Polyelectrolyte Complex Nanoparticles of Poly(ethyleneimine) and Poly(acrylic acid): Preparation and Applications


Abstract
: In this contribution we outline polyelectrolyte (PEL) complex (PEC) nanoparticles, prepared by mixing solutions of the low cost PEL components poly(ethyleneimine) (PEI) and poly(acrylic acid) (PAC). It was found, that the size and internal structure of PEI/PAC particles can be regulated by process, media and structural parameters. Especially, mixing order, mixing ratio, PEL concentration, pH and molecular weight, were found to be sensible parameters to regulate the size (diameter) of spherical PEI/PAC nanoparticles, in the range between 80–1,000 nm, in a defined way. Finally, applications of dispersed PEI/PAC particles as additives for the paper making process, as well as for drug delivery, are outlined. PEI/PAC nanoparticles mixed directly on model cellulose film showed a higher adsorption level applying the mixing order 1. PAC 2. PEI compared to 1. PEI 2. PAC. Surface bound PEI/PAC nanoparticles were found to release a model drug compound and to stay immobilized due to the contact with the aqueous release medium.1. Introduction
Progressively, polymeric nanoparticles are used for the immobilization, storage, carriage and release of proteins or drugs. In that respect, the nano dimension offers a high surface/volume ratio and the correlation with structural sizes of biological components. Widely known polymeric nanoparticular systems are, e.g., polymer liposomes [1,2], or hollow PEL shelled capsules [3,4]. Partly related to these systems, the aim of our work is to prepare nanoparticles on the basis of PEL complexes (PEC) by mixing polycation and polyanion solutions in nonstoichmetric ratios, as it was introduced and reviewed by these authors [5-10], and to explore their potential to interact in a useful manner with pharmaceutically and biomedically relevant compounds. Main issues of our research are dedicated to the reproducibility in the preparation protocol, the size and shape uniformity, the conservation of colloidal stability after binding of compounds and the interaction to surfaces. In typical PEC systems standard cationic and anionic PELs and PELs of natural origin like polypeptides, polysaccharides and their modified analogues are combined. Beside PEL structural parameters, important parameters were found to be the molar mixing ratio of charged units (n−/n+), concentration, pH and ionic strength, which can regulate particle size, distribution and shape [11,12] and the interaction to surfaces [13-16]. Dynamic light scattering (DLS) and colloid titration were applied on the dispersions as well as scanning force microscopy (SFM) and attenutaed total reflection (ATR) Fourier transform infrared (FTIR) spectroscopy on particle layers. Recently, the monomodality of PEC dispersions could be significantly improved applying consecutive centrifugation, separation and redispersion steps of the coacervate phase, which was explained by “accelerated ripening” (Ostwald) of the raw dispersion [12]. This experimental finding was proved by recent simulation studies [17] supporting, that the PEC formation process is subdivided in the rapid formation of molecular or primary complex particles (RH = 5–20 nm), which is followed by aggregation of primary particles to secondary particles. This proposed scenario is schemed in Figure 1.
Primary particles are suggested to consist ideally of only one or realistically a few polycation/polyanion pairs held together by long range electrostatic interactions. Since the whole PEC formation process is claimed to be athermal [5,19], the driving force of the evidently occurring polycation/polyanion pairing is claimed to be the gain of entropy, when the respective counterions are released from their parent PEL backbone. Whereas, secondary particles, the final ones you find in any PEC dispersion, consist of some 100 primary PEC particles held together by short range dispersive interactions. It might be speculated, if this second process might be a slight enthalpic one, since no entropy gain is expected during this process. The purpose of that contribution is to introduce PEC particles restricted to and made of the commercially available low cost PELs branched poly(ethyleneimine) (PEI) and poly(acrylic acid) (PAC), and illustrate the particle size control by process parameters like mixing ratio (n−/n+, charged monomer units) and mixing order and typical wet chemical parameters like PEL concentration (cPEL) and pH and structural parameters like the molecular weight (Mw). Finally, application potential of PEI/PAC particles for paper making and for drug delivery is outlined.
To the best of our knowledge no comparable extensive study has been published on PEC particles of PEI/PAC and the influence of several media parameters, with a special emphasis on the influence of mixing ratio and mixing order. Branched PEI was chosen for several reasons. First of all, branched PEI unlike linear PEI, is commercially available at low cost. Secondly, branched PEI is used for the paper making process, which is related to one of our application examples. Thirdly, PEI is expected to have advantageous properties concerning drug uptake due to its high functional group density. Finally, we have been working for a long time also on PEL multilayers (PEM), composed of PEI/PAC, which serve as an interesting platform to study charge driven protein adsorption [20-22]. Interesting correlations between protein and drug interaction at PEM and at PEC systems are expected in the future.
