
Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers



Abstract
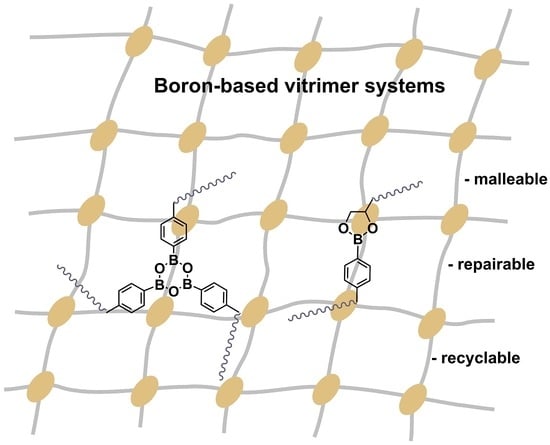
:
1. Introduction
2. Fundamentals of Vitrimers and Boronic Acid-Derived Dynamic Bonds
2.1. Boronic Esters
2.2. Boroxines
2.3. Vitrimers
3. Boronic Acid Esters-Based Vitrimers
3.1. Macromolecules Cross-Linking
3.1.1. Polyolefins
3.1.2. Diene Elastomers
3.1.3. Vinyl Thermoplastics
3.2. Small Molecule Resins
4. Boroxine-Based Vitrimers
4.1. Polysiloxane-Based Systems
4.2. Polyether-Based Systems
4.3. Vinyl Monomers Based Systems
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mark, H.F. Encyclopedia of Polymer Science and Technology, 3rd. ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007; ISBN 978-0470046104. [Google Scholar]
- Matyjaszewski, K. Macromolecular engineering: From rational design through precise macromolecular synthesis and processing to targeted macroscopic material properties. Prog. Polym. Sci. 2005, 30, 858–875. [Google Scholar] [CrossRef]
- Rajak, D.K.; Pagar, D.D.; Kumar, R.; Pruncu, C.I. Recent progress of reinforcement materials: A comprehensive overview of composite materials. J. Mater. Res. Technol. 2019, 8, 6354–6374. [Google Scholar] [CrossRef]
- Ruzette, A.V.; Leibler, L. Block copolymers in tomorrow’s plastics. Nat. Mater. 2005, 4, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Yu, K.; Kuang, X.; Mu, X.; Dunn, C.K.; Dunn, M.L.; Wang, T.; Jerry Qi, H. Recyclable 3D printing of vitrimer epoxy. Mater. Horizons 2017, 4, 598–607. [Google Scholar] [CrossRef]
- Spontak, R.J.; Patel, N.P. Thermoplastic elastomers: Fundamentals and applications. Curr. Opin. Colloid Interface Sci. 2000, 5, 333–340. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef]
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2010, 10, 14–27. [Google Scholar] [CrossRef]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef]
- Skene, W.G.; Lehn, J.M.P. Dynamers: Polyacylhydrazone reversible covalent polymers, component exchange, and constitutional diversity. Proc. Natl. Acad. Sci. USA 2004, 101, 8270–8275. [Google Scholar] [CrossRef] [Green Version]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-like malleable materials from permanent organic networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef]
- Röttger, M.; Domenech, T.; Van Der Weegen, R.; Breuillac, A.; Nicolaÿ, R.; Leibler, L. High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis. Science 2017, 356, 62–65. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, T.; Hao, C.; Wang, L.; Han, J.; Liu, H.; Zhang, J. Preparation of a lignin-based vitrimer material and its potential use for recoverable adhesives. Green Chem. 2018, 20, 2995–3000. [Google Scholar] [CrossRef]
- Imbernon, L.; Norvez, S. From landfilling to vitrimer chemistry in rubber life cycle. Eur. Polym. J. 2016, 82, 347–376. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, B.; Zhang, J. Recent development of repairable, malleable and recyclable thermosetting polymers through dynamic transesterification. Polymer 2020, 194, 122392. [Google Scholar] [CrossRef]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]
- Erice, A.; Ruiz de Luzuriaga, A.; Matxain, J.M.; Ruipérez, F.; Asua, J.M.; Grande, H.J.; Rekondo, A. Reprocessable and recyclable crosslinked poly(urea-urethane)s based on dynamic amine/urea exchange. Polymer 2018, 145, 127–136. [Google Scholar] [CrossRef]
- Zhang, Y.; Ying, H.; Hart, K.R.; Wu, Y.; Hsu, A.J.; Coppola, A.M.; Kim, T.A.; Yang, K.; Sottos, N.R.; White, S.R.; et al. Malleable and Recyclable Poly(urea-urethane) Thermosets bearing Hindered Urea Bonds. Adv. Mater. 2016, 28, 7646–7651. [Google Scholar] [CrossRef]
- Taynton, P.; Yu, K.; Shoemaker, R.K.; Jin, Y.; Jerry Qi, H.; Zhang, W.; Taynton, P.; Shoemaker, R.K.; Jin, Y.; Zhang, W.; et al. Heat- or Water-Driven Malleability in a Highly Recyclable Covalent Network Polymer. Adv. Mater. 2014, 26, 3938–3942. [Google Scholar] [CrossRef]
- Nishimura, Y.; Chung, J.; Muradyan, H.; Guan, Z. Silyl Ether as a Robust and Thermally Stable Dynamic Covalent Motif for Malleable Polymer Design. J. Am. Chem. Soc. 2017, 139, 14881–14884. [Google Scholar] [CrossRef]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Bapat, A.P.; Sumerlin, B.S.; Sutti, A. Bulk network polymers with dynamic B–O bonds: Healable and reprocessable materials. Mater. Horizons 2020, 7, 694–714. [Google Scholar] [CrossRef]
