Cellulosic Bionanocomposites: A Review of Preparation, Properties and Applications


Abstract
:1. Introduction to Natural Fibers and Cellulosic Nanofillers
1.1. Natural Fibers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Fibers | Main Producers Countries | World Production | Production Costs | ||
|---|---|---|---|---|---|
| Million Tons | % | Million US$ | % | ||
| Cellulosic | |||||
| Cotton | China, USA, India, Pakistan, Uzbekistan, Brazil | 25.00 | 78.8 | 31.20 | 85.8 |
| Jute | India, Bangladesh | 2.70 | 8.5 | 0.48 | 1.3 |
| Flax | China, France, Belgium, Belarus, Ukraine | 0.08 | 0.2 | 0.43 | 1.2 |
| Kenaf | Asian Countries | 0.50 | 1.6 | n.a. | n.a. |
| Coir | India, Sri Lanka, Thailand, Malaysia | 0.45 | 1.4 | n.a. | n.a. |
| Sisal, Henequen and other Agaves | Brazil, Tanzania, China, Kenya, Mexico | 0.30 | 0.9 | 0.08 | 0.2 |
| Ramie | China | 0.15 | 0.5 | 0.17 | 0.5 |
| Abaca | Philippines, Equator | 0.08 | 0.3 | 0.03 | 0.1 |
| Hemp | China, Spain, Korea, Russian Federation, Chile | 0.09 | 0.3 | 0.03 | 0.1 |
| Wool | Australia, China, New Zealand | 2.20 | 6.9 | 2.96 | 8.1 |
| Silk | China, India | 0.14 | 0.4 | 0.98 | 2.7 |
| Other animal fibers* | 0.03 | 0.1 | n.a. | n.a. | |
| Total | 31.72 | 100 | 36.35 | 100 | |
| Fiber | Holocellulose (wt%) | Lignin (wt%) | Ash (wt%) | Extractives (wt%) | Ref. | |
|---|---|---|---|---|---|---|
| Cellulose (wt%) | Hemicellulose (wt%) | |||||
| Sugar Cane Bagasse | 71.1–84.9 | 24.3–25.3 | 1.1 | 0.7–3.5 | [2,9] | |
| 54.3–55.2 | 16.8–29.7 | |||||
| Leaflets of Phoenix Dactilyfera Palm | 59.5 | 27 | 6.5 | 2 | [10] | |
| 33.5 | 26 | |||||
| Rachis of Phoenix Dactilyfera Palm | 72 | 14 | 2.5 | 6 | [10] | |
| 44 | 28 | |||||
| Jute | 82.1 | 15.9 | 1 | — | [11,12] | |
| 60 | 22.1 | |||||
| Cotton Lintners | 96 | — | — | 0.40 | [2] | |
| 90 | 6 | |||||
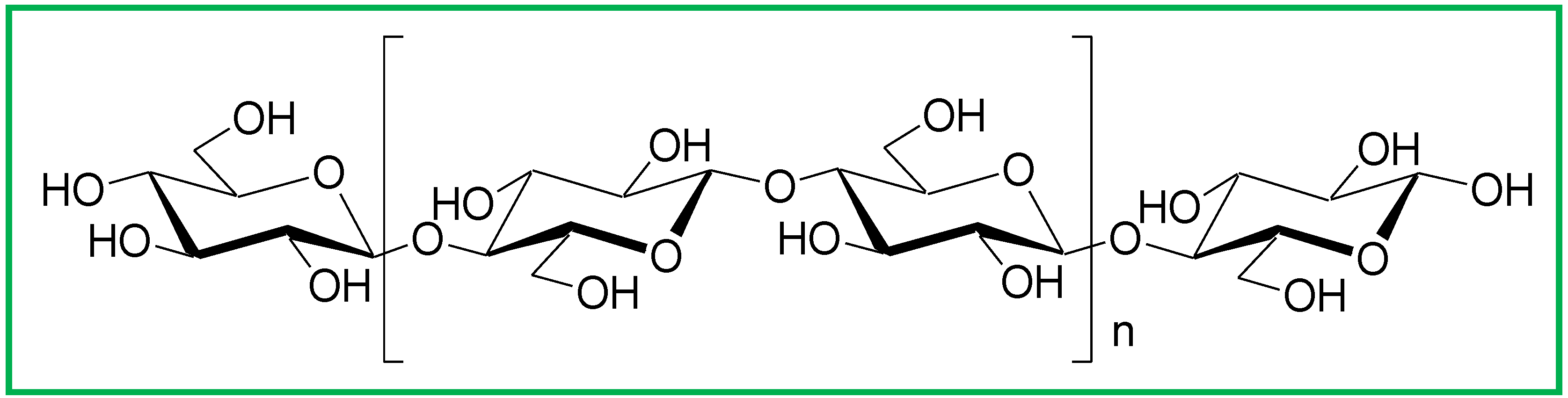