2. Materials and Methods
2.1. Polyelectrolytes
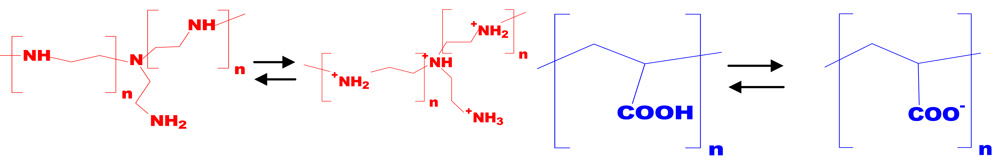
Branched poly(ethyleneimine) (PEI, BASF-SE) of the molecular weights Mw= 1,300, 25,000 and 750,000 g/mol (denoted as PEI-1,300, PEI-25,000, PEI-750,000) were used in the original base form at pH ≈ 10 and in the quarternized form at pH ≈ 7. Generally, these PEI samples are claimed to be highly branched macromolecules containing primary, secondary and tertiary, groups in the ratio of 1:2:1 and that their branching sites are separated mainly by secondary amine groups (one branch for every 3–3.5 N atom within a linear chain [23,24]).
Linear poly(acrylic acid) (PAC, Polysciences) of the molecular weights between Mw = 2,000–5,000,000 g/mol) was used in the original acid form at pH ≈ 4 and the dissociated form at pH ≈ 7, which is shown in Figure 2.
The native PEL solutions before mixing were prepared using deionized refined water (Millipore GmbH, Eschborn, Germany) to give PEL concentrations between cPEL = 0.001–0.01 M without any addition of low molecular salt.
2.2. PEC Nanoparticles
PEL solutions of equal PEL concentration (CPA = CPC) were mixed in a glass beaker using a homebuilt device consisting of a peristaltic pump and a stirring panel. No additional salt was used, if not specifically referred to. The stoichiometric mixing ratios n−/n+ are related to the charged monomer units and were varied from n−/n+ = 0.1–1.8, denoted further as PEC-0.1 –PEC-1.8, respectively. For example, a PEC-0.66 dispersion was prepared by dosing 10 mL of minority component PAC solution dropwise into 15 mL of excess component PEI solution under continuous stirring for 20 min.
2.3. PEC Conjugates
PEC drug conjugates were prepared by dosing a charged drug solution into the oppositely charged equimolar PEL solution applying volume ratios of 1/10 or 1/5 with respect to drug/PEL. Then this solution was either dosed into excess oppositely charged PEL solution or oppositely charged PEL solution was dosed into this solution as excess. Typically, cPEL was 0.002 M.
2.4. Cellulose Film Preparation
Thin cellulose model films were prepared taking reference to a protocol described therein [25] but modified with respect to the type of adhesion promoter, which will be described therein [26]. Generally, instead of a copolymer of maleic acid anhydride herein a polyepoxide compound was used to chemically bind cellulose onto silicon substrates.
2.5. Dynamic Light Scattering
The size of PEC particles was checked by dynamic light scattering (DLS). DLS data were recorded by Zetasizer 3000 (632.8 nm, 10 mW He-Ne Laser, Malvern Instruments, Worcestershire, UK) or by the Jianke Portable Particle Sizer (Jianke Instruments Co. Ltd., Wuhu, P.R. China) applying the scattering angle of 90°. The samples were held either in 10 mm cuvettes with quadratic (Malvern) or circular bottom (Jianke Ltd.). The mean particle radius RH was estimated as hydrodynamic radius using the Stokes-Einstein equation. The shown errors are related to the standard deviation of at least three different measurements. Either the Malvern Software or the ALV-5000/E/EPP-Software of ALV GmbH, Langen, Germany, was used for calculations of DLS parameters.
2.6. ATR-FTIR Spectroscopy
ATR-FTIR measurements were performed on a FTIR spectrometer (Bruker Optik GmbH, Ettlingen, Germany) using the commercial single-beam-sample-reference (SBSR) ATR mirror attachment (OPTISPEC, Zurich, Switzerland) as it is described elsewhere [27]. ATR-FTIR spectra on PEC/drug nanoparticles were performed by casting and slowly drying (50 °C) defined volumes of the respective dispersions onto Ge-ATR crystals and accumulating 200 scans at 2 cm−1 resolution. ATR-FTIR spectra on PEC/cellulose film interaction were recorded on Si-ATR crystals coated by cellulose film (described above) in contact to water. ATR-FTIR spectra on cellulose bound PEC were due to the difference between cellulose film in contact to respective mixtures of PEI and PAC solutions (0.002 M) and the cellulose film in contact to pure deionized water. A more detailed description of ATR-FTIR measurements on the interaction of cellulose films with PEL systems will be reported elsewhere [26].