- Hall, D.G. Structure, Properties, and Preparation of Boronic Acid Derivatives: Overview of Their Reactions and Applications. In Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials (Volume 1 and 2); Wiley-VCH: Hoboken, NJ, USA, 2011; pp. 1–133. ISBN 9783527325986. [Google Scholar]
- Roy, C.D.; Brown, H.C. Stability of boronic esters—Structural effects on the relative rates of transesterification of 2-(phenyl)-1,3,2-dioxaborolane. J. Organomet. Chem. 2007, 692, 784–790. [Google Scholar] [CrossRef]
- Lor, J.P.; Edwards, J.O. Polyol Complexes and Structure of the Benzeneboronate Ion. J. Org. Chem. 2002, 24, 769–774. [Google Scholar] [CrossRef]
- Yoon, J.; Czarnik, A.W. Fluorescent Chemosensors of Carbohydrates. A Means of Chemically Communicating the Binding of Polyols in Water Based on Chelation-Enhanced Quenching. J. Am. Chem. Soc. 1992, 114, 5874–5875. [Google Scholar] [CrossRef]
- Barker, S.A.; Hatt, B.W.; Somers, P.J.; Woodbury, R.R. The use of poly(4-vinylbenzeneboronic acid) resins in the fractionation and interconversion of carbohydrates. Carbohydr. Res. 1973, 26, 55–64. [Google Scholar] [CrossRef]
- Wang, X.; Xia, N.; Liu, L. Boronic Acid-based approach for separation and immobilization of glycoproteins and its application in sensing. Int. J. Mol. Sci. 2013, 14, 20890–20912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaupp, G.; Naimi-Jamal, M.R.; Stepanenko, V. Waste-Free and Facile Solid-State Protection of Diamines, Anthranilic Acid, Diols, and Polyols with Phenylboronic Acid. Chem.—A Eur. J. 2003, 9, 4156–4161. [Google Scholar] [CrossRef]
- Gosecki, M.; Kazmierski, S.; Gosecka, M. Diffusion-Controllable Biomineralization Conducted in Situ in Hydrogels Based on Reversibly Cross-Linked Hyperbranched Polyglycidol. Biomacromolecules 2017, 18, 3418–3431. [Google Scholar] [CrossRef]
- Gosecka, M.; Gosecki, M.; Urbaniak, M. Composite Dynamic Hydrogels Constructed on Boronic Ester Cross-Links with NIR-Enhanced Diffusivity. Biomacromolecules 2022. [Google Scholar] [CrossRef]
- Dufort, B.M.; Tibbitt, M.W. Design of moldable hydrogels for biomedical applications using dynamic covalent boronic esters. Mater. Today Chem. 2019, 12, 16–33. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, Y. Boronic acid-containing hydrogels: Synthesis and their applications. J. Cite Chem. Soc. Rev. 2013, 42, 8106. [Google Scholar] [CrossRef]
- Yang, P.; Zhu, F.; Zhang, Z.; Cheng, Y.; Wang, Z.; Li, Y. Stimuli-responsive polydopamine-based smart materials. Chem. Soc. Rev. 2021, 50, 8319. [Google Scholar] [CrossRef]
- Hong, S.H.; Kim, S.; Park, J.P.; Shin, M.; Kim, K.; Ryu, J.H.; Lee, H. Dynamic Bonds between Boronic Acid and Alginate: Hydrogels with Stretchable, Self-Healing, Stimuli-Responsive, Remoldable, and Adhesive Properties. Biomacromolecules 2018, 19, 2053–2061. [Google Scholar] [CrossRef]
- Springsteen, G.; Wang, B. A detailed examination of boronic acid-diol complexation. Tetrahedron 2002, 58, 5291–5300. [Google Scholar] [CrossRef]
- Gamoh, K.; Brooks, C.J.W. Stability and Reversed-Phase Liquid Chromatographic Studies of Cyclic Boronates. Anal. Sci. 1993, 9, 549–552. [Google Scholar] [CrossRef] [Green Version]
- Sheepwash, E.; Luisier, N.; Krause, M.R.; Noé, S.; Kubik, S.; Severin, K. Supramolecular polymers based on dative boron-nitrogen bonds. Chem. Commun. 2012, 48, 7808–7810. [Google Scholar] [CrossRef]
- Ono, K.; Shimo, S.; Takahashi, K.; Yasuda, N.; Uekusa, H.; Iwasawa, N. Dynamic Interconversion between Boroxine Cages Based on Pyridine Ligation. Angew. Chemie—Int. Ed. 2018, 57, 3113–3117. [Google Scholar] [CrossRef]
- Campillo-Alvarado, G.; Vargas-Olvera, E.C.; Höpfl, H.; Herrera-España, A.D.; Sánchez-Guadarrama, O.; Morales-Rojas, H.; Macgillivray, L.R.; Rodríguez-Molina, B.; Farfán, N. Self-Assembly of Fluorinated Boronic Esters and 4,4′-Bipyridine into 2:1 N→B Adducts and Inclusion of Aromatic Guest Molecules in the Solid State: Application for the Separation of o, m, p -Xylene. Cryst. Growth Des. 2018, 18, 2726–2743. [Google Scholar] [CrossRef]
- Höpfl, H. The tetrahedral character of the boron atom newly defined—A useful tool to evaluate the N → B bond. J. Organomet. Chem. 1999, 581, 129–149. [Google Scholar] [CrossRef]
