

DFT Calculation and MD Simulation Studies on Gemini Surfactant Corrosion Inhibitor in Acetic Acid Media


 , and
, and
Abstract
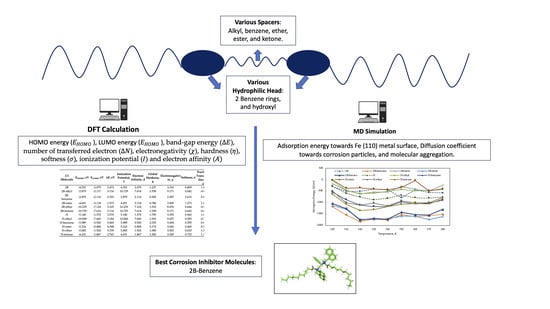
:
1. Introduction
2. Methodology
2.1. DFT Calculation
2.2. MD Simulation
3. Results and Discussion
3.1. Method Validation
3.2. DFT Calculation
3.3. MD Simulation
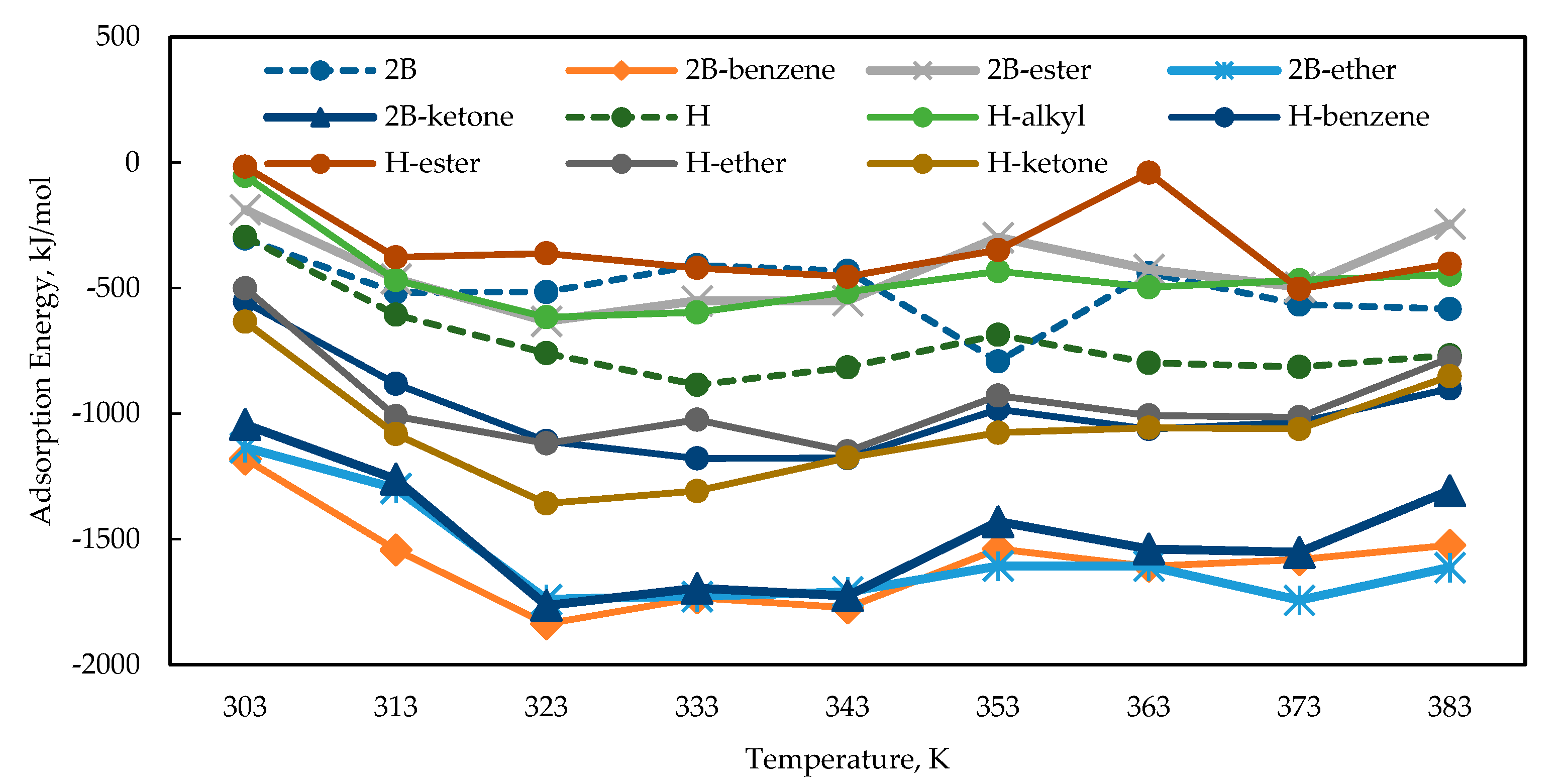
3.3.1. Adsorption Energy
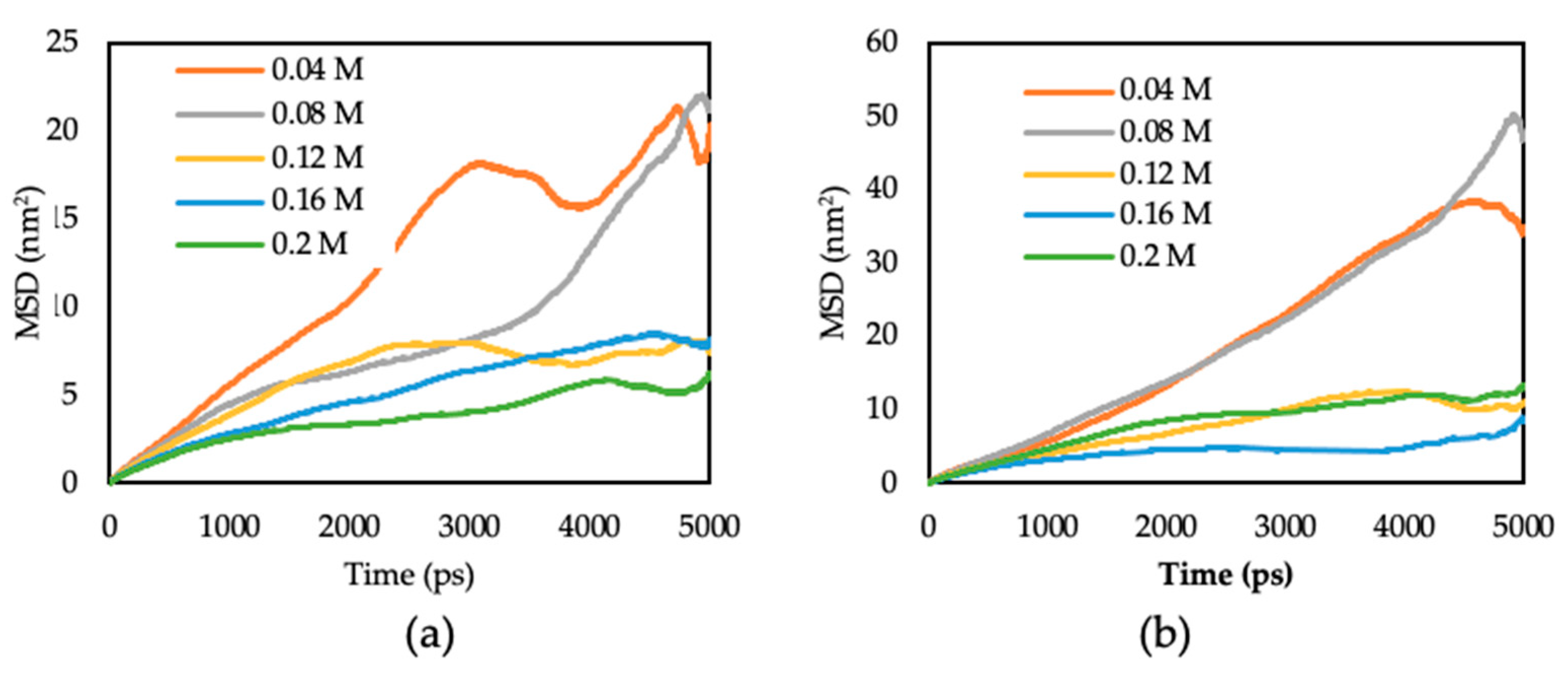
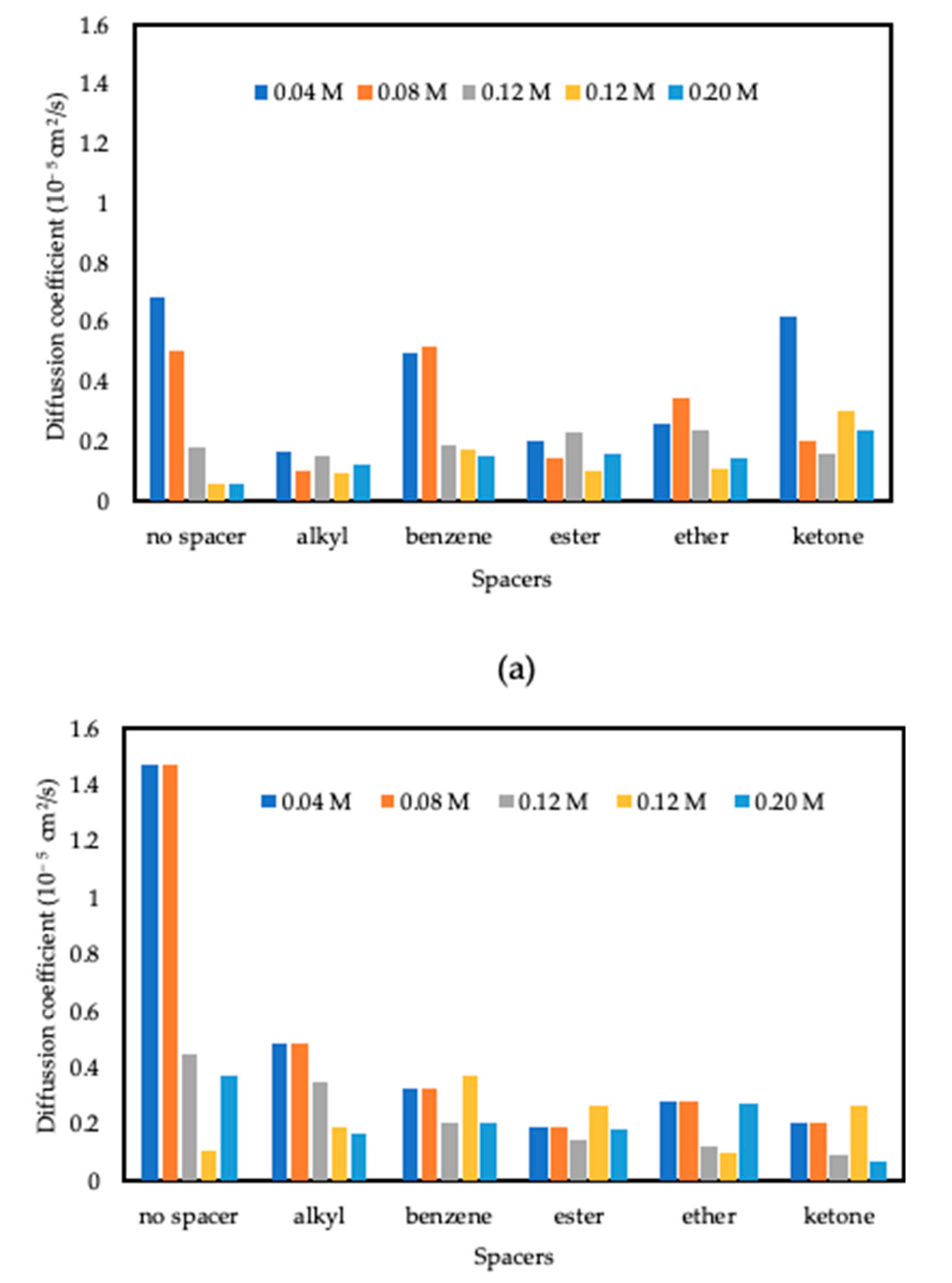
3.3.2. Diffusion Coefficient
3.3.3. Molecular Aggregation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Champion, N. Corrosion Mitigation for Complex Environments, Houston, 2012. Available online: https://issuu.com/championtechnologies/docs/champion_corrosion (accessed on 19 February 2023).