3. Results and Discussion
3.1. Influence of Mixing Order and Mixing Ratio
3.1.1. Size, Polydispersity, Count Rate
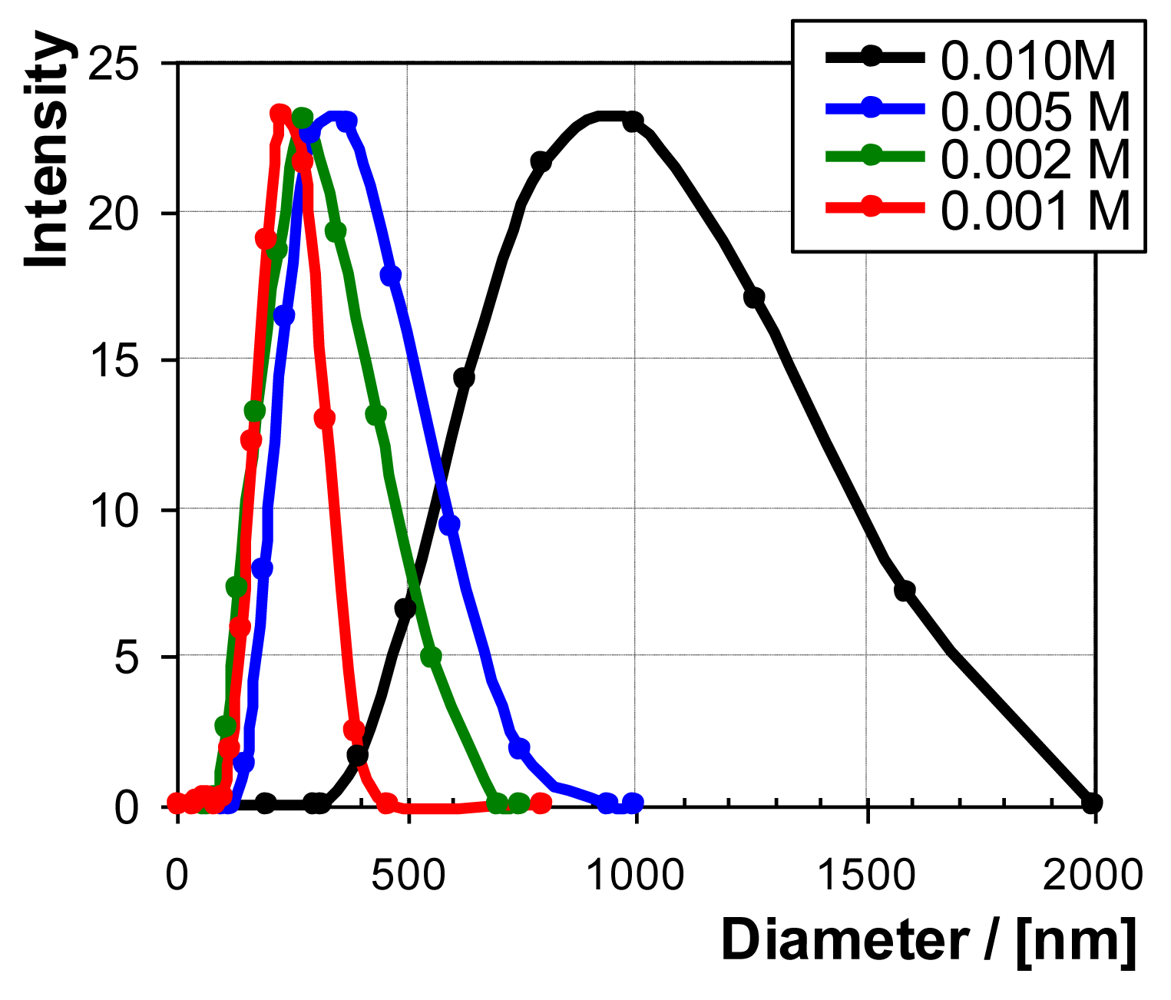
In Figure 3, as DLS raw data, two typical intensity distributions versus particle size are given. PEI with Mw = 750,000 g/mol and PAC with Mw = 50,000 g/mol were used in that series. The red curve represents a PEC dispersion prepared by dosing PAC into PEI solution (0.001 M), denoted further as 1. PEI 2. PAC and the blue curve relates to dosing PAC into PEI solution, denoted further as 1. PAC 2. PEI. In both cases, the same stoichiometric mixing ratio n−/n+ = 0.6 was applied. Such PEC particles or dispersions are further denoted as PEC-0.6 ones. The main DLS parameters are summarized in Table 1.