- Brus, J.; Czernek, J.; Urbanova, M.; Kobera, L.; Jegorov, A. An efficient 2D 11B-11B solid-state NMR spectroscopy strategy for monitoring covalent self-assembly of boronic acid-derived compounds: The transformation and unique architecture of bortezomib molecules in the solid state. Phys. Chem. Chem. Phys. 2017, 19, 487–495. [Google Scholar] [CrossRef]
- Yang, X.; Lee, M.C.; Sartain, F.; Pan, X.; Lowe, C.R. Designed Boronate Ligands for Glucose-Selective Holographic Sensors. Chem.—A Eur. J. 2006, 12, 8491–8497. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.L.A.; Sumerlin, B.S. Synthesis and Applications of Boronic Acid-Containing Polymers: From Materials to Medicine. Chem. Rev. 2015, 116, 1375–1397. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, O.R.; Chung, J.; Guan, Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137, 6492–6495. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G.; Lauer, M.; Böhnke, H. Rapid Proton Transfer as Cause of an Unusually Large Neighboring Group Effect. Angew. Chemie Int. Ed. Eng. 1984, 23, 741–742. [Google Scholar] [CrossRef]
- Yang, Y.; Du, F.S.; Li, Z.C. Thermally healable and reprocessable polymethacrylate networks based on diol-mediated metathesis of 6-membered boronic esters. Polym. Chem. 2020, 11, 1860–1870. [Google Scholar] [CrossRef]
- Brunet, J.; Collas, F.; Humbert, M.; Perrin, L.; Brunel, F.; Lacôte, E.; Montarnal, D.; Raynaud, J. High Glass-Transition Temperature Polymer Networks Harnessing the Dynamic Ring Opening of Pinacol Boronates. Angew. Chemie.—Int. Ed. 2019, 58, 12216–12222. [Google Scholar] [CrossRef]
- Kua, J.; Iovine, P.M. Formation of para-substituted triphenylboroxines: A computational study. J. Phys. Chem. A 2005, 109, 8938–8943. [Google Scholar] [CrossRef]
- Brock, C.P.; Minton, R.P.; Niedenzu, K. Structure and thermal motion of triphenylboroxin. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1987, 43, 1775–1779. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, Y. Boroxine chemistry: From fundamental studies to applications in supramolecular and synthetic organic chemistry. Heterocycles 2013, 87, 991–1021. [Google Scholar] [CrossRef] [Green Version]
- Kua, J.; Fletcher, M.N.; Iovine, P.M. Effect of para-substituents and solvent polarity on the formation of triphenylboroxine·amine adducts. J. Phys. Chem. A 2006, 110, 8158–8166. [Google Scholar] [CrossRef]
- Matteson, D.S. The Preparation of Di-tert-butyl Ethyleneboronate via the Ethyleneboronic Anhydride—Pyridine Complex. J. Org. Chem. 1962, 27, 3712. [Google Scholar] [CrossRef]
- Lai, J.C.; Mei, J.F.; Jia, X.Y.; Li, C.H.; You, X.Z.; Bao, Z. A Stiff and Healable Polymer Based on Dynamic-Covalent Boroxine Bonds. Adv. Mater. 2016, 28, 8277–8282. [Google Scholar] [CrossRef]
- Iovine, P.M.; Gyselbrecht, C.R.; Perttu, E.K.; Klick, C.; Neuwelt, A.; Loera, J.; DiPasquale, A.G.; Rheingold, A.L.; Kua, J. Hetero-arylboroxines: The first rational synthesis, X-ray crystallographic and computational analysis. Dalt. Trans. 2008, 3791–3794. [Google Scholar] [CrossRef]
- Ogden, W.A.; Guan, Z. Recyclable, Strong, and Highly Malleable Thermosets Based on Boroxine Networks. J. Am. Chem. Soc. 2018, 140, 6217–6220. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Nicolaÿ, R. Vitrimers: Permanently crosslinked polymers with dynamic network topology. Prog. Polym. Sci. 2020, 104, 101233. [Google Scholar] [CrossRef]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. Temperature Dependence of Relaxation Mechanisms The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-forming Liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Ngai, K.L.; Plazek, D.J. Relation of internal rotational isomerism barriers to the flow activation energy of entangled polymer melts in the high-temperature Arrhenius region. J. Polym. Sci. Polym. Phys. Ed. 1985, 23, 2159–2180. [Google Scholar] [CrossRef]
- Liu, C.Y.; He, J.; Keunings, R.; Bailly, C. New Linearized Relation for the Universal Viscosity−Temperature Behavior of Polymer Melts. Macromolecules 2006, 39, 8867–8869. [Google Scholar] [CrossRef]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic control of the vitrimer glass transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef]
- Chen, M.; Si, H.; Zhang, H.; Zhou, L.; Wu, Y.; Song, L.; Kang, M.; Zhao, X.-L. The Crucial Role in Controlling the Dynamic Properties of Polyester-Based Epoxy Vitrimers: The Density of Exchangeable Ester Bonds (υ). Macromolecules 2021, 54, 10110–10117. [Google Scholar] [CrossRef]