- Popoola, L.T.; Grema, A.S.; Latinwo, G.K.; Gutti, B.; Balogun, A.S. Corrosion problems during oil and gas production and its mitigation. Int. J. Ind. Chem. 2013, 4, 35. [Google Scholar] [CrossRef]
- Koch, G.; Varney, J.; Thompson, N.; Moghissi, O.; Gould, M.; Payer, J. Corrosion Cost and Prevention Strategies in The United States. In Proceedings of the Road Safety on Five Continents Conference, Houston, TX, USA, March 2002. [Google Scholar]
- Koch, G.; Varney, J.; Thompson, N.; Moghissi, O.; Gould, M.; Payer, J. International Measures of Prevention, Application, and Economics of Corrosion Technologies Study; Jacobson, G., Ed.; NACE International: Houston, TX, USA, 2016. [Google Scholar]
- Kurapati, Y. A Molecular Dynamics Study to Understand Behavior of Corrosion Inhibitors in Bulk Aqueous Phase and Near Metal-Water Interface. Master’s Thesis, Ohio University, Russ College of Engineering and Technology, Athens, OH, USA, 2018. [Google Scholar]
- Papavinasm, P. Evaluation and Selection of Corrosion Inhibitors. In Uhlig’s Corrosion Handbook, 2nd ed.; John Wiley & Sons: Toronto, ON, Canada, 1999. [Google Scholar]
- El-Hamdani, N.; Fdil, R.; Tourabi, M.; Jama, C.; Bentiss, F. Alkaloids extract of Retama monosperma (L.) Boiss. seeds used as novel eco-friendly inhibitor for carbon steel corrosion in 1 M HCl solution: Electrochemical and surface studies. Appl. Surf. Sci. 2015, 357, 1294–1305. [Google Scholar] [CrossRef]
- Faustin, M.; Maciuk, A.; Roos, C.; Lebrini, M. Corrosion inhibition of C38 steel by alkaloids extract of Geissospermum laeve in 1 M hydrochloric acid: Electrochemical and phytochemical studies. Corros. Sci. 2015, 92, 287–300. [Google Scholar] [CrossRef]
- Numin, M.S.; Hassan, A.; Jumbri, K.; Kee, K.E.; Borhan, N.; Daud, N.M.R.N.M.; Nor, A.M.N.; Suhor, F.; Wahab, R.A. A recent review on theoretical studies of Gemini surfactant corrosion inhibitors. J. Mol. Liq. 2022, 368, 120649. [Google Scholar] [CrossRef]
- Mahdavian, M.; Tehrani-Bagha, A.R.; Holmberg, K. Comparison of a Cationic Gemini. Surfactant J. Surfactant Deterg. 2011, 14, 605–613. [Google Scholar] [CrossRef]
- Zhou, T.; Yuan, J.; Zhang, Z.; Xin, X.; Xu, G. The comparison of imidazolium Gemini surfactant [C14-4-C14im] Br2 and its corresponding monomer as corrosion inhibitors for A3 carbon steel in hydrochloric acid solutions: Experimental and quantum chemical studies. Colloids Surf. A Physicochem. Eng. Asp. 2019, 575, 57–65. [Google Scholar] [CrossRef]
- Kuznetsov, Y.I.; Redkina, G.V. Thin Protective Coatings on Metals Formed by Organic Corrosion Inhibitors in Neutral Media. Coatings 2022, 12, 149. [Google Scholar] [CrossRef]
- Verma, D.K. Density Functional Theory (DFT) as a Powerful Tool for Designing Corrosion Inhibitors in Aqueous Phase; Advanced Engineering Testing; InTechOpen: London, UK, 2018; pp. 87–105. [Google Scholar]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional theory. III. The role of exact exchange. J. Chem. Phys. 2003, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. Density funtional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524–4529. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Zurek, T.V.E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Chem. 2012, 4, 17. [Google Scholar] [CrossRef]