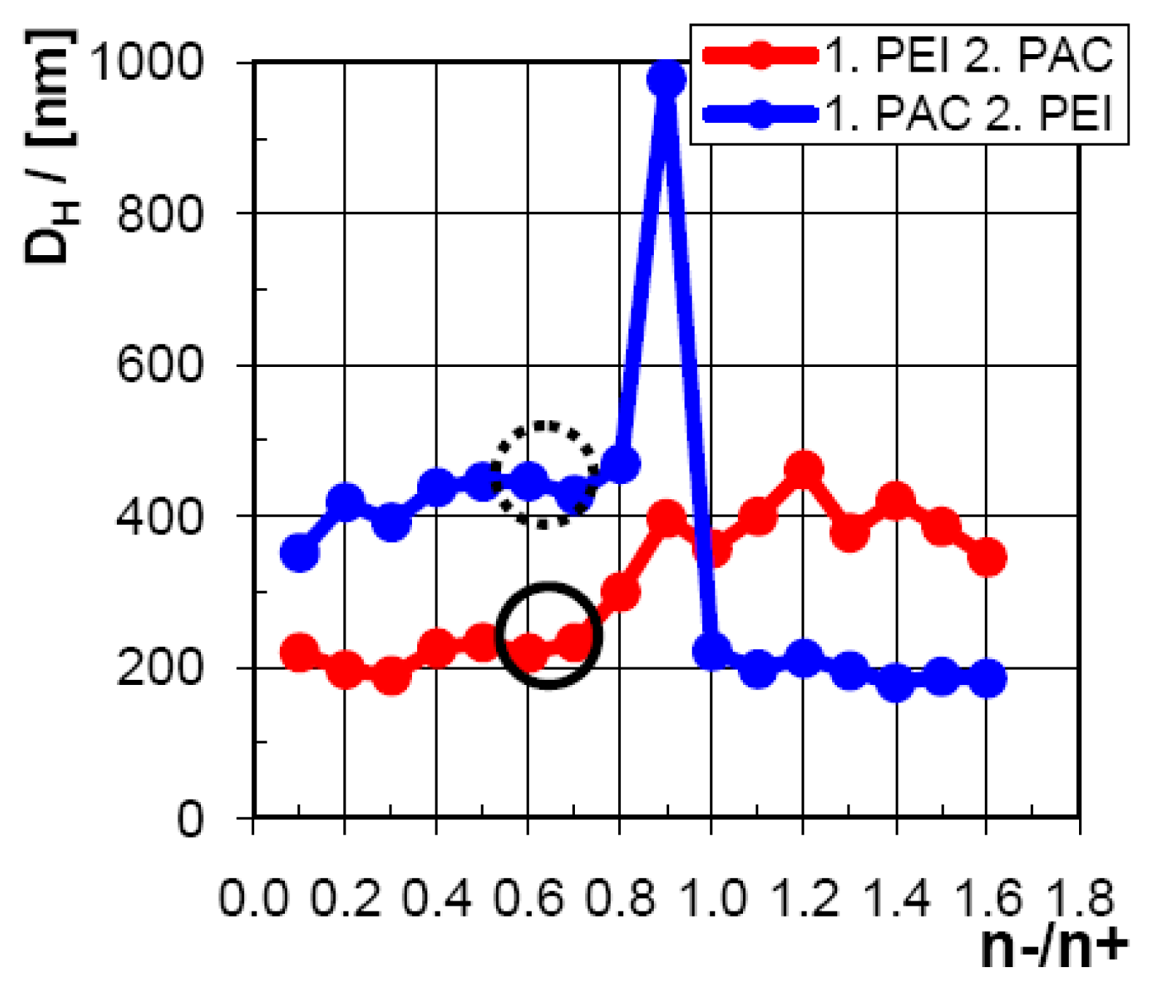
Significantly, from DH = 210 nm for 1. PEI 2. PAC and DH = 450 nm for 1. PAC 2. PEI obvious differences in the size of the formed particles were obtained, although the overall composition of PEI/PAC is the same in both samples. Furthermore, while the polydispersity index (PDI) was around the same for 1. PEI 2. PAC (PDI = 0.07) compared to 1. PAC 2. PEI (PDI = 0.08), the count rate (CR) of around 177 KHz for 1. PEI 2. PAC was very different from CR = 55 KHz for 1. PAC 2. PEI. Additionally, from visual inspection, opaque milk like appearance was observed for 1. PEI 2. PAC and a more transparent microgel-like one for 1. PAC 2. PEI. The whole mixing ratio (n−/n+) profile of the particle size is given in Figure 4.
Obviously, there is a dramatic difference, whether the minority component is given to the majority component, which is related to the interval n−/n+ = 0.1–0.7 for 1. PEI 2. PAC or n−/n+ = 1.0–1.6 for 1. PAC 2. PEI, or the majority is given to the minority component, which is related to the interval n−/n+ = 0.9–1.6 for 1. PEI 2. PAC or n−/n+ = 0.1–0.8 for 1. PAC 2. PEI. For the former case (“minority into majority”) we suggest a more equilibrated consumption of the “dosed in PEL”, since the point of 1:1 stoichiometry (not necessarily exactly at n−/n+ = 1) must not be exceeded, while in the latter case (“majority into minority”), consumption of the “dosed in PEL” is suggested to be rather in a nonequilibrium state, since the 1:1-point must be exceeded. Similar trends have also been found by Schatz [28,29] and Delair [30] for the chitosan/dextrane sulfate system. This step-like behavior of the particle size in dependence of n−/n+, which is diametrally different for 1. PEI 2. PAC compared to 1. PAC 2. PEI, was also obtained for the mixing ratio profile of the count rate, as is shown in Figure 5.
Dosing minority into majority component solution, the count rate increases continuously up to 250–300 KHz at the more or less defined range of 1:1 stoichometry (n−/n+ = 0.8–1.0), which holds for both 1. PEI 2. PAC and 1. PAC 2. PEI scenarios. Exceeding this point (range) of 1:1 stoichometry, or in other words starting to dose majority into minority component solution, the count rate falls off step-like to around 50–100 KHz and more or less keeps this level. Obviously, the step-like rise of the particle size evidenced above is paralleled by a respective drop of the count rate. This could be explained by either a decreasing concentration or structural density of the particles. This lower density of the particles obviously correlates with larger particle sizes.