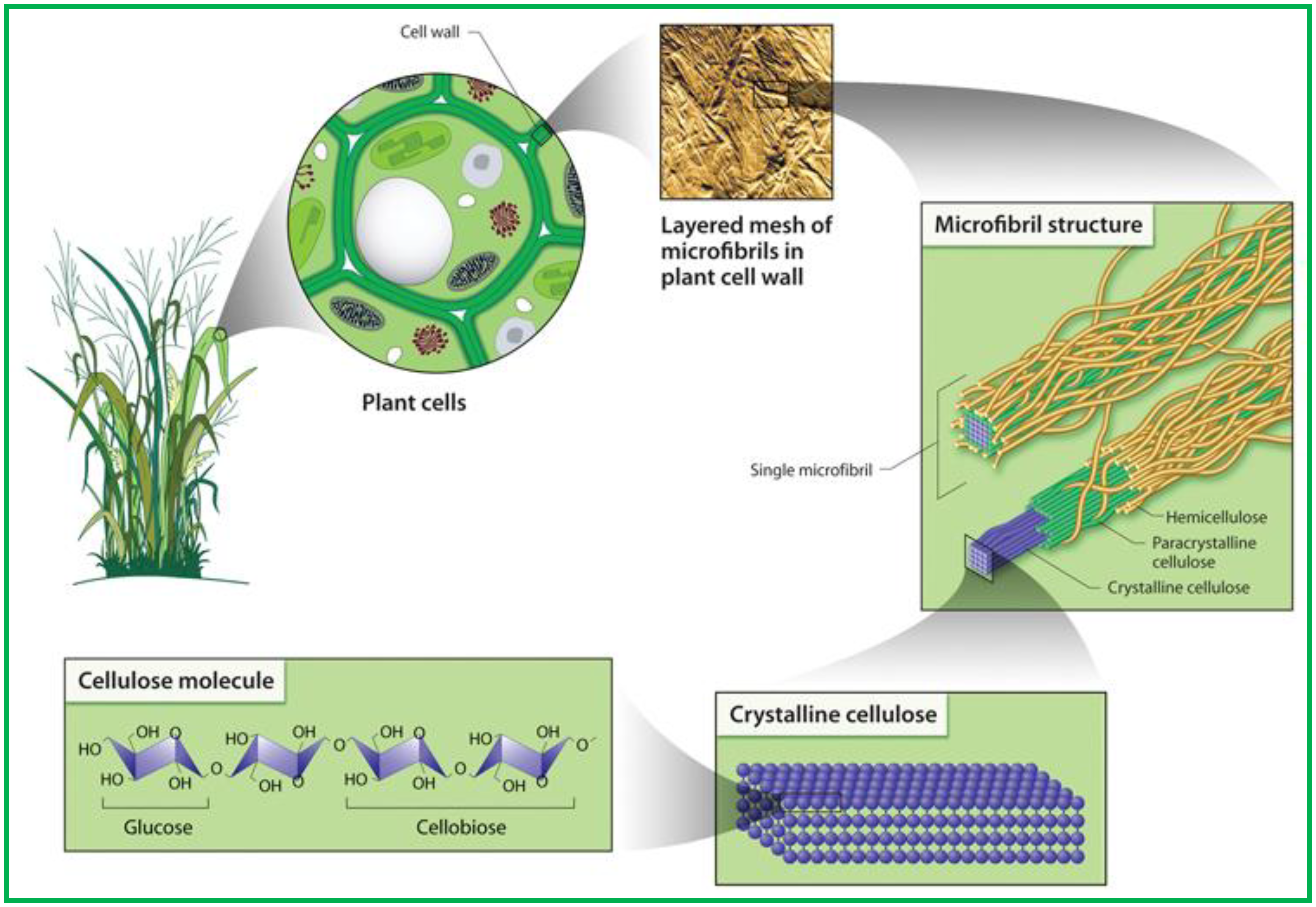
1.2. Cellulose


| Acronyms | Name | Source | Process | Reference |
|---|---|---|---|---|
| CNW | Cellulose nanowhiskers | Ramie | H2SO4 hydrolysis | [36] |
| MCC | H2SO4 hydrolysis | [37] | ||
| MCC | H2SO4 hydrolysis | [38] | ||
| Grass fiber | H2SO4 hydrolysis | [39] | ||
| MCC | LiCl:DMAc | [40] | ||
| CNXL | Cellulose Nanocrystals | Cotton Whatman filter paper | H2SO4 hydrolysis | [41] |
| [42] | ||||
| Bacterial cellulose | H2SO4 hydrolysis | [43] | ||
| Cotton (cotton wool) | H2SO4 hydrolysis | [44] | ||
| MCC | H2SO4 hydrolysis | [45] | ||
| MCC | Sonication | [46] | ||
| CNW-HCl | Cellulose nanowhiskers | Cotton linters | HCl hydrolysis | [47] |
| Wh | Whiskers | Cellulose fibers | H2SO4 hydrolysis | [8,48] |
| NF | Nanofibers | Wheat straw | HCl + Mechanical Treatment | [49] |
| NCC | Nanocrystalline cellulose | MCC | H2SO4 hydrolysis | [50] |
| MFC | Microfibrillated Cellulose | Pulp Gaulin | Homogenizer | [51] |
| Pulp Daicel | - | [52] | ||
| Pulp Daicel | - | [53] | ||
| NFC | Nanofibrillated cellulose Cellulose nanofibrils | Sulfite pulp | Mechanical | [54,55] |
| MCC | Microcrystalline cellulose | Alpha-cellulose fibers | Hydrolysis | [56] |
| - | Cellulose Crystallites | Cotton Whatman filter paper | H2SO4 hydrolysis | [57] |
| - | Nanocellulose | Sisal fibers | H2SO4 hydrolysis | [58] |
| - | Cellulose Microcrystal | Cotton Whatman filter paper | HCl hydrolysis | [59] |
| - | Nanofibers | Soybean pods | Chemical treatment + high pressure defibrilator | [60] |

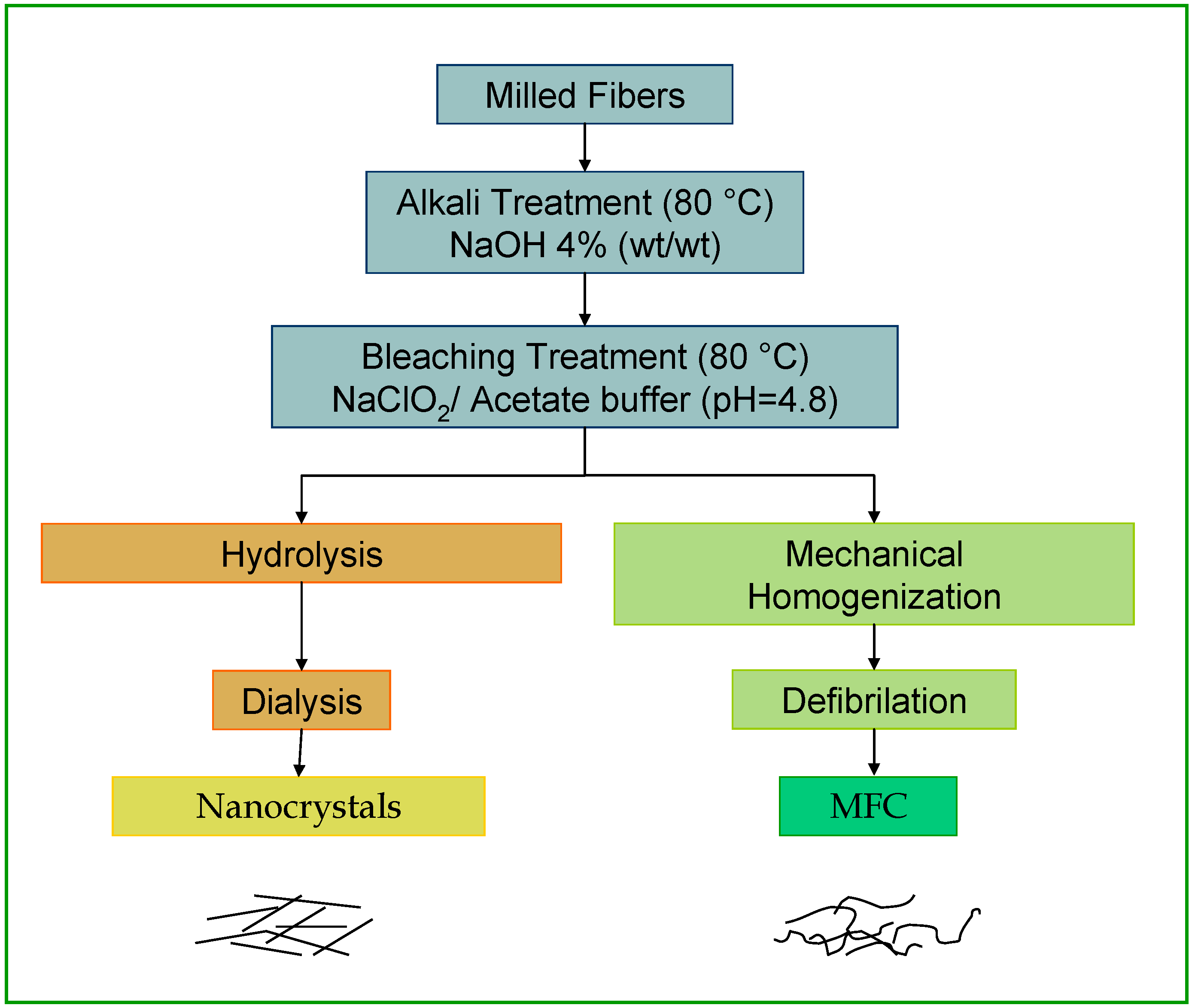
1.3. Cellulose Whiskers

1.4. Microfibrillated Cellulose
2. Bionanocomposites
2.1. Nanocomposites Definition
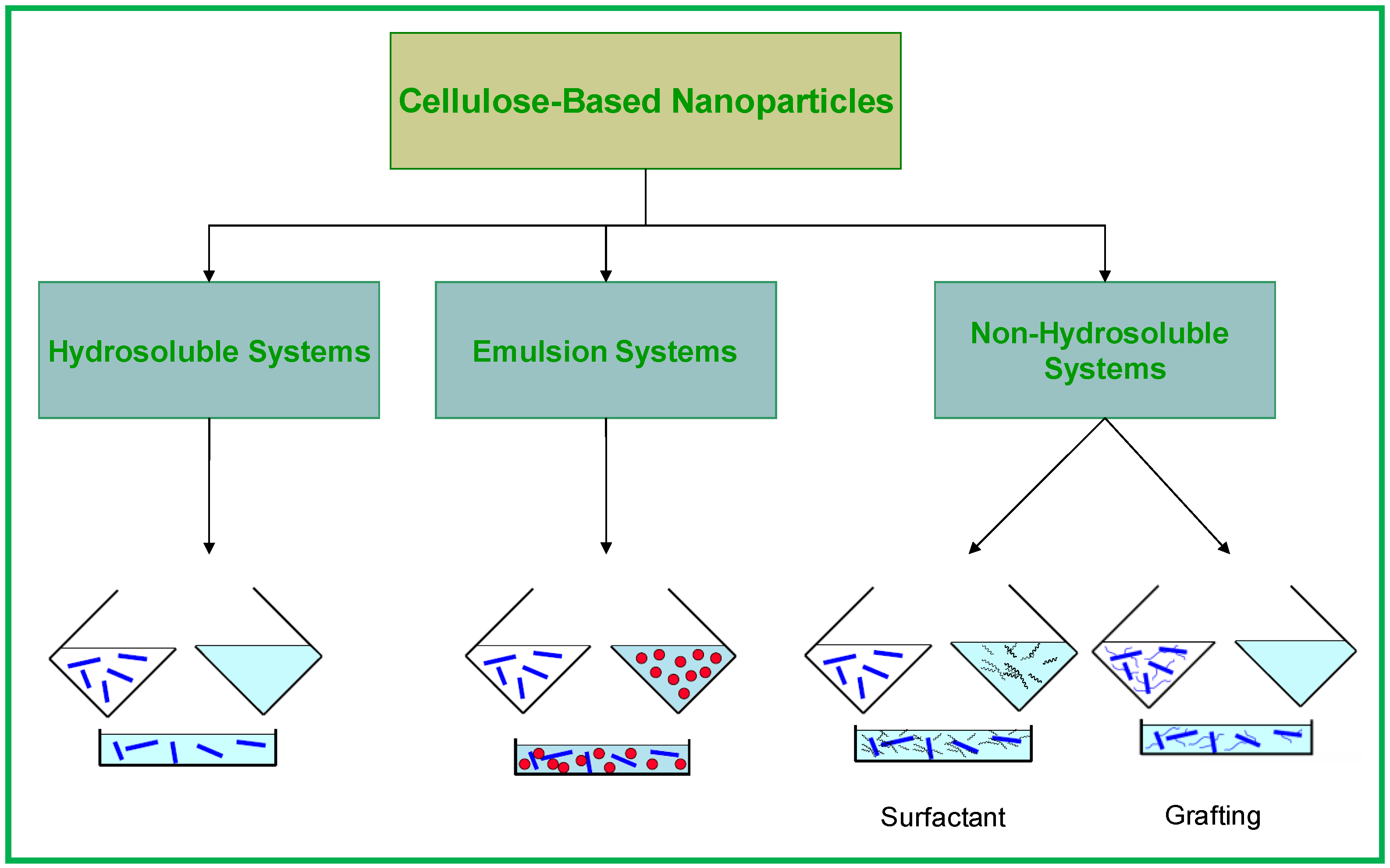
2.2. Cellulose Based Nanocomposites
- -
- Water or organic solvent evaporation by solvent casting;
- -
- Extrusion with freeze-dried cellulose nanoparticles.