- Steffen, C.; Thomas, K.; Huniar, U.; Hellweg, A.; Rubner, O.; Schroer, A. Software news and updates TmoleX-a graphical user interface for TURBOMOLE. J. Comput. Chem. 2012, 31, 2967–2970. [Google Scholar]
- Khan, M.U.; Khalid, M.; Khera, R.A.; Akhtar, M.N.; Abbas, A.; Rehman, M.F.U.; Braga, A.A.C.; Alam, M.M.; Imran, M.; Wang, Y.; et al. Influence of acceptor tethering on the performance of nonlinear optical properties for pyrene-based materials with A-π-D-π-D architecture. Arab. J. Chem. 2022, 15, 103673. [Google Scholar] [CrossRef]
- Siddiqui, W.A.; Khalid, M.; Ashraf, A.; Shafiq, I.; Parvez, M.; Imran, M.; Irfan, A.; Hanif, M.; Khan, M.U.; Sher, F.; et al. Antibacterial metal complexes of o-sulfamoylbenzoic acid: Synthesis, characterization, and DFT study. Appl. Organomet. Chem. 2021, 36, e6464. [Google Scholar] [CrossRef]
- Khalid, M.; Shafiq, I.; Hani, U.; Khalid, M.; Hussain, R.; Rehman, M.F.U.; Assiri, M.A.; Imran, A.; Akram, M.S. Effect of different end-capped donor moieties on non-fullerenes based non-covalently fused-ring derivatives for achieving high-performance NLO properties. Sci. Rep. 2023, 13, 1395. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Pearson, R.G. Negativity and hardness: Application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. [Google Scholar] [CrossRef]
- Henriquez-Roman, J.H.; Padilla-Campos, L.; Paez, M.A.; Zagal, J.H.; Rubio, M.A.; Rangel, C.M.; Costamagna, J.; Cardenas-Jiron, G. The influence of aniline and its derivatives on the corrosion behaviour of copper in acid solution: A theoretical approach. J. Mol. Struct. THEOCHEM 2005, 757, 1–7. [Google Scholar] [CrossRef]
- Sastri, V.S.; Perumareddi, J.R. Molecular Orbital Theoretical Studies of Some Organic Corrosion Inhibitors. Corros. Sci. 1997, 53, 617–622. [Google Scholar] [CrossRef]
- Lukovits, L.; Kalman, E.; Zucchi, F. Corrosion Inhibitors-Correlation between Electronic Structure and Efficiency. Corrosion 2001, 57, 3–8. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Pall, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–1224. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Fuhrmans, M.; Sanders, B.P.; Marrink, S.J.; Vries, A.H.D. Effects of bundling on the properties of the SPC water model. Theor. Chem. Acc. 2010, 125, 335–344. [Google Scholar] [CrossRef]
- Hossain, M.A.; Jewaratnam, J.; Ramalingam, A.; Sahu, J.N.; Ganesan, P. A DFT method analysis for formation of hydrogen rich gas from acetic acid by steam reforming process. Fuel 2018, 212, 49–60. [Google Scholar] [CrossRef]
- Turi, L.; Dannenberg, J.J. Molecular orbital study of acetic acid aggregation. 1. Monomers and dimers. J. Phys. Chem. 1993, 97, 12197–12204. [Google Scholar] [CrossRef]
- Wazzan, N.A.; Mahgoub, F.M. DFT Calculations for Corrosion Inhibition of Ferrous Alloys by Pyrazolopyrimidine Derivatives. Open J. Phys. Chem. 2014, 4, 6–14. [Google Scholar] [CrossRef]
- Al-Sabagh, A.M.; Abd-El-Bary, H.M.; El-Ghazawy, R.A.; Mishrif, M.R.; Hussein, B.M. Corrosion inhibition efficiency of linear alkyl benzene derivatives for carbon steel pipelines in 1M HCl. Egypt. J. Pet. 2011, 20, 33–45. [Google Scholar] [CrossRef]
- Zinad, D.S.; Jawad, Q.A.; Hussain, M.A.M.; Mahal, A.; Mohamed, L.; Al-Amiery, A.A. Adsorption, temperature and corrosion inhibition studies of a coumarin derivatives corrosion inhibitor for mild steel in acidic medium: Gravimetric and theoretical investigations. Int. J. Corros. Scale Inhib. 2020, 9, 134–151. [Google Scholar]