Based on these findings we suggest a more compact structure for PEI/PAC particles mixed in the “minority-to-majority” compared to the “majority-to-minority” scenario. An explanation is still speculative. However, based on the PEC formation scheme given in Figure 1, according to which the observed secondary PEC particles are aggregates of primary particles, one could rationalize the two scenarios. For the scenario “minority-to-majority”, either cationic (1. PEI 2. PAC) or anionic (1. PAC 2. PEI) secondary PEC particles are “electrosterically” stabilized by the respective excess like charged PEI or PAC component. This is not the case for the scenario “majority-to-minority”, where immediately after exceeding the critical 1:1 stoichometry the respective excess oppositely charged PAC or PEI component can “crosslink” the secondary particles to colloidal networks with lower structural density. We would like to emphasize, that for both scenarios the suggested PEC structures are in nonequilibrium and local and kinetic factors play a substantial role. The scenario “minority-to-majority” might result in more “equilibrated” PEC structures, since the charge sign is never reversed.
3.1.2. Charge
Additionally, the two mixing orders reveal slightly different points of zero charge (PZC) in dependence of the mixing ratio n-/n+, which can be evidenced from Figure 6.
While for 1. PEI 2. PAC the PZC is reached for n−/n+ = 0.7–0.8, for 1. PAC 2. PEI the PZC is shifted to n−/n+ = 0.6. This means that starting with PAC in the beaker, less PEI is needed to compensate to zero charge, while starting with PEI, more PAC is needed for compensation. Presumably, this finding can be seen in line with the results concerning the influence of mixing order on the internal structure of PEI/PAC dispersions, as seen by the count rate and size effects. Loose PEI/PAC particles found in the case of dosing majority into minority component might have outermost PEL shells with different dimensions compared to those formed by the reverse mixing order.
3.2. Influence of PEL Concentration
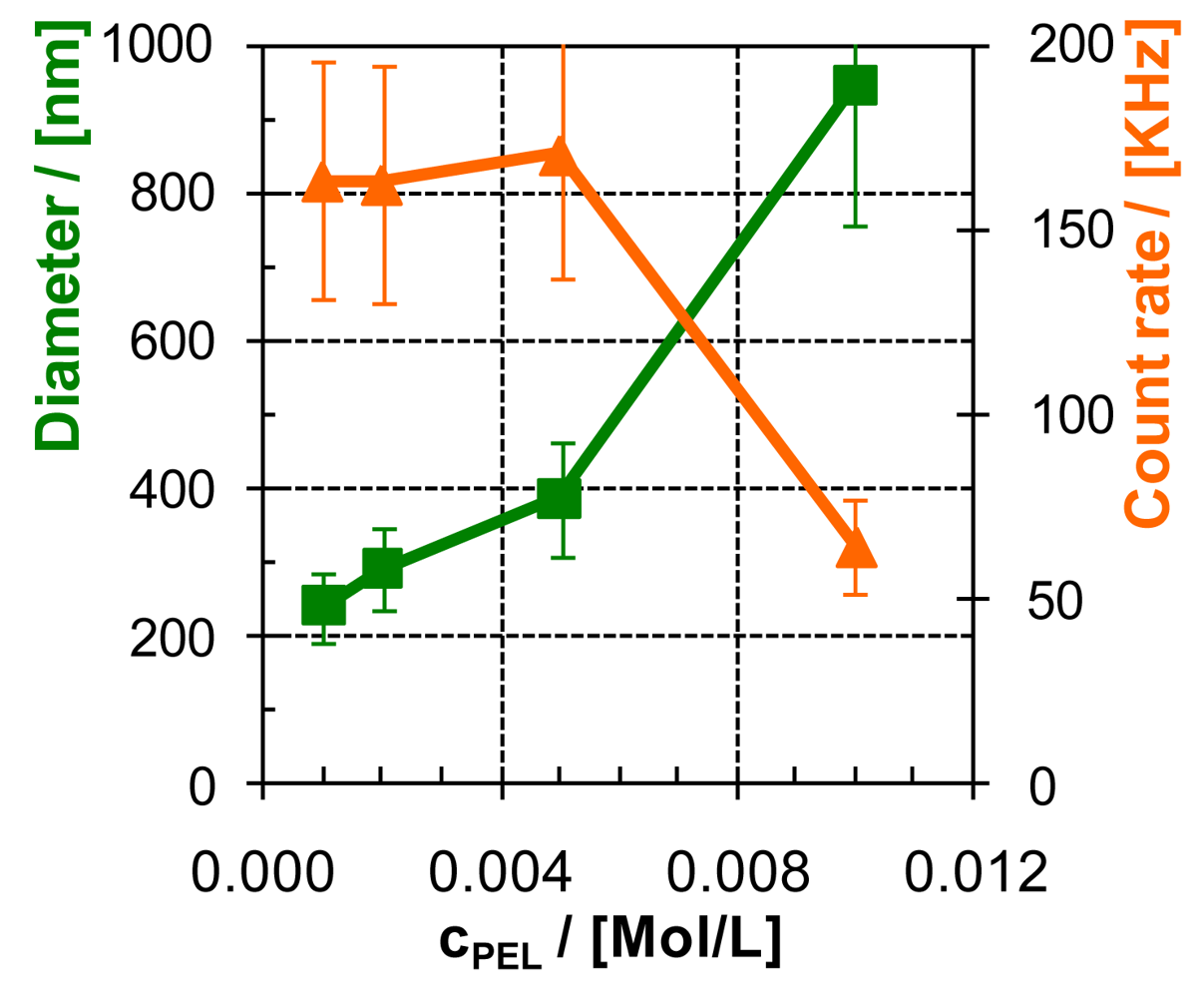
In Figure 7, the dependence of PEC particle size on PEL concentration (cPEL) is given for PEC-0.6 of PEI/PAC as raw DLS intensity distribution data and in Figure 8 as direct plots of particle size and count rate versus cPEL. PEI with Mw = 750,000 g/mol and PAC with Mw = 50,000 g/mol were used in that series.