- Coating of the surface of the cellulose nanocrystals with surfactants having polar heads and long hydrophobic tails;
- Grafting of hydrophobic chains at the surface of cellulose nanocrystals.

2.2.1. Hydrosoluble Systems
2.2.2. Emulsion Systems
2.2.3. Non-Hydrosoluble Systems


3. Properties of Cellulose-Based Nanocomposites
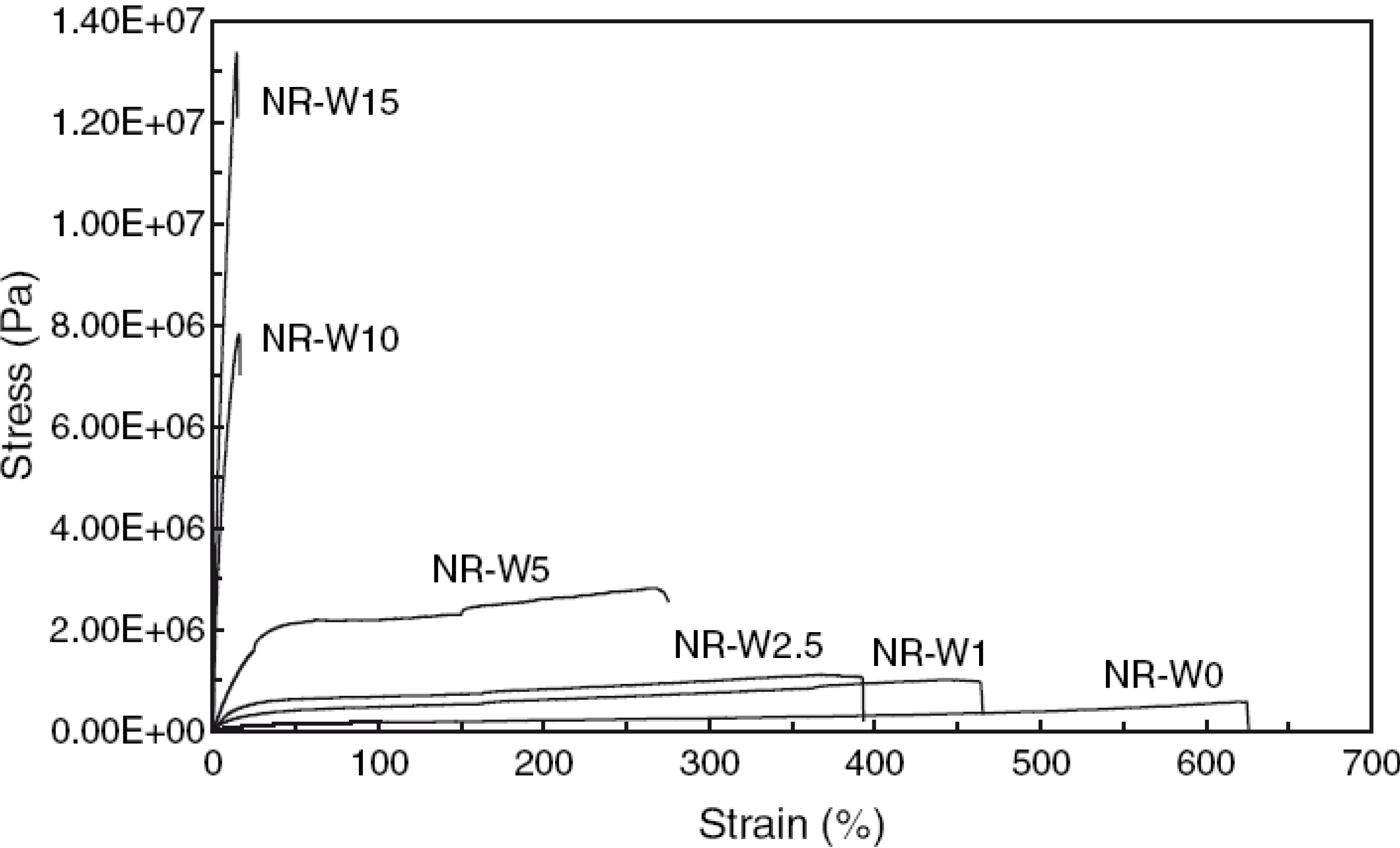
3.1. Mechanical Properties
3.1.1. Mechanical Modeling

| Source | L (nm) | D (nm) | L/d | Φc | Reference |
|---|---|---|---|---|---|
| Cotton | 171.6 | 14.6 | 11.8 | 5.9 | [105] |
| Ramie | 200 | 7 | 28.6 | 2.5 | [36] |
| MCC | 200 | 5 | 40 | 1.75 | [104] |
| Sugar beet pulp | 210 | 5 | 42 | 1.7 | [73] |
| Palm tree | 260 | 6.1 | 43 | 1.6 | [10] |
| Wheat straw | 225 | 5 | 45 | 1.6 | [103] |