- Soman, B.; Evans, C.M. Effect of precise linker length, bond density, and broad temperature window on the rheological properties of ethylene vitrimers. Soft Matter 2021, 17, 3569–3577. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Winne, J.M.; Du Prez, F.E. Vitrimers: Directing chemical reactivity to control material properties. Chem. Sci. 2020, 11, 4855–4870. [Google Scholar] [CrossRef] [Green Version]
- Smallenburg, F.; Leibler, L.; Sciortino, F. Patchy particle model for vitrimers. Phys. Rev. Lett. 2013, 111. [Google Scholar] [CrossRef]
- Singh, G.; Sundararaghavan, V. Modeling self-healing behavior of vitrimers using molecular dynamics with dynamic cross-linking capability. Chem. Phys. Lett. 2020, 760, 137966. [Google Scholar] [CrossRef]
- Perego, A.; Khabaz, F. Effect of bond exchange rate on dynamics and mechanics of vitrimers. J. Polym. Sci. 2021, 59, 2590–2602. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Shanbhag, S. Unentangled Vitrimer Melts: Interplay between Chain Relaxation and Cross-link Exchange Controls Linear Rheology. Macromolecules 2021, 54, 3304–3320. [Google Scholar] [CrossRef]
- Ciarella, S.; Sciortino, F.; Ellenbroek, W.G. Dynamics of Vitrimers: Defects as a Highway to Stress Relaxation. Phys. Rev. Lett. 2018, 121, 058003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Terentjev, E.M. Transient Network at Large Deformations: Elastic–Plastic Transition and Necking Instability. Polymers 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perego, A.; Khabaz, F. Volumetric and Rheological Properties of Vitrimers: A Hybrid Molecular Dynamics and Monte Carlo Simulation Study. Macromolecules 2020, 53, 8406–8416. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- McGregor, R.R.; Warrick, E.L. Treating Dimethyl Silicone Polymer with Boric Oxide. US Patent 2,431, 878 1947. [Google Scholar]
- Wright, J. Process for Making Puttylike Elastic Plastic, Siloxane Derivative Composition Containing Zinc Hydroxide. U.S. Patent 2,541,851, 13 February 1951. [Google Scholar]
- Boland, C.S.; Khan, U.; Ryan, G.; Barwich, S.; Charifou, R.; Harvey, A.; Backes, C.; Li, Z.; Ferreira, M.S.; Möbius, M.E.; et al. Sensitive electromechanical sensors using viscoelastic graphene-polymer nanocomposites. Science 2016, 354, 1257–1260. [Google Scholar] [CrossRef]
- Chen, X.; Dam, M.A.; Ono, K.; Mal, A.; Shen, H.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef]
- Niu, W.; O’Sullivan, C.; Rambo, B.M.; Smith, M.D.; Lavigne, J.J. Self-repairing polymers: Poly(dioxaborolane)s containing trigonal planar boron. Chem. Commun. 2005, 4342–4344. [Google Scholar] [CrossRef]
- Govorun, E.N.; Chertovich, A.V. Microphase Separation in Random Multiblock Copolymers. Cit. J. Chem. Phys. 2017, 146, 34903. [Google Scholar] [CrossRef] [Green Version]
- Chum, P.S.; Swogger, K.W. Olefin polymer technologies—History and recent progress at The Dow Chemical Company. Prog. Polym. Sci. 2008, 33, 797–819. [Google Scholar] [CrossRef]
- Collins, B.E.; Metola, P.; Anslyn, E.V. On the rate of boronate ester formation in ortho-aminomethyl-functionalised phenyl boronic acids. Supramol. Chem. 2013, 25, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Korley, L.S.T.J.; Epps, T.H.; Helms, B.A.; Ryan, A.J. Toward polymer upcycling-adding value and tackling circularity. Science 2021, 373, 66–69. [Google Scholar] [CrossRef]
- Caffy, F.; Nicolaÿ, R. Transformation of polyethylene into a vitrimer by nitroxide radical coupling of a bis-dioxaborolane. Polym. Chem. 2019, 10, 3107–3115. [Google Scholar] [CrossRef]
- Yang, F.; Pan, L.; Ma, Z.; Lou, Y.; Li, Y.; Li, Y. Highly elastic, strong, and reprocessable cross-linked polyolefin elastomers enabled by boronic ester bonds. Polym. Chem. 2020, 11, 3285–3295. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.; Zhang, X.; Liu, Y.; Wu, S.; Guo, B. Covalently Cross-Linked Elastomers with Self-Healing and Malleable Abilities Enabled by Boronic Ester Bonds. ACS Appl. Mater. Interfaces 2018, 10, 24224–24231. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.; Liu, Y.; Wu, S.; Guo, B. Mechanically Robust, Self-Healable, and Reprocessable Elastomers Enabled by Dynamic Dual Cross-Links. Macromolecules 2019, 52, 3805–3812. [Google Scholar] [CrossRef]
- Chung, J.; Kushner, A.M.; Weisman, A.C.; Guan, Z. Direct correlation of single-molecule properties with bulk mechanical performance for the biomimetic design of polymers. Nat. Mater. 2014, 13, 1055–1062. [Google Scholar] [CrossRef]