Generally by cPEL variation, PEC-0.6 particles of PEI/PAC with defined diameters between DH = 200–1,000 nm could be generated. Generally, the PEC particle size increases with cPEL. However, for cPEL > 0.01 M, the PEC dispersions tend to instability, as can be seen from the respective drop in the count rate. As an explanation of the increasing PEC particle size with increasing cPEL, we assume an influence of the Debye length, which is a measure of electrostatic reach. As was pointed out by Wandrey [31], not only increasing salt but also PEL concentration decreases the Debye length of a PEL system. Hence, based on the model of aggregation of primary PEC to secondary PEC particles due to short range dispersive interactions, we suggest that increasing cPEL results in the reduced electrostatic repulsion between like charged primary PEC particles and thus in their elevated dispersive attraction. Furthermore, an increase of cPEL might also result in a larger number of primary PEC particles per volume. Both could lead to larger secondary PEC particle sizes, but exceeding certain cPEL values also to precipitation.
3.3. Influence of pH
In Figure 9 again as raw DLS intensity data, and in Figure 10 the influence of pH combination on PEC particle size, is shown for the PEC system consisting of the weak PELs PEI (750,000 g/mol) and PAC (50,000 g/mol) at the molar mixing ratio n−/n+ = 1.50. Significantly, a decrease of particle size from DH ≈ 400 to ≈ 160 nm with decreasing values of pH = 10, 8.5, 7, 4 of PEI solution at constant pH = 4 of PAC solution is obtained. This trend can be explained by the graded charging up of the PEI, resulting in a rather stretched conformation. Moreover for pH = 4/10 a further size drop was seen, since both PEI and PAC were fully charged.
Again an explanation could be derived based on the accepted model of aggregation of small primary PEC particles to larger secondary particles [17]. Dependent on the model and system, the lower charge screening at lower ionic strength led to the repulsion of primary PEC particles and therefore to lower coagulation tendency and lower particle size. Analogously, the high charged primary particles of PEI/PAC at pH = 4/10 may have also a lower coagulation tendency due to mutual electrostatic repulsion, compared to the lower charged primary particles formed at pH = 10/4 due to electrostatic attraction.
3.4. Influence of the Molecular Weight
In Figure 11 representative raw DLS data for PEC-0.6 mixed at pH = 7/7 varying PEI molecular weight (Mw) and in Figure 12, the influence of molecular weight of PEI and PAC on the size of PEC-0.6 and PEC-1.5 particles, respectively, of PEI/PAC, are shown. In the case of PEC-0.6 particles, only the Mw of PEI was varied, and of PEC-1.5 particles only the Mw of PAC, since, based on the PEC model of Figure 1, we assume the respective excess PEL dominating the shell region and therefore being more effective for particle size changes.
Only for the pH combination of pH = 7/7, and only in the case of PEI, a significant particle size enlargement was obtained from DH = 120 nm (PEI-1,300) to DH = 380 nm (PEI-750,000) upon increasing MW of PEI (Figure 12) for PEC-0.6 particles. For pH = 10/4, both PEC-0.6 particles did not show a significant dependence on the Mw of PEI, for which we have no straight forward explanation. Presumably, in the more compact state of PEI at pH = 10/4, the formed PEI/PAC particles are not so sensitive to Mw variation. Furthermore, neither for pH = 10/4 nor for pH = 7/7, a significant enlargement of particle size with increasing Mw of PAC was obtained. Even for pH = 10/4 rather a decrease of particle size with increasing MW of PAC was obtained. Furthermore, it should be noted that in our studies on PEI/PAC particle size parameters, the smallest particle size of DH = 80 nm was obtained for PEC-1.5, mixed at pH = 7/7 at a Mw = 2,000 g/mol (PAC), which is evident in Figure 12 (first open blue square data point in bottom series).
3.5. Applications
Finally, two potential application fields of PEC particles will be introduced in the following. At first we address PEI/PAC particles, which were used as surface modifiers for cellulose systems being relevant for the papermaking process. In that frame we present recent data on the interaction of PEI/PAC complexes with cellulose model films. Secondly, PEI/PAC particles are addressed as drug delivery particles. In that respect we present recent data on the release of a model drug compound from a deposited PEI/PAC complex film.