| Tunicin | 1,000 | 15 | 67 | 1.0 | [71] |
3.1.2. Reinforcement with Cellulose Based Nanoparticles

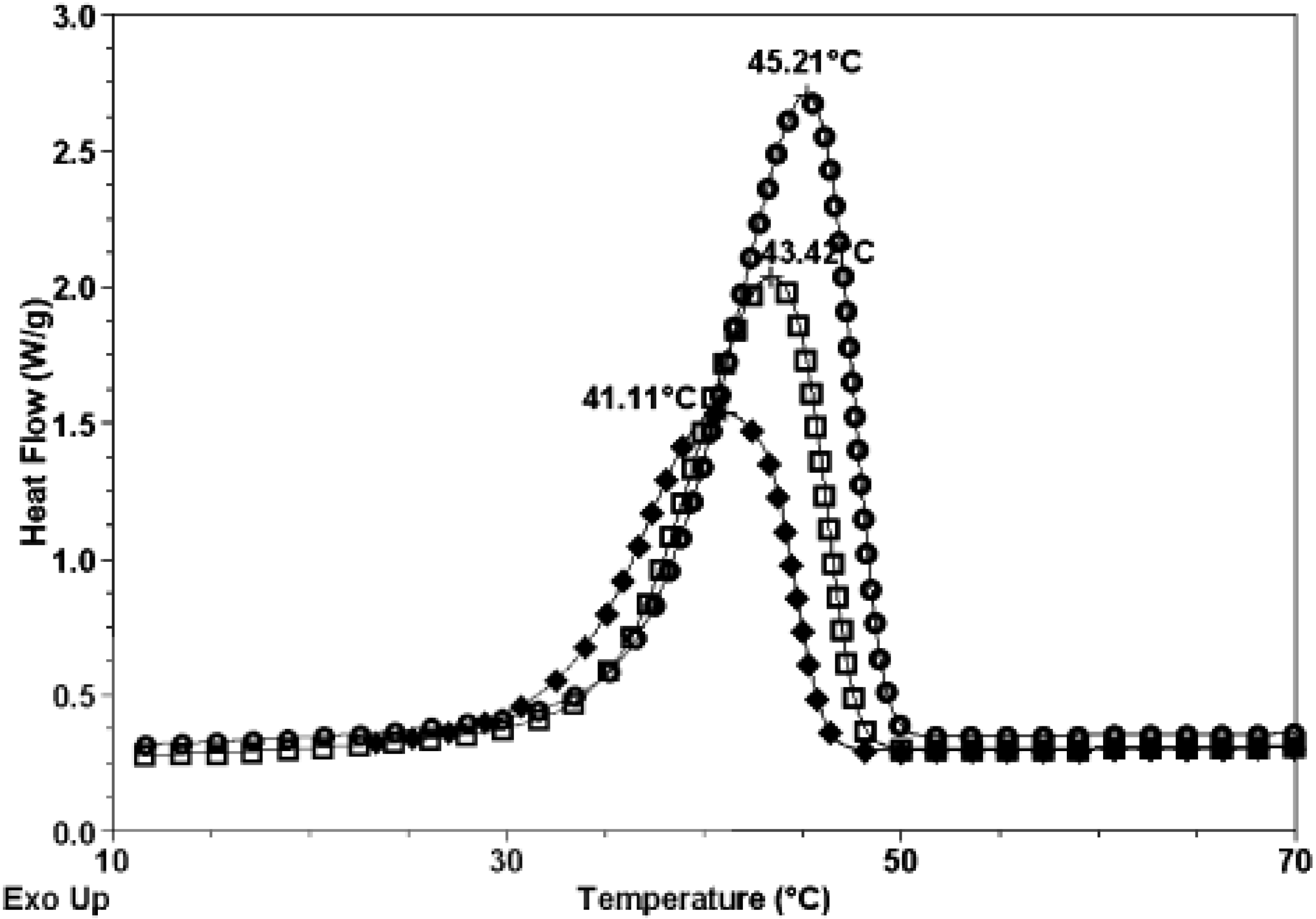
3.2. Thermal Properties

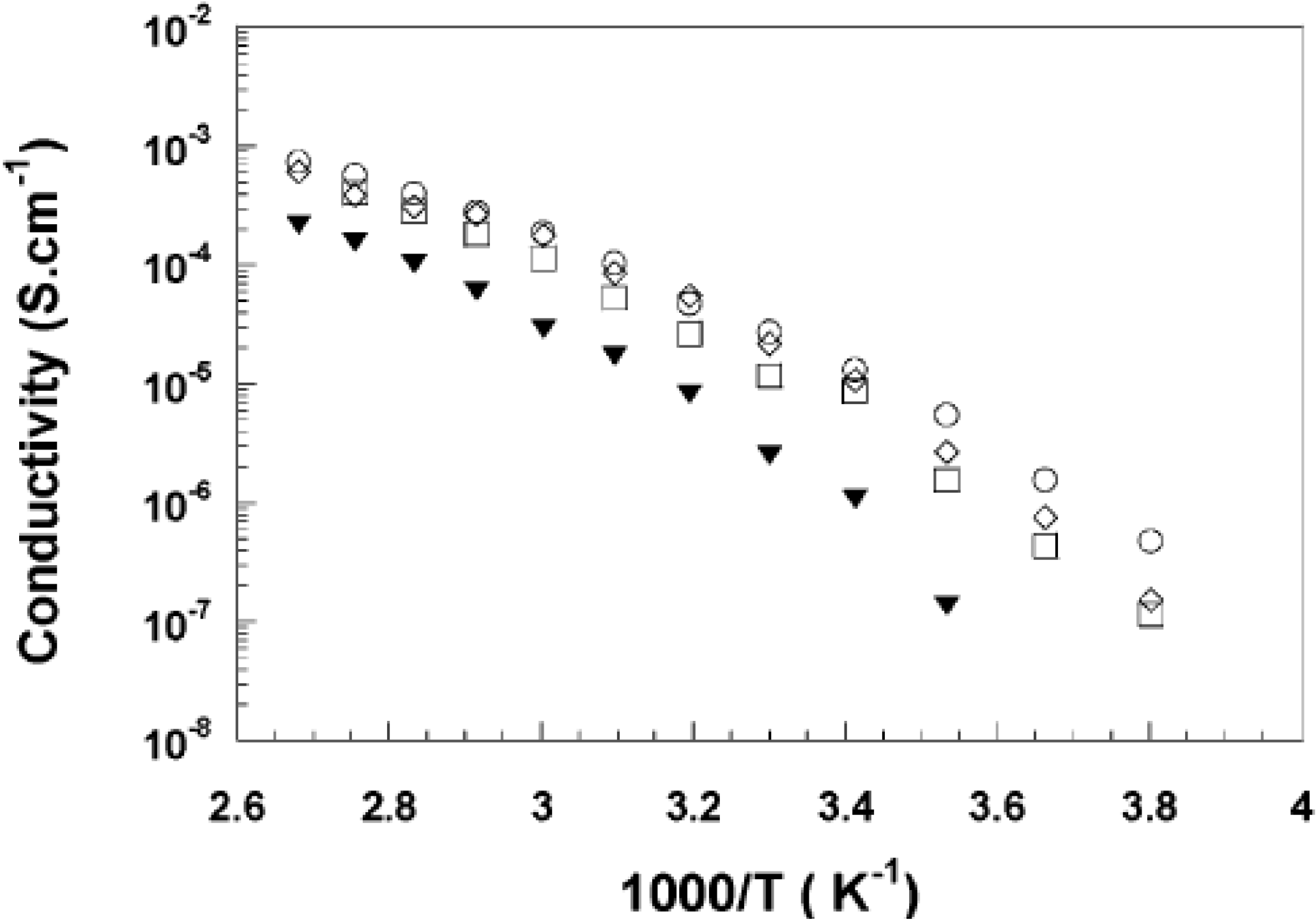
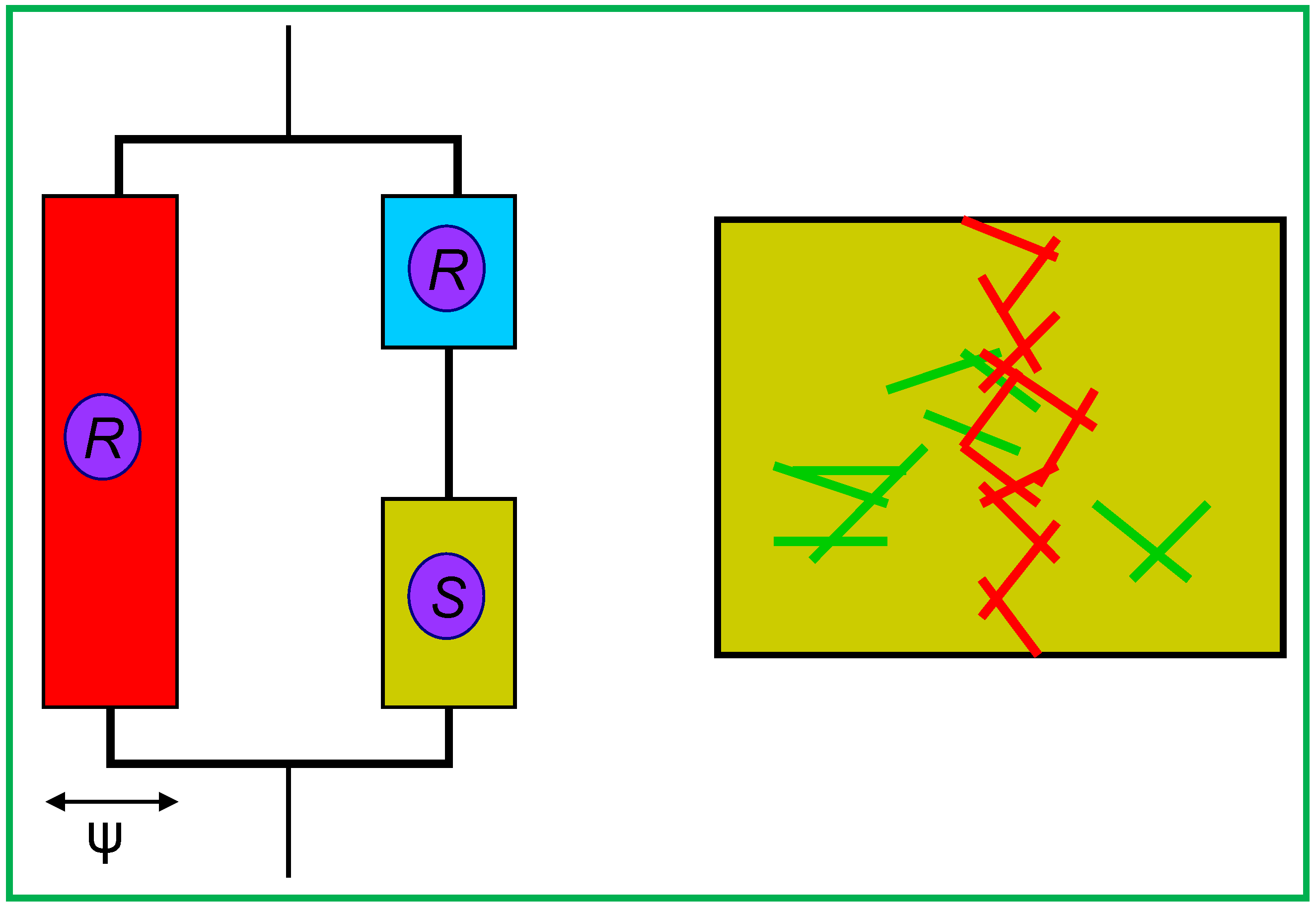
3.3. Barrier Properties
4. Conclusions
Acknowledgments
References
- FAO. Available online: www.naturalfibres2009.org (accessed on 8 December 2010).
- Satyanarayana, K.G.; Guilmaraes, J.L.; Wypych, F. Studies on lignocellulosic fibers of Brazil. Part I: Source, production, morphology, properties and applications. Compos. A-Appl. Sci. Manufact. 2007, 38, 1694–1709. [Google Scholar] [CrossRef]
- FAOSTAT. Available online: www.faostat.fao.org (accessed on 8 December 2010).