- Nabavi, S.S.; Harrington, M.J.; Fratzl, P.; Hartmann, M.A. Influence of sacrificial bonds on the mechanical behaviour of polymer chains. Bioinspired Biomim. Nanobiomaterials 2014, 3, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tang, Z.; Wang, D.; Wu, S.; Guo, B. Biomimetic design of elastomeric vitrimers with unparalleled mechanical properties, improved creep resistance and retained malleability by metal-ligand coordination. J. Mater. Chem. A 2019, 7, 26867–26876. [Google Scholar] [CrossRef]
- Kim, C.; Ejima, H.; Yoshie, N. Non-swellable self-healing polymer with long-term stability under seawater. RSC Adv. 2017, 7, 19288–19295. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ejima, H.; Yoshie, N. Polymers with autonomous self-healing ability and remarkable reprocessability under ambient humidity conditions. J. Mater. Chem. A 2018, 6, 19643–19652. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Duong, N.T.; Li, X.; Nishiyama, Y.; Wu, Q.; Zhang, R.; Sun, P. Using Dynamic Bonds to Enhance the Mechanical Performance: From Microscopic Molecular Interactions to Macroscopic Properties. Macromolecules 2019, 52, 5014–5025. [Google Scholar] [CrossRef]
- Wang, Z.; Gu, Y.; Ma, M.; Chen, M. Strong, Reconfigurable, and Recyclable Thermosets Cross-Linked by Polymer-Polymer Dynamic Interaction Based on Commodity Thermoplastics. Macromolecules 2020, 53, 956–964. [Google Scholar] [CrossRef]
- Ricarte, R.G.; Tournilhac, F.; Leibler, L. Phase Separation and Self-Assembly in Vitrimers: Hierarchical Morphology of Molten and Semicrystalline Polyethylene/Dioxaborolane Maleimide Systems. Macromolecules 2019, 52, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, B.; Zhang, X.; Wang, L.; Fan, W.; Li, H.; Bian, C.; Jing, X. Fully recyclable and reprocessable polystyrene-based vitrimers with improved thermal stability and mechanical properties through nitrogen-coordinating cyclic boronic ester bonds. Appl. Surf. Sci. 2021, 570, 151157. [Google Scholar] [CrossRef]
- Song, K.; Ye, W.; Gao, X.; Fang, H.; Zhang, Y.; Zhang, Q.; Li, X.; Yang, S.; Wei, H.; Ding, Y. Synergy between dynamic covalent boronic ester and boron-nitrogen coordination: Strategy for self-healing polyurethane elastomers at room temperature with unprecedented mechanical properties. Mater. Horiz. 2021, 8, 216–223. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Bapat, A.P.; Sumerlin, B.S. Room-temperature self-healing polymers based on dynamic-covalent boronic esters. Macromolecules 2015, 48, 2098–2106. [Google Scholar] [CrossRef]
- Cash, J.J.; Kubo, T.; Dobbins, D.J.; Sumerlin, B.S. Maximizing the symbiosis of static and dynamic bonds in self-healing boronic ester networks. Polym. Chem. 2018, 9, 2011–2020. [Google Scholar] [CrossRef]
- Robinson, L.L.; Self, J.L.; Fusi, A.D.; Bates, M.W.; Read De Alaniz, J.; Hawker, C.J.; Bates, C.M.; Sample, C.S. Chemical and Mechanical Tunability of 3D-Printed Dynamic Covalent Networks Based on Boronate Esters. ACS Macro Lett. 2021, 10, 857–863. [Google Scholar] [CrossRef]
- Zych, A.; Tellers, J.; Bertolacci, L.; Ceseracciu, L.; Marini, L.; Mancini, G.; Athanassiou, A. Biobased, Biodegradable, Self-Healing Boronic Ester Vitrimers from Epoxidized Soybean Oil Acrylate. ACS Appl. Polym. Mater. 2021, 3, 1135–1144. [Google Scholar] [CrossRef]
- Zeng, Y.; Liu, S.; Xu, X.; Chen, Y.; Zhang, F. Fabrication and curing properties of o-cresol formaldehyde epoxy resin with reversible cross-links by dynamic boronic ester bonds. Polymer 2020, 211, 123116. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Jiang, Z.; Li, Y.; Jing, X. Boronic Ester Based Vitrimers with Enhanced Stability via Internal Boron-Nitrogen Coordination. J. Am. Chem. Soc. 2020, 142, 21852–21860. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, A.; Lin, Y. Reprocessability of dynamic polydioxaborolane networks activated by heat, moisture and mechanical force. Polymer 2020, 209, 123037. [Google Scholar] [CrossRef]
- Côté, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Chemistry: Porous, crystalline, covalent organic frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.S.; Bein, T. Covalent Organic Frameworks: Structures, Synthesis, and Applications. Adv. Funct. Mater. 2018, 28. [Google Scholar] [CrossRef] [Green Version]