- Wade, L.G. Organic Chemistry, 8th ed.; Pearson: Harlow, UK, 2014; p. 794. [Google Scholar]
- Al-Sabagh, A.M.; Abd-El-Bary, H.M.; El-Ghazawy, R.A.; Mishrif, M.R.; Hussein, B.M. Corrosion inhibition efficiency of heavy alkyl benzene derivatives for carbon steel pipelines in 1 M HCl. Egypt. J. Pet. 2012, 21, 89–100. [Google Scholar] [CrossRef]
- Hassan, A.; Numin, M.S.; Jumbri, K.; Kee, K.E.; Borhan, N. Review on the Recent Development of Fatty Hydrazide as Corrosion Inhibitor in Acidic Medium: Experimental and Theoretical Approaches. Metals 2022, 12, 1058. [Google Scholar] [CrossRef]
- Brycki, B.; Szulc, A. Gemini surfactants as corrosion inhibitors. A review. J. Mol. Liq. 2021, 344, 117686. [Google Scholar] [CrossRef]
- Zhu, H.; Li, X.; Lu, X.; Wang, J.; Hu, Z.; Ma, X. Efficiency of Gemini surfactant containing semi-rigid spacer as microbial corrosion inhibitor for carbon steel in simulated seawater. Bioelectrochemistry 2021, 140, 107809. [Google Scholar] [CrossRef]
- Mazlan, N.; Jumbri, K.; Kassim, M.A.; Wahab, R.A.; Rahman, M.B.A. Density functional theory and molecular dynamics simulation studies of bio-based fatty hydrazide-corrosion inhibitors on Fe (110) in acidic media. J. Mol. Liq. 2022, 347, 118321. [Google Scholar] [CrossRef]
- Boughoues, Y.; Benamira, M.; Messaadia, L.; Bouider, N.; Abdelaziz, S. Experimental and theoretical investigations of four amine derivatives as effective corrosion inhibitors for mild steel in HCl medium. RSC Adv. 2020, 10, 24145–24158. [Google Scholar] [CrossRef]
- Damej, M.; Kaya, S.; Ibrahimi, B.E.; Lee, H.S.; Molhi, A.; Serdaroglu, G.; Benmessaoud, M.; Ali, I.H.; Hajjaji, S.E.; Lgaz, H. The corrosion inhibition and adsorption behavior of mercaptobenzimidazole and bis-mercaptobenzimidazole on carbon steel in 1.0 M HCl: Experimental and computational insights. Surf. Interfaces 2021, 24, 101095. [Google Scholar] [CrossRef]
- Hu, S.Q.; Guo, A.L.; Yan, Y.G.; Jia, X.L.; Geng, Y.F.; Guo, W.Y. Computer Simulation of diffusion of corrosive particle in corrosion inhibitor membrane. Comput. Theor. Chem. 2011, 964, 176–181. [Google Scholar] [CrossRef]
- Sharma, S.; Ko, X.; Kurapati, Y.; Singh, H.; Nesic, S. Adsorption Behavior of Organic Corrosion Inhibitors on Metal Surfaces—Some New Insights from Molecular Simulation. Corrosion 2019, 75, 90–105. [Google Scholar] [CrossRef]
- Singh, H.; Kurapati, Y.; Sharma, S. Aggregation and Adsorption Behavior of Organic Corrosion Inhibitors Studied Using Molecular Simulations. In Corrosion 2019; OnePetro: Nashville, TN, USA, 2019. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CI Molecule | Optimized | HOMO | LUMO |
|---|---|---|---|
| 2B |  |  |  |
| 2B-alkyl |  |  |  |
| 2B-benzene |  |  |  |
| 2B-ester |  |  |  |
| 2B-ether |  |  |  |
| 2B-ketone |  |  |  |
| Bond Lentgh (Å) | Calculated Value | Literature 1 [32] | Percentage Error (%) | Literature 2 [33] | Percentage Error (%) |
|---|---|---|---|---|---|
| C1-C2 | 1.522 | 1.508 | 0.066 | 1.507 | 0.995 |
| C1-H4 | 1.099 | 1.090 | 0.275 | 1.093 | 0.549 |
| C1-H5 | 1.106 | 1.095 | 0.274 | 1.098 | 0.729 |
| C1-H3 | 1.106 | 1.095 | 0.274 | 1.098 | 0.729 |
| C2=O6 | 1.198 | 1.210 | 3.223 | 1.249 | 4.083 |
| C2-O7 | 1.360 | 1.359 | 3.238 | 1.403 | 3.065 |
| O7-H8 | 0.973 | 0.976 | 0.922 | 0.985 | 1.218 |
| Energy (kJ/mol) | −228.773 | −229.082 | 2.785 | −222.701 | 2.727 |