- Moir, B.; Plastina, A. 2009 international year of natural fibers. J. Int. Forum Cotton Promotion 2010, 26. Available online: http://www.cottonpromotion.org/features/international_year_of_natural_fibers/ (accessed on 8 December 2010). [Google Scholar]
- Bledzki, A.K.; Gassan, J. Composites reinforced with cellulose based fibres. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Satyanarayana, K.G.; Sukumaran, K.; Mukherjee, P.S.; Pavithran, C.; Pillai, S.G.K. Natural fibre-polymer composites. Cem. Concr. Compos. 1990, 12, 117–136. [Google Scholar] [CrossRef]
- Rong, M.Z.; Zhang, M.Q.; Liu, Y.; Yang, G.C.; Zeng, H.M. The effect of fiber treatment on the mechanical properties of unidirectional sisal-reinforced epoxy composites. Compos. Sci. Technol. 2001, 61, 1437–1447. [Google Scholar] [CrossRef]
- Dufresne, A. Cellulose-based composites and nanocomposites. In Monomers, Polymers and Composites from Renewable Resources, 1st ed.; Gandini, A., Belgacem, M.N., Eds.; Elsevier: Oxford, UK, 2008; pp. 401–418. [Google Scholar]
- Satyanarayana, K.G.; Wypych, F. Characterization of natural fibers. In Handbook of Engineering Biopolymers: Homopolymers, Blends and Composites; Fakirov, S., Bhattacharyya, D., Eds.; Carl Hanser Verlag: Munchen, Germany, 2007; pp. 4–47. [Google Scholar]
- Bendahou, A.; Habibi, Y.; Kaddami, H.; Dufresne, A. Physico-chemical characterization of palm from Phoenix Dactylifera-L, preparation of cellulose whiskers and natural rubber-based nanocomposites. J. Biobased Mat. Bioenerg. 2009, 3, 81–90. [Google Scholar] [CrossRef]
- Razera, I.A.T.; Frollini, E. Composites based on jute fibers and phenolic matrices: Properties of fibers and composites. J. Appl. Polym. Sci. 2004, 91, 1077–1085. [Google Scholar] [CrossRef]
- Rowell, R.M.; Han, J.S.; Rowell, J.S. Characterization and factors effecting fiber properties. In Natural Polymers and Agrofibers Composites; Frollini, E., Leão, A.L., Mattoso, L.H.C., Eds.; Embrapa Instrumentação Agropecuária: Sao Carlos, Brazil, 2000. [Google Scholar]
- Dufresne, A. Polymer nanocomposites from Biological Sources. In Encyclopedia of Nanoscience and Nanotechnology, 2nd ed.; Nalwa, H.S., Ed.; American Scientific Publisher: Valencia, CA, USA, in press.
- Heux, L.; Dinand, E.; Vignon, M.R. Structural aspects in ultrathin cellulose microfibrils followed by 13C CP-MAS NMR. Carbohydr. Polym. 1999, 40, 115–124. [Google Scholar] [CrossRef]
- Brännvall, E. Aspect on Strength Delivery and Higher Utilisation of Strength Potential of Soft Wood Kraft Pupl Fibres. Ph.D. Thesis, KTH, Royal Institute of Technology, Stockholm, Sweden, 2007. [Google Scholar]
- John, M.J.; Thomas, S. Biofibres and biocomposites. Carbohydr. Polym. 2008, 71, 343–364. [Google Scholar] [CrossRef]
- Sjöström, E. Wood Chemistry Fundamentals and Applications; Academic Press: New York, NY, USA, 1981. [Google Scholar]
- Daniel, J.R. Cellulose structure and properties. In Encyclopedia of Polymer Science and Engineering; Kroschwitz, J.I., Ed.; Wiley-Interscience Publication John Wiley & Sons: New York, NY, USA, 1985; Volume 3, pp. 86–123. [Google Scholar]
- Andresen, M.; Johansson, L.S.; Tanem, B.S.; Stenius, P. Properties and characterization of hydrophobized microfibrillated cellulose. Cellulose 2006, 13, 665–677. [Google Scholar] [CrossRef]
- Dufresne, A.; Cavaille, J.Y.; Vignon, M.R. Mechanical behavior of sheets prepared from sugar beet cellulose microfibrils. J. Appl. Polym. Sci. 1997, 64, 1185–1194. [Google Scholar] [CrossRef]
- Stenstad, P.; Andresen, M.; Tanem, B.S.; Stenius, P. Chemical surface modifications of microfibrillated cellulose. Cellulose 2008, 15, 35–45. [Google Scholar] [CrossRef]
- Azizi Samir, M.A.S.; Alloin, F.; Dufresne, A. Review of recent research into cellulosic whiskers, their properties and their application in nanocomposite field. Biomacromolecules 2005, 6, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Askeland, P.; Drzal, L.T. Surface modification of microfibrillated cellulose for epoxy composite applications. Polymer 2008, 49, 1285–1296. [Google Scholar] [CrossRef]
- Montanari, S.; Roumani, M.; Heux, L.; Vignon, M.R. Topochemistry of carboxylated cellulose nanocrystals resulting from TEMPO-mediated oxidation. Macromolecules 2005, 38, 1665–1671. [Google Scholar] [CrossRef]
- Dinand, E.; Vignon, M.; Chanzy, H.; Heux, L. Mercerization of primary wall cellulose and its implication for the conversion of cellulose I → cellulose II. Cellulose 2002, 9, 7–18. [Google Scholar] [CrossRef]
- Sugiyama, J.; Persson, J.; Chanzi, H. Combined infrared and electron diffraction study of polymorphism of native cellulose. Macromolecules 1991, 24, 2461–2466. [Google Scholar] [CrossRef]
- French, A.D.; Bertoniere, N.R.; Brown, R.M.; Chanzy, H.; Gray, D.; Hattori, K.; Glasser, W. Cellulose. In Encyclopedia of Polymler Science and Technology; Kroschwitz, J.I., Ed.; Wiley Interscience Publication John Wiley & Sons: New Jersey, NJ, USA, 2003; Volume 5, pp. 473–507. [Google Scholar]
- Saxena, I.M.; Brown, R.M.J. Cellulose Biosynthesis: Current views and envolving Concepts. Ann. Bot. 2005, 96, 9–21. [Google Scholar] [CrossRef]
- U.S. Department of Energy, Office of Science. Genomics: GTL Roadmap. Available online: http://genomicsgtl.energy.gov/roadmap/ (accessed on 8 December 2010).