- Mehta, M.A.; Fujinami, T.; Yang, Y.; Inoue, T. Ionic conductivity and interfacial properties of polymer electrolytes based on PEO and boroxine ring polymer. J. Appl. Polym. Sci. 2002, 84, 17–21. [Google Scholar] [CrossRef]
- Li, Y.; Ding, J.; Day, M.; Tao, Y.; Lu, J.; D’Iorio, M. Novel Stable Blue-Light-Emitting Oligofluorene Networks Immobilized by Boronic Acid Anhydride Linkages. Chem. Mater. 2003, 15, 4936–4943. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, L.; Wu, R.; Fan, W.; Dai, Q.; He, J.; Bai, C. Assembling of Reprocessable Polybutadiene-Based Vitrimers with High Strength and Shape Memory via Catalyst-Free Imine-Coordinated Boroxine. ACS Appl. Mater. Interfaces 2020, 12, 33305–33314. [Google Scholar] [CrossRef]
- Yang, X.; Guo, M.; Wu, Y.; Xue, S.; Li, Z.; Zhou, H.; Smith, A.T.; Sun, L. Biomimetic Boroxine-Based Multifunctional Thermosets via One-Pot Synthesis. ACS Appl. Mater. Interfaces 2020, 12, 56445–56453. [Google Scholar] [CrossRef]
- Hoogenboom, R. Hard autonomous self-healing supramolecular materials-A contradiction in terms? Angew. Chemie - Int. Ed. 2012, 51, 11942–11944. [Google Scholar] [CrossRef]
- Chen, L.; Chu, D.; Cheng, Z.A.; Wang, M.; Huang, S. Designing seamless-welded liquid-crystalline soft actuators with a “glue-free” method by dynamic boroxines. Polymer 2020, 208, 122962. [Google Scholar] [CrossRef]
- Yu, H.; Feng, Y.; Gao, L.; Chen, C.; Zhang, Z.; Feng, W. Self-Healing High Strength and Thermal Conductivity of 3D Graphene/PDMS Composites by the Optimization of Multiple Molecular Interactions. Macromolecules 2020, 53, 7161–7170. [Google Scholar] [CrossRef]
- Liang, S.; Huang, Y.; Yuan, Y.; Wang, Y.; Yang, B.; Zhao, X.; Liu, L. Development of a Strong, Recyclable Poly(dimethylsiloxane) Elastomer with Autonomic Self-Healing Capabilities and Fluorescence Response Properties at Room Temperature. Macromol. Mater. Eng. 2021, 306, 1–10. [Google Scholar] [CrossRef]
- Delpierre, S.; Willocq, B.; De Winter, J.; Dubois, P.; Gerbaux, P.; Raquez, J.M. Dynamic Iminoboronate-Based Boroxine Chemistry for the Design of Ambient Humidity-Sensitive Self-Healing Polymers. Chem.—A Eur. J. 2017, 23, 6730–6735. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Jiang, Y.J.; Zhang, H.; Lu, X.; Sun, J. Room-Temperature Self-Healing and Recyclable Tough Polymer Composites Using Nitrogen-Coordinated Boroxines. Adv. Funct. Mater. 2018, 28, 1800560. [Google Scholar] [CrossRef]
- Bao, C.; Guo, Z.; Sun, H.; Sun, J. Nitrogen-Coordinated Boroxines Enable the Fabrication of Mechanically Robust Supramolecular Thermosets Capable of Healing and Recycling under Mild Conditions. ACS Appl. Mater. Interfaces 2019, 11, 9478–9486. [Google Scholar] [CrossRef] [PubMed]
- Delpierre, S.; Willocq, B.; Manini, G.; Lemaur, V.; Goole, J.; Gerbaux, P.; Cornil, J.; Dubois, P.; Raquez, J.M. Simple Approach for a Self-Healable and Stiff Polymer Network from Iminoboronate-Based Boroxine Chemistry. Chem. Mater. 2019, 31, 3736–3744. [Google Scholar] [CrossRef]
- Yuan, D.; Delpierre, S.; Ke, K.; Raquez, J.M.; Dubois, P.; Manas-Zloczower, I. Biomimetic Water-Responsive Self-Healing Epoxy with Tunable Properties. ACS Appl. Mater. Interfaces 2019, 11, 17853–17862. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Bao, C.; Xie, P.; Guo, Z.; Sun, J. Solution-Processable and Thermostable Super-Strong Poly(aryl ether ketone) Supramolecular Thermosets Cross-Linked with Dynamic Boroxines. Adv. Funct. Mater. 2021, 31, 2103061. [Google Scholar] [CrossRef]
- Andyopadhyay, A.; Ao, J.; Bandyopadhyay, A.; Gao, J. Iminoboronate Formation Leads to Fast and Reversible Conjugation Chemistry of α-Nucleophiles at Neutral pH. Chem.—A Eur. J. 2015, 21, 14748–14752. [Google Scholar] [CrossRef] [Green Version]
- Li, S.S.; Lv, X.H.; Sun, X.L.; Wan, W.M.; Bao, H. Well-controlled polymerization of tri-vinyl dynamic covalent boroxine monomer: One dynamic covalent boroxine moiety toward a tunable penta-responsive polymer. Polym. Chem. 2020, 11, 2914–2922. [Google Scholar] [CrossRef]
- Huang, Q.; Tang, Z.; Wang, D.; Wu, S.; Guo, B. Engineering Segregated Structures in a Cross-Linked Elastomeric Network Enabled by Dynamic Cross-Link Reshuffling. ACS Macro Lett. 2021, 10, 231–236. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Properties | References |
|---|---|---|
| cyclooctene, cyclooct-5-ene-1,2-diol copolymer (20% diol copolymer) crossilineg with (((hexane-1,6-diylbis(methylazanediyl))bis(methylene))bis(2,1-phenylene))diboronic acid 1,2-dihydroxypropane ester (3) | YM 4.68 MPa, TS, 1.85 MPa, SatB 345%; self-healing at 50 °C, 95% HE; remoldable at 80 °C | [45] |