3.5.1. Additives for Papermaking
For longer times, one component polycation systems are used in the papermaking industry to increase critical parameters retention and wet/dry strength [32]. In recent times also mixed polycation/polyanion (PEL complex) systems were used for that purpose, which was scientifically studied by, e.g., Wagberg [34] and Pelton [34]. Here we present some related results on the interaction of model cellulose films with mixed systems of PEI and PAC. Exemplarily, in Figure 13 ATR-FTIR spectra are given on PEI/PAC complexes, which were formed on a cellulose film applying similar mixing ratios of n−/n+ = 0.5 but under different mixing orders.
While the bottom ATR-FTIR spectrum (A) is due to dosing 0.001 M PAC solution to 0.001 M PEI solution (1. PEI 2. PAC), the top ATR-FTIR spectrum (B) is due to dosing PEI into PAC solution (1. PAC 2. PEI). A significant difference in the signal intensity of the asymmetric stretching band of carboxylate groups (νa(COO−)) due to cellulose bound PAC was found: The mixing order 1. PAC 2. PEI resulted into around 5-fold higher adsorbed amounts compared to 1. PEI 2. PAC. In Figure 14, the whole profile of the integrated band areas of the νa(COO−) due to bound PAC versus the mixing ratio, is given for the two mixing orders.
Significantly, after exceeding the point of zero charge (PZC), which is located at n−/n+ of around 1.00–1.25, the adsorbed PAC amount drastically increases for 1. PAC 2. PEI, which was not the case for 1. PEI 2. PAC. An explanation for that result is not straightforward. We assume that for 1. PEI 2. PAC initially an adsorbed PEI layer is formed onto the cellulose film, which was slightly negatively charged [35]. Then, upon adding PAC, subsequently positively charged PEI/PAC complexes are formed, which are repelled by the equally charged PEI layer. In contrast, for 1. PAC 2. PEI, initially no PAC layer is formed onto cellulose film due to electrostatic repulsion, which is also not the case for subsequently dosing PEI into PAC solution. However, exceeding the PZC suddenly loose positively charged PEI/PAC particles are formed, as it is claimed in the discussion of Figure 4–6 previously, which can readily adsorb at the cellulose film due to electrostatic attraction. Since we concentrate here only on the νa(COO−) band diagnostic for PAC, we cannot detect the total i.e., PEI and PAC amount. However, from the ATR-FTIR spectra on PEI/PAC complexes of n−/n+ = 0.5 deposited under different mixing orders a qualitative information can be obtained by the negative v(OH) band due to desorbed water [36]. Obviously, the amount of desorbed water, which correlates with the sorbed PEI/PAC amount, is also higher for 1. PAC 2. PEI compared to 1. PEI 2. PAC.
3.5.2. PEC/drug Conjugates
We selected malachite green (MLG) as a model charged drug compound to be incorporated into PEI/PAC particles, since the release of MLG from surface bound PEC particles can be nicely probed by ATR IR spectroscopy via depletion in the PEC layer, as well as by UV-VIS spectroscopy via the enrichment in the release medium. Preliminary ATR-FTIR results on the uptake and release of malachite green as a model drug compound at immobilized PEC-0.8 particles of PEI/PAC are given in Figure 15.
Therein ATR-FTIR spectra of solution cast dry PEC films of PEI/PAC (n−/n+ = 0.8, cationic) onto the Ge ATR crystal loaded with MLG (A) and after 15 h exposure to H2O followed by drying (B) are shown. At first, the difference spectrum (C) between (B) and (A) shows negative bands (1,585, 1,370, 1,170 cm−1: full lines) diagnostic to MLG, whose positions can be compared to those from spectrum (D) of the dry pure MLG film, from which the release of MLG can be rationalized. Secondly, no negative IR bands from the pure PEI/PAC complex (1,550, 1,400 cm−1: broken lines) can be found in the difference spectrum (C), but IR peaks at these wavenumber positions can be found in both the start (A) and end (B) dry spectrum. This finding is very important, since it is the proof that the releasing matrix stays intact, while the drug is released. Further studies will address the stimulation and optimization of release properties of PEI/PAC particles, the usage of a variety of other more relevant drug compounds, and the biocompatibilty of PEI/PAC particle films.
4. Conclusions
In this contribution we outline PEC nanoparticles of PEI/PAC, prepared by mixing solutions of oppositely charged PELs, whose size and shape can be regulated by process, media and structural parameters like mixing order, mixing ratio, cPEL, cS, pH and Mw, in a defined way from 80 nm up to 1,000 nm.
A significant dependence of PEI/PAC particle size on the mixing order was obtained. Dosing minority into majority PEL solution and not exceeding n−/n+ = 1 compact particle sizes of around DH = 200 nm were obtained, and dosing majority into minority PEL solution loose particles with lower structural density and larger sizes of around DH = 400 nm were obtained.
PEI/PAC particles showed a significant dependence on the pH values of both PEI and PAC solutions, so that increasing the charge density of PEI and PAC, respectively, resulted in smaller particle sizes.