- Atalla, R.H.; VanderHart, L.D. Native cellulose: A composite of two distinct crystalline forms. Science 1984, 223, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, J.; Okano, T.; Yamamoto, H.; Horii, F. Transformation of Valonia cellulose crystals by an alkaline hydrothermal treatment. Macromolecules 1990, 23, 3196–3198. [Google Scholar] [CrossRef]
- Sugiyama, J.; Vuong, R.; Chanzi, H. Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 1991, 24, 4168–4175. [Google Scholar] [CrossRef]
- de Souza Lima, M.M.; Borsali, R. Rodlike cellulose microcrystals: Structure, properties and applications. Macromol. Rapid Commun. 2004, 25, 771–787. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Sugiyama, J.; Chanzy, H.; Langan, P. Crystal structure and hydrogen bonding system in cellulose 1(alpha), from synchrotron X-ray and neutron fiber diffraction. J. Am. Chem. Soc. 2003, 125, 14300–14306. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Askeland, P.; Drzal, L.T. Surface modification of microfibrillated cellulose for epoxy composite applications. Polymer 2008, 49, 1285–1298. [Google Scholar] [CrossRef]
- Habibi, Y.; Goffin, A.L.; Schiltz, N.; Duquesne, E.; Dubois, P.; Dufresne, A. Bionanocomposites based on poly(epsilon-caprolactone)-grafted cellulose nanocrystals by ring-opening polymerization. J. Mater. Chem. 2008, 18, 5002–5010. [Google Scholar] [CrossRef]
- Petersson, L.; Kvien, I.; Oksman, K. Structure and thermal properties of poly(lactic acid)/cellulose whiskers nanocomposites materials. Compos. Sci. Technol. 2007, 67, 2535–2544. [Google Scholar] [CrossRef]
- Rojas, O.J.; Montero, G.A.; Habibi, Y. Electronspun nancomposites from polystyrene loaded with cellulose nanowhiskers. Appl. Polym. Sci. 2009, 113, 927–935. [Google Scholar]
- Pandey, J.K.; Kim, S.C.; Chu, C.S.; Lee, C.S.; Jang, D.J.; Ahn, S.H. Evaluation of morphological architecture of cellulose chains in grass during conversion from macro to nano dimensions. e-Polymer 2009, 102, 1–15. [Google Scholar]
- Oksman, K.; Mathew, A.P.; Bondeson, D.; Kvien, I. Manufacturing process of cellulose whiskers/polylactic acid nanocomposites. Compos. Sci. Technol. 2006, 66, 2776–2784. [Google Scholar] [CrossRef]
- Paralikar, S.A.; Simonsen, J.; Lombardi, J. Poly(vinyl alcohol)/cellulose nanocrystals barrier membranes. J. Membr. Sci. 2008, 320, 248–258. [Google Scholar] [CrossRef]
- Mangalam, A.P.; Simonsen, J.; Benight, A. Cellulose/DNA hybrid nanomaterials. Biomacromolecules 2009, 10, 497–504. [Google Scholar] [PubMed]
- Grunert, M.; Winter, W.T. Nanocomposites of cellulose acetate butyrate reinforced with cellulose nanocrystals. J. Polym. Envir. 2002, 10, 27–30. [Google Scholar] [CrossRef]
- Morandi, G.; Heath, L.; Thielemans, W. Cellulose nanocrystals grafted with polystyrene chains through surface-initiated atom transfer radical polymerization (SI-ATRP). Langmuir 2009, 25, 8280–8286. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.; Mathew, A.; Oksman, K. Optimization of the isolation of nanocrystals from microcrystalline cellulose by acid hydrolysis. Cellulose 2006, 13, 171–180. [Google Scholar] [CrossRef]
- Filson, P.B.; Dawson-Andoh, B.E. Sono-chemical preparation of cellulose nanocrystals from lignocellulose derived materials. Biores. Technol. 2009, 100, 2259–2264. [Google Scholar] [CrossRef]
- Braun, B.; Dorgan, J.R.; Chandler, J.P. Cellulosic nanowhiskers. Theory and application of light scattering from polydisperse spheroids in the Rayleigh-Gans-Debye regime. Biomacromoleucules 2008, 9, 1255–1263. [Google Scholar] [CrossRef]
- Dufresne, A.; Cavaille, J.Y.; Helbert, W. Thermoplastic nanocomposites filled with wheat straw cellulose whiskers. Part II. Effect of processing and modeling. Polym. Compos. 1997, 18, 198–210. [Google Scholar] [CrossRef]
- Alemdar, A.; Sain, M. Biocomposites from wheat straw nanofibers: Morphology, thermal and mechanical properties. Comp. Sci. Technol. 2008, 68, 557–565. [Google Scholar] [CrossRef]
- Bai, W.; Holbery, J.; Li, K. A techinique for production of nanocrystalline cellulose with a narrow size distribution. Cellulose 2009, 16, 455–465. [Google Scholar] [CrossRef]
- Andresen, M.; Stenius, P. Water-in-oil emulsions stabilized by hydrophobized microfibrillated cellulose. J. Disp. Sci. Technol. 2007, 28, 837–844. [Google Scholar] [CrossRef]
- Lu, J.; Wang, T.; Drzal, L.T. Preparation and properties of microfibrillated cellulose polyvinyl alcohol composite materials. Compos. A Appl. Sci. Manuf. 2008, 39, 738–746. [Google Scholar] [CrossRef]
- Suryanegara, L.; Nakagaito, A.N.; Yano, H. The effect of crystallization of PLA on the thermal and mechanical properties of microfibrillated cellulose-reinforced PLA composites. Comp. Sci. Technol. 2009, 69, 1187–1192. [Google Scholar] [CrossRef]
- Mörseburg, K.; Chinga-Carrasco, G. Assessing the combined benefits of clay and nanofibrillated cellulose in layered TMP-based sheets. Cellulose 2009, 16, 795–806. [Google Scholar] [CrossRef]
- Chinga-Carrasco, G.; Syverud, K. Computer-assisted quantification of the multi-scale structure of films made of nanofibrillated cellulose. J. Nanopart. Res. 2010, 12, 841–851. [Google Scholar] [CrossRef]
- Battista, O.A.; Hill, D.; Smith, P.A. Microcrystalline cellulose. Ind. Eng. Chem. 1962, 20, 54–59. [Google Scholar]
- Dong, X.M.; Kimura, T.; Revol, J.F.; Gray, D.G. Effects of ionic strength on the isotropic-chiral nematic phase transition of suspensions of cellulose crystallites. Langmuir 1996, 12, 2076–2082. [Google Scholar] [CrossRef]
- Moran, J.I.; Alvarez, V.A.; Cyras, V.P.; Vazquez, A. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 2008, 15, 149–159. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S. Steric stabilization of a cellulose microcrystal suspension by poly(ethylene glycol) grafting. Langmuir 2001, 17, 21–27. [Google Scholar] [CrossRef]
- Wang, B.; Mohini, S. Isolation of nanofibers from soybean source and their reinforcing capability on synthetic polymers. Comp. Sci. Technol. 2007, 67, 2521–2527. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose nanocrystals: Chemistry, self-assembly, and applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Thielemans, W.; Warbey, C.R.; Walsh, D.A. Permselective nanostructured membranes based on cellulose nanowhiskers. Green Chem. 2009, 11, 531–537. [Google Scholar] [CrossRef]
- Beck-Candanedo, S.; Roman, M.; Gray, D. Effect of conditions on the properties behavior of wood cellulose nanocrystals suspensions. Biomacromolecules 2005, 6, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.M.; Revol, J.F.; Gray, D. Effect of microcrystallite preparation conditions on the formation of colloid crystals of cellulose. Cellulose 1998, 5, 19–32. [Google Scholar] [CrossRef]
- Araki, J.; Wada, M.; Kuga, S.; Okano, T. Low properties of microcrystalline cellulose suspension prepared by acid treatement of native cellulose. Colloids Surf. A 1998, 142, 75–82. [Google Scholar] [CrossRef]
- Angellier, H.; Putaux, J.L.; Molina-Boisseau, S.; Dupeyre, D.; Dufresne, A. Starch nanocrystal fillers in a acrylic polymer matrix. Macromol. Symp. 2005, 221, 95–104. [Google Scholar] [CrossRef]
- Hirai, A.; Inui, O.; Horii, F.; Tsuji, M. Phase separation behavior in aqueous suspensions of bacterial cellulose nanocrystals prepared by sulfuric acid treatment. Langmuir 2009, 25, 497–502. [Google Scholar] [PubMed]
- Sugiyama, J.; Chanzi, H.; Revol, J.F. On the polarity of cellulose in the cell wall of Valonia. Planta 1994, 193, 260–265. [Google Scholar] [CrossRef]
- Cao, X.; Chen, Y.; Chang, P.R.; Stumborg, M.; Huneault, M.A. Green Composites Reinforced with Hemp Nanocrystals in Plasticized Starch. J. Appl. Polym. Sci. 2008, 109, 3804–3810. [Google Scholar] [CrossRef]
- Bonini, C.; Heux, L.; Cavaille, J.Y.; Lindner, P.; Dewhurst, C.; Terech, P. Rodlike cellulose whiskers coated with surfactant: A small-angle neutron scattering characterization. Langmuir 2002, 18, 3311–3314. [Google Scholar] [CrossRef]
- Favier, V.; Chanzy, H.; Cavaille, J.Y. Polymer nanocomposites reinforced by cellulose whiskers. Macromolecules 1995, 28, 6365–6367. [Google Scholar] [CrossRef]
- Garcia de Rodriguez, N.L.; Thielemans, W.; Dufresne, A. Sisal cellulose whiskers reinforced polyvinyl acetate nanocomposites. Cellulose 2006, 13, 261–270. [Google Scholar] [CrossRef]