| HDPE modified with 1-[(2-phenyl-1,3,2-dioxaborolan-4-yl)methyl]-1H-pyrrole-2,5-dione, dioxaborolane functionalized maleimid, cross-linked with 2,2′-(1,4-phenylene)bis[4-methyl-1,3,2-dioxaborolane] (2) | YM 0.5 GPa, TS 20 MPa, SatB 700%; extrudable | [12] |
| HDPE cross-linked with 4,4′-(((1,4-phenylenebis(1,3,2-dioxaborolane-2,4-diyl))bis-(methylene))bis(oxy))bis(2,2,6,6-tetramethyl-piperidin-1-oxyl) (7) | YM 444 MPa, TS 19.5 MPa, SatB 675%; extrudable | [82] |
| polyolefin elastomers: ethylene, 1-tetradecane, 9-(but-3-en-1-yl)anthracene copolymer modified with dioxaborolane maleimide, cross-linked with 2,2′-(1,4-phenylene)bis[4-methyl-1,3,2-dioxaborolane] (2) | YM 4.2 MPa, TS 15.6 MPa, EatB 1270%; remoldable at 200 °C | [83] |
| styrene-btutadiene rubber cross-linked with 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (6) | YM 1.68–2,43 MPa, TS 1.70–2.68 MPa, SatB 498–215%, at 1 to 5% cross-linker fraction; over 80% HE at 80 °C after 24 h; remoldable at 160 °C under 10 MPa | [84] |
| epoxidized natural rubber cross-linked with 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (6) in the presence of DMAP | YM 0.85–4.45 MPa, TS 1.60–14.63 MPa, SatB 811–475%, at 1–10% cross-linker fraction; over 80% HE at 80 °C after 24 h; the addition of Zn2+ ions to form sacrificial bonds, remoldable at 160 °C under 10 MPa load | [85] |
| SBR modified with 2-(2-benzimidazolyl)ethanethiol (3.3%), cross-linked with 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (6), with the addition Zn2+ ions | YM 1.1–3.9 MPa, TS, 4.0–18.6 MPa, SatB 404–355%, at 0 to 1.6% Zn; remoldable at 160 °C under 10 MPa | [88] |
| 1: styrene, (5,6-dioxaborolane)hexyl methacrylate copolymers 2: methyl methacrylate, (5,6-dioxaborolane)hexyl methacrylate copolymers cross-linked with is 2,2′-(1,4-phenylene)bis[4-methyl-1,3,2-dioxaborolane] (3) | 1: YM ~ 1.5 GPa, TS ~ 25 MPa2: YM > 1.5 GPa, TS > 50 MPa; extrudable | [12] |
| poly(dopamine acrylamide-co-n-butyl acrylate) 10 mol % dopamine units, Mn = 79,000, cross-linked with benzene-1,4-diboronic acid (1) in the presence of triethylamine | YM 13.1 MPa, TS 6.7 0.1 MPa, SatB 230 30%; H2O triggered healing at RT, or at 60 °C under 10 MPa without activation | [90] |
| buthyl methacrylate, 2-(2-ureido-4[1H]-6-methylpyrimidinone)ethyl methacrylate, 2,3-dihydroxypropyl methacrylate and benzene-1,4-diboronic acid bis(2,3-dihydroxypropyl methacrylate) ester (5) | YM 13 MPa, TS 5.5 MPa, SatB 230%; 90%HE at 100 °C after 48 h; remoldable at 110 °C | [91] |
| mixture of poly(styrene-co-dioxaborolane 2,3-dihydroxypropyl methacrylate) and poly(styrene-co-4-vinylphenylboronic acid 1,2-propanediol ester) | YM 2 GPa, TS 65 MPa, EatB, 4%; remoldalble at 200 °C under 2 MPa | [92] |
| poly(butyl methacrylate) and butyl methacrylate copolymers with diol-containing methracylic monomers cross-linked with (5-ethyl-2-(4-((methacryloyloxy)methyl)phenyl)-1,3,2-dioxaborinan-5-yl) methyl methacrylate (9) | YM 301.7 MPa, TS 7.7 MPa, SatB 86%; remoldable at 150 °C under 8 MPa; addition of 5 mol % 2,3-dihydroxypropyl methacrylate: YM 572.4 MPa, TS 19.8 MPa, SatB 93%; remoldable at 120 °C under 8 MPa | [47] |
| poly(styrene-co-HEMA) cross-linked with 4-(6-((6-isocyanatohexyl)carbamoyl)-1,3,6,2-dioxazaborocan-2-yl)benzyl (6-isocyanatohexyl)carbamate (12) | YM 1.86 GPa, TS 44.7 MPa. SatB 4.51%; remoldable at 180 °C, under 1 MPa | [94] |
| Composition | Properties | References |
|---|---|---|
| 4-((allyloxy)methyl)-2-(4-vinylphenyl)-1,3,2-dioxaborolane (8) pentaerythritol tetrakis(3-mercaptopropionate) 3,6-dioxa-1,8-octanedithiol | TS 1.5–3 MPa SatB 40–50% H2O triggered healing at RT | [96] |
| hexamethylene diisocyanate trimer and: 1: 4-hydroxyethylphenylboronic acid triethanolamine ester (10) 2: 4-hydroxyethylphenylboronic acid diethanolamine ester (11) | 1: YM 0.73 GPa, TS 41.21 MPa, SatB 12.44%; 2: YM 1.10 GPa, TS 38.93 MPa, SatB 4.70% | [101] |
| divinyltetramethyldisiloxane and 1,3,5,7-tetravinyl- 1,3,5,7- tetramethylcyclotetrasiloxane cross-linked with benzene-1,4-diboronic acid (1) | YM 11.4–545 MPa, TS 1.81–30 MPa, SatB 717–12%; soft vitrimers remoldable at 35 °C under 30 MPa, stiff ones at 120 °C under 20 MPa; | [102] |