| CI Molecule | Optimized | HOMO | LUMO |
|---|---|---|---|
| H |  |  |  |
| H-alkyl |  |  |  |
| H-benzene |  |  |  |
| H-ester |  |  |  |
| H-ether |  |  |  |
| H-ketone |  |  |  |
| CI Molecule | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2B | −4.552 | −2.079 | 2.473 | 4.552 | 2.079 | 1.237 | 3.316 | 0.809 | 1.490 |
| 2B-alkyl | −2.879 | −2.117 | 3.116 | 10.729 | 7.614 | 1.558 | 9.171 | 0.642 | −0.697 |
| 2B-benzene | −2.879 | −2.114 | 0.765 | 2.879 | 2.114 | 0.382 | 2.497 | 2.616 | 5.890 |
| 2B-ester | −4.691 | −3.118 | 1.573 | 4.691 | 3.118 | 0.786 | 3.905 | 1.272 | 1.968 |
| 2B-ether | −10.229 | −7.124 | 3.105 | 10.229 | 7.124 | 1.552 | 8.676 | 0.644 | −0.540 |
| 2B-ketone | −10.729 | −7.614 | 3.116 | 10.729 | 7.614 | 1.558 | 9.171 | 0.642 | −0.697 |
| H | −5.140 | −1.570 | 3.570 | 5.140 | 1.570 | 1.785 | 3.355 | 0.560 | 1.021 |
| H-alkyl | −10.849 | −7.665 | 3.184 | 10.849 | 7.665 | 1.592 | 9.257 | 0.395 | −0.709 |
| H-benzene | −5.989 | −0.920 | 5.069 | 5.989 | 0.920 | 2.535 | 3.454 | 0.395 | 0.699 |
| H-ester | −5.216 | −0.868 | 4.348 | 5.216 | 0.868 | 2.174 | 3.042 | 0.460 | 0.910 |
| H-ether | −5.083 | −1.924 | 3.159 | 5.083 | 1.924 | 1.580 | 3.503 | 0.633 | 1.107 |
| H-ketone | −4.631 | −1.867 | 2.765 | 4.631 | 1.867 | 1.382 | 3.249 | 0.723 | 1.357 |
| Molecule | Bandgap Energy (eV) | Adsorption Energy (kJ/mol) | Inhibition Efficiency (%) |
|---|---|---|---|
| 2B-Benzene | 0.756 | 1837.33 | - |
| N,N′-(((1,4-phenylenebis(methylene)) bis(oxy)) bis(ethane-2,1-diyl)) bis (N,N- dimethyldodecan-1-aminium) dibromide [41] | 1.62 | 1195.54 | 98.3 |
| 4-{[(2E)-3-phenylprop-2-en-1-yl] amino} phenol [43] | 3.99 | 541.55 | 96.72 |
| Bis-Mercaptobenzimidazole [44] | 3.34 | 748.43 | 92 |
| CI Molecule | Cluster Aggregation | ||||
|---|---|---|---|---|---|
| 0.04 M | 0.08 M | 0.12 M | 0.16 M | 0.20 M | |
| 2B |  |  |  |  |  |
| 2B-alkyl |  |  |  |  |  |
| 2B-benzene |  |  |  |  |  |
| 2B-ester |  |  |  |  |  |
| 2B-ether |  |  |  |  |  |
| 2B-ketone |  |  |  |  |  |
| CI Molecule | Cluster Aggregation | ||||
|---|---|---|---|---|---|
| 0.04 M | 0.08 M | 0.12 M | 0.16 M | 0.20 M | |
| H |  |  |  |  |  |
| H-alkyl |  |  |  |  |  |
| H-benzene |  |  |  |  |  |
| H-ester |  |  |  |  |  |
| H-ether |  |  |  |  |  |
| H-ketone |  |  |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Numin, M.S.; Jumbri, K.; Kee, K.E.; Hassan, A.; Borhan, N.; Matmin, J. DFT Calculation and MD Simulation Studies on Gemini Surfactant Corrosion Inhibitor in Acetic Acid Media. Polymers 2023, 15, 2155. https://doi.org/10.3390/polym15092155
Numin MS, Jumbri K, Kee KE, Hassan A, Borhan N, Matmin J. DFT Calculation and MD Simulation Studies on Gemini Surfactant Corrosion Inhibitor in Acetic Acid Media. Polymers. 2023; 15(9):2155. https://doi.org/10.3390/polym15092155
Chicago/Turabian StyleNumin, Mohd Sofi, Khairulazhar Jumbri, Kok Eng Kee, Almila Hassan, Noorazlenawati Borhan, and Juan Matmin. 2023. "DFT Calculation and MD Simulation Studies on Gemini Surfactant Corrosion Inhibitor in Acetic Acid Media" Polymers 15, no. 9: 2155. https://doi.org/10.3390/polym15092155
APA StyleNumin, M. S., Jumbri, K., Kee, K. E., Hassan, A., Borhan, N., & Matmin, J. (2023). DFT Calculation and MD Simulation Studies on Gemini Surfactant Corrosion Inhibitor in Acetic Acid Media. Polymers, 15(9), 2155. https://doi.org/10.3390/polym15092155