- Azizi Samir, M.A.S.; Alloin, F.; Paillet, M.; Dufresne, A. Tangling effect in fibrillated cellulose reinforced nanocomposites. Macromolecules 2004, 37, 4313–4316. [Google Scholar] [CrossRef]
- Favier, V. Etude de Nouveaux Matériaux Composites Obtenus à Partir de Latex Filmogènes et de Whiskers de Cellulose: Effect de Percolation Mechanique. Ph.D. Thesis, Joseph Fourier University, Grenoble, France, 1995. [Google Scholar]
- Kvien, I.; Tanem, B.S.; Oksman, K. Characterization of cellulose whiskers and their nanocomposites by atomic force and electron microscopy. Biomacromolecules 2005, 6, 3160–3165. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, I.; Nukushina, Y.I.T. Experimental determination of the elastic modulus of crystalline regions in oriented polymers. J. Polym. Sci. 1962, 57, 651–659. [Google Scholar] [CrossRef]
- Zimmermann, T.; Pohler, E.; Geiger, T. Cellulose fibrils for polymer reinforcement. Adv. Eng. Mat. 2004, 6, 754–761. [Google Scholar] [CrossRef]
- Herrick, F.W.; Casebier, R.L.; Hamilton, J.K.; Sandberg, K.R. Microfibrillated cellulose: Morphology and accessibility. J. Appl. Polym. Sci. Appl. Polym. Symp. 1983, 37, 797–813. [Google Scholar]
- Turbak, A.F.; Snyder, F.W.; Sandberg, K.R. Microfibrillated cellulose, a new cellulose product: Properties, uses, and commercial potential. J. Appl. Polym. Sci. Appl. Polym. Symp. 1983, 37, 815–827. [Google Scholar]
- Nakagaito, A.N.; Yano, H. Novel high-strength biocomposites based on microfibrillated cellulose having nano-order-unit web-like network structure. Appl. Phys. A-Mat. Sci. Process. 2005, 80, 155–159. [Google Scholar] [CrossRef]
- Iwamoto, S.; Kai, W.; Isogai, A.; Iwata, T. Elastic modulus of single cellulose microfibrils from tunicate measured by atomic force microscopy. Biomacromolecules 2009, 10, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, M.; Henriksson, G.; Berglund, L.A.; Lindstrom, T. An environmentally friendly method for enzyme-assisted preparation of microfibrillated cellulose (MFC) nanofibers. Europ. Polym. J. 2007, 43, 3434–3441. [Google Scholar] [CrossRef]
- Nakagaito, A.N.; Yano, H. The effect of morphological changes from pulp fiber towards nano-scale fibrillated cellulose on the mechanical properties of high-strength plant fiber based composites. Appl. Phys. A-Mat. Sci. Process. 2004, 78, 547–552. [Google Scholar] [CrossRef]
- Ankerfors, M.P.; Kosonen, H.; Nyknen, A.; Ahola, S.; Sterberg, M.; Roukolainen, J.; Laine, J.; Larsson, P.T.; Ikkala, O.; Lindstrm, T. Enzymatic hydrolisis combined with mechanical shearing and high-pressure homogenization for nanoscale cellulose fibrils and strong gels. Biomacromolecules 2007, 8, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishiyama, Y.; Putaux, J.L.; Vignon, M.; Isogai, A. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 2006, 7, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S. Nanotechnology and its application in lignocellulosics composites, a mini review. Exp. Polym. Lett. 2007, 1, 546–575. [Google Scholar]
- Khademhosseini, A.; Langer, R. Nanobiotechnology—Drug delivery and tissue engineering. Chem. Engin. Progress 2006, 102, 38–42. [Google Scholar]
- Dalmas, F.; Cavaille, J.Y.; Gauthier, C.; Chazeau, L.; Dendievel, R. Viscoelastic behavior and electrical properties of flexible nanofiber filled polymer nanocomposites. Influence of processing conditions. Comp. Sci. Technol. 2007, 67, 829–839. [Google Scholar] [CrossRef]
- Esteves, A.C.C.; Barros-Timmons, A.; Trindade, T. Polymer based nanocomposites: Synthetic strategies for hybrid materials. Quim. Nova 2004, 27, 798–806. [Google Scholar] [CrossRef]
- Azeredo, M.C. Nanocomposites for food packaging applications. Food Res. Int. 2009, 42, 1240–1253. [Google Scholar] [CrossRef]
- Schadler, L.S.; Brinson, L.C.; Sawyer, W.G. Polylmer nanocomposites: A small part of the story. Nanocompos. Mat. 2007, 59, 53–60. [Google Scholar]
- Kovacs, T.; Naish, V.; O’Connor, B.; Blaise, C.; Gagné, F.; Hall, L.; Trudeau, V.; Martel, P. An ecotoxicological characterization of nanocrystalline cellulose (NCC). Nanotoxicology 2010, 4, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.K.; Misra, M.; Drzal, L.T.; Selke, S.E.; Harte, B.R.; Hinrichsen, G. Natural fibers, biopolymers, and biocomposites: An introduction. In Natural Fibers, Biopolymers, and Biocomposites; Mohanty, A.K., Misra, M., Drzal, L.T., Eds.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Darder, M.; Aranda, P.; Ruiz-Hitzky, E. Bionanocomposites: A new concept of ecological, bioinspired, and functional hybrid materials. Adv. Mat. 2007, 19, 1309–1319. [Google Scholar] [CrossRef]
- Espert, A.; Vilaplana, F.; Karlsson, S. Comparison of water absorption in natural cellulosic fibres from wood and one-year crops in polypropylene composites and its influence on their mechanical properties. Compos. A-Appl. Sci. Manuf. 2004, 35, 1267–1276. [Google Scholar] [CrossRef]
- Gandini, A.; Belgacem, M.N. Chemical Modification of Wood. In Monomers, Polymers and Composites from Renewable Resources, 1st ed.; Gandini, A., Belgacem, M.N., Eds.; Elsevier: Oxford, UK, 2008; pp. 419–432. [Google Scholar]
- Gandini, A.; Belgacem, M.N. The state of the art. In Monomers, Polymers and Composites from Renewable Resources, 1st ed.; Gandini, A., Belgacem, M.N., Eds.; Elsevier: Oxford, UK, 2008; pp. 1–16. [Google Scholar]
- Pasquini, D.; Belgacem, M.N.; Gandini, A.; Curvelo, A.A.D. Surface esterification of cellulose fibers: Characterization by DRIFT and contact angle measurements. J. Colloid Interface Sci. 2006, 295, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Tserki, V.; Zafeiropoulos, N.E.; Simon, F.; Panayiotou, C. A study of the effect of acetylation and propionylation surface treatments on natural fibres. Compos. A-Appl. Sci. Manuf. 2005, 36, 1110–1118. [Google Scholar] [CrossRef]
- Wambua, P.; Ivens, J.; Verpoest, I. Natural fibres: Can they replace glass in fibre reinforced plastics? Compos. Sci. Technol. 2003, 63, 1259–1264. [Google Scholar] [CrossRef]
- Favier, V.; Canova, G.R.; Cavaillé, J.-Y.; Chanzy, H.; Dufresne, A.; Gauthier, C. Nanocomposites materials from latex and cellulose whiskers. Polym. Adv. Technol. 1995, 6, 351–355. [Google Scholar] [CrossRef]
- Helbert, W.; Cavaille, J.Y.; Dufresne, A. Thermoplastic nanocomposites filled with wheat straw cellulose whiskers. Part I. Processing and mechanical behavior. Polym. Compos. 1996, 17, 604–611. [Google Scholar]
- Kvien, I.; Oksman, K. Orientation of cellulose nanowhiskers in polyvinyl alcohol. Appl. Phys. A-Mat. Sci. Process. 2007, 87, 641–643. [Google Scholar] [CrossRef]
- Roohani, M.; Habibi, Y.; Belgacem, N.M.; Ebrahim, G.; Karimi, A.N.; Dufresne, A. Cellulose whiskers reinforced polyvinyl alcohol copolymers nanocomposites. Europ. Polym. J. 2008, 44, 2489–2498. [Google Scholar]
- Johnson, K.R.; Zink-Sharp, A.; Renneckar, S.H.; Glasser, W.G. A new bio-based nancomposite: fibrillated TEMPO-oxidazed celluloses in hydroxypropylcellulose matrix. Cellulose 2008, 16, 227–238. [Google Scholar] [CrossRef]
- Dubief, D.; Samain, E.; Dufresne, A. Polysaccharide microcrystals reinforced amorphous poly(beta-hydroxyoctanoate) nanocomposite materials. Macromolecules 1999, 32, 5765–5771. [Google Scholar]
- Dufresne, A.; Kellerhals, M.B.; Witholt, B. Transcrystallization in Mcl-PHAs/cellulose whiskers composites. Macromolecules 1999, 32, 7396–7401. [Google Scholar]
- Chazeau, L.; Cavaille, J.Y.; Canova, G.; Dendievel, R.; Boutherin, B. Viscoelastic properties of plasticized PVC reinforced with cellulose whiskers. J. Appl. Polym. Sci. 1999, 71, 1797–1808. [Google Scholar] [CrossRef]
- Chazeau, L.; Cavaille, J.Y.; Terech, P. Mechanical behaviour above T-g of a plasticised PVC reinforced with cellulose whiskers; A SANS structural study. Polymer 1999, 40, 5333–5344. [Google Scholar]
- Chazeau, L.; Paillet, M.; Cavaille, J.Y. Plasticized PVC reinforced with cellulose whiskers. I. Linear viscoelastic behavior analyzed through the quasi-point defect theory. J. Polym. Sci. B 1999, 37, 2151–2164. [Google Scholar] [CrossRef]
- Terech, P.; Chazeau, L.; Cavaille, J.Y. A small-angle scattering study of cellulose whiskers in aqueous suspensions. Macromolecules 1999, 32, 1872–1875. [Google Scholar] [CrossRef]
- Ruiz, M.M.; Cavaille, J.Y.; Dufresne, A.; Graillat, C.; Gerard, J.F. New waterborne epoxy coatings based on cellulose nanofillers. Macromol. Symp. 2001, 169, 211–222. [Google Scholar] [CrossRef]