| soybean oil acrylate and [2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (6) | stretches to fibers; self-healing at RT; remoldable at 120 °C | [99] |
| o-Cresol Formaldehyde Epoxy Resin cross-linked with [2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (6) | YM 1.25 MPa, TS 3.99 MPa, SatB 3.2%; 95%-HE after 12 h at 160 °C; remoldable at 200 °C under 10 MPa, | [100] |
| Composition | Properties | References |
|---|---|---|
| NH2-terminated PDMS (Mn = 700–900) cross-linked with 4,4′,4″-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzoyl chloride | YM 182 MPa, TS 9.46 MPa, SatB 9.72%; H2O triggered healing at 70 °C, HE 95% | [54] |
| polymethylhydrosiloxane modified with 4-methoxyphenyl-4-(1-buteneoxy) benzoate mesogen and cross-linked with 2,4,6-tris(4-(but-3-en-1-yloxy)phenyl)-1,2,3,4,5,6-trioxatriborinane | YM 0.53 MPa, TS 0.29 MPa, SatB 58.2%; weldable at 45 °C | [110] |
| NH2-terminated PDMS (Mn = 800−900) modified with 2-ureido-4[1H]-pyrimidinone, cross-linked with 4,4′,4″-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl) tribenzoyl chloride | YM 130.46;TS 7.33; SatB 12%; HE 98%; composite with UPy modified grapheme aerogel: YM 425.65; TS 39.63, SatB 7.5%; HE 79%; H2O triggerd healing at 40 °C | [111] |
| PDMS with aminopropyl pendant groups cross-linked with 4,4′,4″-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzoyl chloride | YM 11.1 ± 0.52 MPa, TS 1.61 MPa, SatB 307%; 91% healing efficiency at RT | [112] |
| polybutadiene (Mn = 1200) modified with 2-aminoethanethiol and 2-formylphenylboronic acid, cross-linked by dehydration | YM 10–241 MPa, TS 0.62–12.35 MPa, SatB 216–29%, at cross-linking density from 6 to 12% remoldable at 70 °C under 4 MPa; | [107] |
| NH2-terminated PPG (Mn = 400 or 2000) cross-linked with 2,2′,2″-((1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzaldehyde | YM 61 MPa TS ~ 4 MPa, SatB ~ 300% H2O triggered healing, HE 93% | [113] |
| NH2-terminated PPG (Mn 2000) modified with 2-formylphenylboronic acid with subsequent imine bonds reduction, mixed with poly(acrylic acid) (Mn 450,000), cross-linked by dehydration. | YM 2.72–11,45 MPa, TS 1,75–9.16 MPa, SatB 659–182%, at 6 to 40% PAA content; H2O triggered healing at RT; remoldalbe at RT under 0.4 kPa for 24 h | [114] |
| linear and tri-arm NH2-terminated PPG (Mn ~ 400) modified with 2-formylphenylboronic acid and reduced imine bonds, cross-linked by dehydration | YM 63.9–298.5 MPa, TS 5.95–31.96 MPa, SatB 376–36%; H2O triggered healing at 55 °C; remoldalbe at 60 °C under 4 MPa | [115] |
| NH2 -terminated PPG-based polyhydroxyurethane cross-linked with 2,2′,2″-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzaldehyde | YM 551 MPa, TS 11 MPa, SatB 3%; H2O triggered healing at RT or without H2O at 70 °C; remoldable at 80 °C under 1 MPa | [116] |
| NH2 -terminated PPG cross-linked with Bisphenol A diglycidyl ether and 2,2′,2″-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tribenzaldehyde | YM 1059 MPa, TS 37 MPa, SatB 6.77%; H2O triggered healing at 80 °C; 80% HE | [117] |
| ((((oxybis(ethane-2,1-diyl))bis(oxy))bis(methylene))bis(4,1-phenylene))diboronic acid bisphenylboronic acid bind with diethylene glycol) cross-linked by dehydration in the presence of pyridine | YM 559 MPa, TS 17.8 MPa; remoldable at 80 °C | [56] |
| poly(aryl ether ketone) (Mn 5100–17,600) end-capped with 4-hydroxyphenylboronic acid, cross-linked by dehydration | YM 1.59–4.1 GPa, TS 60.5.95–97.8 MPa, SatB 7–0.7%; solvent (THF/ethanol) assisted processsing | [118] |
| 4-vinylphenylboronic acid and octadecanoxy polyethylene glycol methacrylate free radical copolymerization | YM 255.0 MPa, TS 27.5 MPa, SatB 21%; H2O triggered healing at 70 °C 98% HE; strong adhesion to steel and titanium | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gosecki, M.; Gosecka, M. Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers. Polymers 2022, 14, 842. https://doi.org/10.3390/polym14040842
Gosecki M, Gosecka M. Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers. Polymers. 2022; 14(4):842. https://doi.org/10.3390/polym14040842
Chicago/Turabian StyleGosecki, Mateusz, and Monika Gosecka. 2022. "Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers" Polymers 14, no. 4: 842. https://doi.org/10.3390/polym14040842
APA StyleGosecki, M., & Gosecka, M. (2022). Boronic Acid Esters and Anhydrates as Dynamic Cross-Links in Vitrimers. Polymers, 14(4), 842. https://doi.org/10.3390/polym14040842