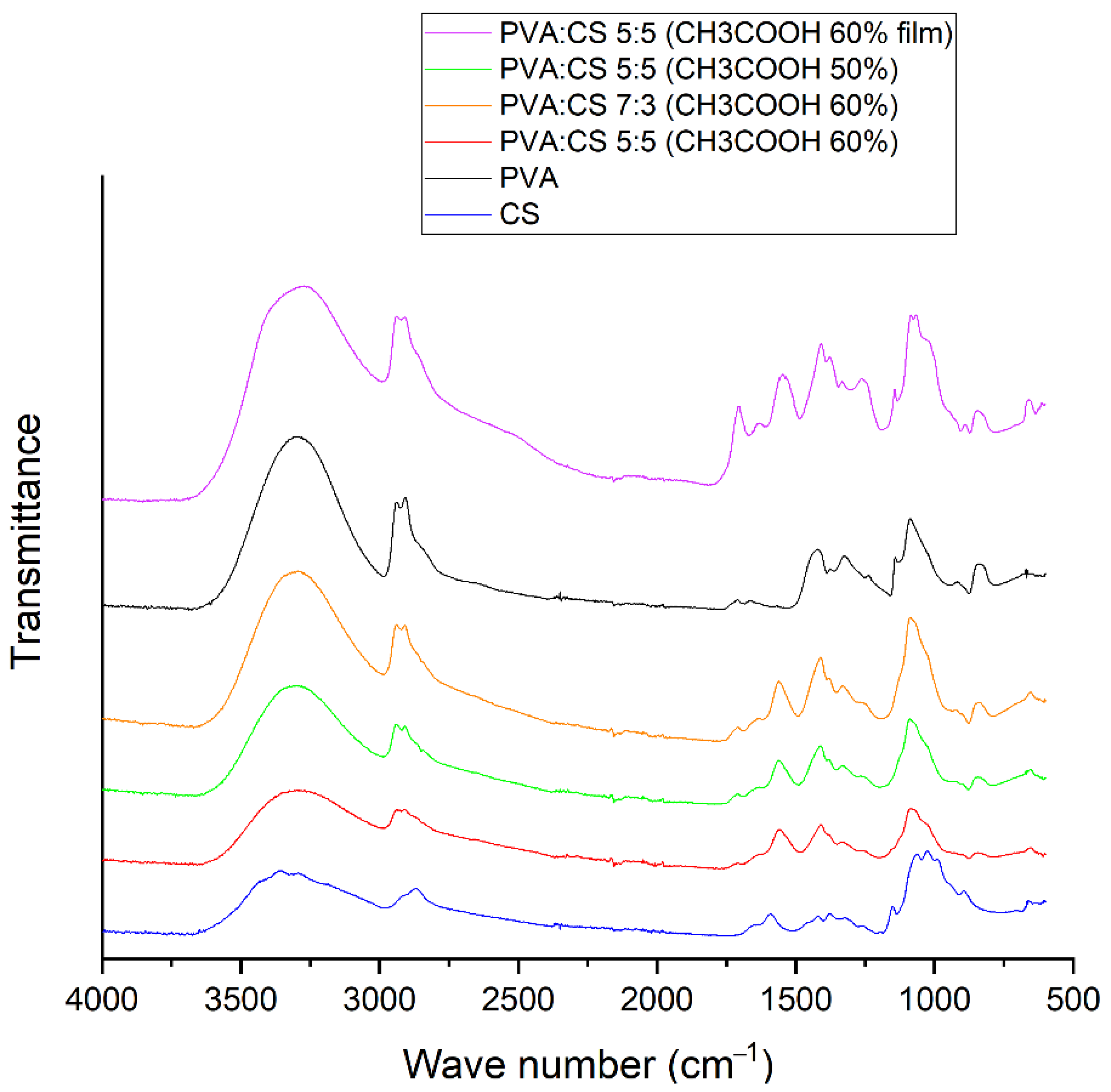
Infrared spectroscopy confirmed the complete separation of the acetic acid from the nanofibers, ensuring its safety for future drug integration purposes. The results of infrared spectrum analysis also showed that hydrogen bonds were the only type of bond formed between the polymers in the nanofibers.
PVA–CS nanofibers were successfully fabricated from a solution containing 5% (w/w) PVA; 5% (w/w) CS; and 60% (w/w) CH3COOH. The following electrospinning parameters were used: voltage 30 kV, feed rate 0.2 mL/h, needle-collector distance 140 mm. The obtained fibers exhibited diameters that were in the range of 77–292 nm, with an average diameter of 153 nm. These nanofiber systems had relatively few defects and could be used for the integration of drugs.
The formed PVA–CS nanofiber system holds promise as a potential material for the integration of therapeutic molecules.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}