- Heux, L.; Bonini, C. Dispersion de microfibrilles et/ou de microcristaux, notamment de cellulose, dans un solvant organique. French Patent FR2794762, 14 June 1999. [Google Scholar]
- Marchessault, R.H.; Morehead, F.F.; Walter, N.M. Liquid crystal systems from fibrillar polysaccharides. Nature 1959, 184, 632–633. [Google Scholar] [CrossRef]
- Heux, L.; Chauve, G.; Bonini, C. Nonflocculating and chiral-nematic self-ordering of cellulose microcrystals suspensions in nonpolar solvents. Langmuir 2000, 16, 8210–8212. [Google Scholar] [CrossRef]
- Ljungberg, N.; Bonini, C.; Bortolussi, F.; Boisson, C.; Heux, L.; Cavaille, J.Y. New nanocomposite material reinforced with cellulose whiskers in atactic polypropylene: Effect of surface and dispersion cheracteristics. Biomacromolecules 2005, 6, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, N.; Cavaille, J.Y.; Heux, L. Nanocomposites of isotactic polypropylene reinforced with rod-like cellulose whiskers. Polymer 2006, 47, 6285–6292. [Google Scholar] [CrossRef]
- Azizi Samir, M.A.S.; Alloin, F.; Sanchez, J.Y.; El Kissi, N.; Dufresne, A. Preparation of cellulose whiskers reinforced nanocomposites from an organic medium suspension. Macromolecules 2004, 37, 1386–1393. [Google Scholar]
- Azizi Samir, M.A.S.; Alloin, F.; Sanchez, J.Y.; Dufresne, A. Cross-linked nanocomposites polymer eletrolytes reinforced with cellulose whiskers. Macromolecules 2004, 37, 4839–4844. [Google Scholar] [CrossRef]
- Viet, D.; Beck-Candanedo, S.; Gray, D.G. Dispersion of cellulose nanocrystals in polar organic solvents. Cellulose 2007, 14, 109–113. [Google Scholar] [CrossRef]
- van den Berg, O.; Capadona, J.R.; Weder, C. Preparation of homogeneous dispersions of tunicate cellulose whiskers in organic solvents. Biomacromolecules 2007, 8, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Ly, B.; Thielemans, W.; Dufresne, A.; Chaussy, D.; Belgacem, M.N. Surface functionalization of cellulose fibres and their incorporation in renewable polymeric matrices. Compos. Sci. Technol. 2008, 68, 3193–3201. [Google Scholar] [CrossRef]
- Nair, K.G.; Dufresne, A.; Gandini, A.; Belgacem, M.N. Crab shell chitin whiskers reinforced natural rubber nanocomposites. 3. Effect of chemical modification of chitin whiskers. Biomacromolecules 2003, 4, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Gousse, C.; Chanzy, H.; Excoffier, G.; Soubeyrand, L.; Fleury, E. Stable suspensions of partially silylated cellulose whiskers dispersed in organic solvents. Polymer 2002, 43, 2645–2651. [Google Scholar] [CrossRef]
- Gousse, C.; Chanzy, H.; Cerrada, M.L.; Fleury, E. Surface silylation of cellulose microfibrils: Preparation and rheological properties. Polymer 2004, 45, 1569–1575. [Google Scholar] [CrossRef]
- Lin, N.; Chen, G.; Huang, J.; Dufresne, A.; Chang, P.R. Effects of polymer-grafted natural nanocrystals on the structure and mechanical properties of poly(lactic acid): A case of cellulose whiskers-graft-polycaprolactone. J. Appl. Polym. Sci. 2009, 113, 3417–3425. [Google Scholar] [CrossRef]
- Lönnberg, H.; Fogelström, L.; Samir, S.; Berglund, L.A.; Malmström, E.; Hult, A. Surface grafting of microfibrillated cellulose with poly(ε-caprolactone)—Synthesis and characterization. Europ. Polym. J. 2008, 44, 2991–2997. [Google Scholar] [CrossRef]
- Berlioz, S.; Molina-Boisseau, S.; Nishiyama, Y.; Heux, L. Gas-phase surface esterification of cellulose microfibrils and whiskers. Biomacromolecules 2009, 10, 2144–2151. [Google Scholar] [CrossRef]
- Halpin, J.C.; Kardos, J.L. Moduli of crystalline polymers emploing composites theory. J. Appl. Phys. 1972, 43, 2235–2241. [Google Scholar] [CrossRef]
- Takayanagi, M.; Uemura, S.; Minami, S. Application of equivalent model method to dynamic rheo-optical properties of a crystalline polymer. J. Polym. Sci. C 1964, 5, 113–122. [Google Scholar]
- Ouali, N.; Cavaille, J.Y.; Perez, J. Elastic, viscoelastic and plastic behavior of multiphase polymer blends. Plast. Rubber Comp. Process. Appl. 1991, 16, 55–60. [Google Scholar]
- Dufresne, A. Comparing the mechanical properties of high performances polymer nanocomposites from biological sources. J. Nanosci. Nanotechnol. 2006, 6, 322–330. [Google Scholar] [PubMed]
- Dufresne, A.; Vignon, M.R. Improvement of starch film performances using cellulose microfibrils. Macromolecules 1998, 31, 2693–2696. [Google Scholar] [CrossRef]
- Leitner, J.; Hinterstoisser, B.; Wastyn, M.; Keckes, J.; Gindl, W. Sugar beet cellulose nanofibril-reinforced composites. Cellulose 2007, 14, 419–425. [Google Scholar] [CrossRef]
- Seydibeyoglu, M.O.; Oksman, K. Novel nanocomposites based on polyurethane and micro fibrillated cellulose. Compos. Sci. Technol. 2008, 68, 908–914. [Google Scholar] [CrossRef]
- Wu, Q.J.; Henriksson, M.; Liu, X.; Berglund, L.A. A high strength nanocomposite based on microcrystalline cellulose and polyurethane. Biomacromolecules 2007, 8, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Nakagaito, A.N.; Fujimura, A.; Sakai, T.; Hama, Y.; Yano, H. Production of microfibrillated cellulose (MFC)-reinforced polylactic acid (PLA) nanocomposites from sheets obtained by a papermaking-like process. Compos. Sci. Technol. 2009, 69, 1293–1297. [Google Scholar] [CrossRef]
- Angles, M.N.; Dufresne, A. Plasticized starch/tunicin whiskers nanocomposites. 1. Structural analysis. Macromolecules 2000, 33, 8344–8353. [Google Scholar] [CrossRef]
- Azizi Samir, M.A.S.; Alloin, F.; Sanchez, J.Y.; Dufresne, A. Cellulose nanocrystals reinforced poly(oxyethylene). Polymer 2004, 45, 4149–4157. [Google Scholar] [CrossRef]
- Lu, Y.S.; Weng, L.H.; Cao, X.D. Biocomposites of plasticized starch reinforced with cellulose crystallites from cottonseed linter. Macromol. Biosci. 2005, 5, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.P.; Dufresne, A. Morphological investigation of nanocomposites from sorbitol plasticized starch and tunicin whiskers. Biomacromolecules 2002, 3, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Morin, A.; Dufresne, A. Nanocomposites of chitin whiskers from Riftia tubes and poly(caprolactone). Macromolecules 2002, 35, 2190–2199. [Google Scholar] [CrossRef]
- Petersson, L.; Mathew, A.P.; Oksman, K. Dispersion and properties of cellulose nanowhiskers and layered silicates in cellulose acetate butyrate nanocomposites. J. Appl. Polym. Sci. 2009, 112, 2001–2009. [Google Scholar] [CrossRef]
- Syverud, K.; Stenius, P. Strench and barrier properties of MFC films. Cellulose 2009, 16, 75–85. [Google Scholar] [CrossRef]
- Svagan, A.J.; Hedenqvist, M.S.; Berglund, L. Reduced water vapor sorption in cellulose nanocomposites with starch matrix. Comp. Sci. Technol. 2009, 69, 500–506. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and high gas barrier films of cellulose nanofibers prepared by TEMPO-mediated oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Petersson, L.; Oksman, K. Biopolymer based nanocomposites: Comparing layered silicates and microcrystalline cellulose as nanoreinforcement. Comp. Sci. Technol. 2006, 66, 2187–2196. [Google Scholar] [CrossRef]
- Kristo, E.; Biliaderis, C.G. Physical properties of starch nanocrystal-reinforced pullulan films. Carbohyd. Polym. 2007, 68, 146–158. [Google Scholar] [CrossRef]
- Saxena, A.; Ragauskas, A.J. Water transmission barrier properties of biodegradable films based on cellulosic whiskers and xylan. Carbohyd. Polym. 2009, 78, 357–360. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Siqueira, G.; Bras, J.; Dufresne, A. Cellulosic Bionanocomposites: A Review of Preparation, Properties and Applications. Polymers 2010, 2, 728-765. https://doi.org/10.3390/polym2040728
Siqueira G, Bras J, Dufresne A. Cellulosic Bionanocomposites: A Review of Preparation, Properties and Applications. Polymers. 2010; 2(4):728-765. https://doi.org/10.3390/polym2040728
Chicago/Turabian StyleSiqueira, Gilberto, Julien Bras, and Alain Dufresne. 2010. "Cellulosic Bionanocomposites: A Review of Preparation, Properties and Applications" Polymers 2, no. 4: 728-765. https://doi.org/10.3390/polym2040728
APA StyleSiqueira, G., Bras, J., & Dufresne, A. (2010). Cellulosic Bionanocomposites: A Review of Preparation, Properties and Applications. Polymers, 2(4), 728-765. https://doi.org/10.3390/polym2040728