Only in the case of PEI, a significant increase of particle size from 120 nm (PEI-1,300) to 380 nm (PEI-750,000), was found dependent on Mw.
PEI/PAC particles mixed directly on model cellulose film showed a higher adsorption level applying the mixing order 1. PAC 2. PEI compared to 1. PEI 2. PAC, which is of relevance for paper making additives based on mixed (“dual”) or complexed PEL systems.
Surface bound PEI/PAC nanoparticles were found to release a model drug compound and to stay immobilized due to the contact with the aqueous release medium, which is of relevance for PEC particles as surface bound drug delivery systems.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample PEI/PAC for mixing order | DH / [nm] | PDI | CR / [KHz] |
|---|---|---|---|
| 1. PEI 2. PAC | 210 | 0.07 | 177 |
| 1. PAC 2. PEI | 450 | 0.08 | 55 |
Acknowledgments
A part of this work is supported by German Research Foundation (DFG) within Transregio SFB TRR79 (M7) involving the universities and cooperating research institutes located in Giessen (Head University), Heidelberg and Dresden.
References
- Lasic, D.D. Sterically stabilized vesicles. Angew. Chem. Int. Ed. 1994, 33, 1685–1698. [Google Scholar]
- Antonietti, M.; Förster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar]
- Donath, E.; Sukhorukov, G.B.; Caruso, F.; Davis, S.; Möhwald, H. Novel hollow polymer shells by colloid-templated assembly of polyelectrolytes. Angew. Chem. Int. Ed. 1998, 37, 2202–2205. [Google Scholar]
- Sukhorukov, G.B.; Rogach, A.L.; Zebli, B.; Liedl, T.; Skirtach, A.G.; Köhler, K.; Antipov, A.A.; Gaponik, N.; Susha, A.S.; Winterhalter, M.; Parak, W.J. Nanoengineered polymer capsules: Tools for detection, controlled delivery, and site-specific manipulation. Small 2005, 182, 194–200. [Google Scholar]
- Michaels, A.S. Polyelectrolyte complexes. Ind. Eng. Chem. 1965, 57, 32. [Google Scholar]
- Kabanov, V.A.; Zezin, A.B. Soluble interpolymeric complexes as a new class of synthetic polyelectrolytes. Pure Appl. Chem. 1984, 56, 343. [Google Scholar]
- Philipp, B.; Dautzenberg, H.; Linow, K.J.; Kötz, J.; Dawydoff, W. Polyelectrolyte complexes— Recent developments and open problems. Progr. Polym. Sci. 1989, 14, 91. [Google Scholar]
- Dubin, P.; Bock, J.; Davies, R.M.; Schulz, D.N.; Thies, C. Macromolecular Complexes in Chemistry and Biology; Springer-Verlag: Berlin, Germany, 1994. [Google Scholar]
- Harada, A.; Kataoka, K. Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely charged block-copolymers with poly(ethylene glycol) segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar]
- Harada-Shiba, M.; Yamauchi, K.; Harada, A.; Takamisawa, I.; Shimokado, K.; Kataoka, K. Polyion complex micelles as vectors in gene therapy–pharmacokinetics and in vivo gene transfer. Gene Therapy 2002, 9, 407–414. [Google Scholar]
- Müller, M.; Keßler, B.; Richter, S. Needlelike and spherical polyelectrolyte complex nanoparticles of poly(L-lysine) and copolymers of maleic acid. Langmuir 2005, 21, 7044–7051. [Google Scholar]
- Müller, M.; Reihs, T.; Ouyang, W. Preparation of monomodal polyelectrolyte complex nanoparticles of PDADMAC/poly(maleic acid-alt-alpha-methylstyrene) by consecutive centrifugation. Langmuir 2005, 21, 465–469. [Google Scholar]
- Oertel, U.; Buchhammer, H.M.; Müller, M.; Nagel, J.; Braun, H.G.; Eichhorn, K.J.; Sahre, K. Surface modification by polyelectrolytes: Studies on model systems. Macromol. Symp. 1999, 145, 39–47. [Google Scholar]
- Thünemann, A.F.; Müller, M.; Dautzenberg, H.; Joanny, J.F.; Löwen, H. Polyelectrolyte complexes. Adv. Polym. Sci. 2004, 166, 113–171. [Google Scholar]
- Reihs, T.; Müller, M.; Lunkwitz, K. Preparation and adsorption of refined polyelectrolyte complex nanoparticles. J. Coll. Interf. Sci. 2004, 271, 69–79. [Google Scholar]
- Reihs, T.; Müller, M.; Lunkwitz, K. Deposition of polylelectrolyte complex nano-particles at silica surfaces characterized by ATR-FTIR and SEM. Coll. Surf. A 2003, 212, 79–95. [Google Scholar]
- Starchenko, V.; Müller, M.; Lebovka, N. Growth of polyelectrolyte complex nanoparticles: Computer Simulations and experiments. J. Phys. Chem. C 2008, 112, 8863–8869. [Google Scholar]
- Müller, M.; Starchenko, V.; Lebovka, N.; Ouyang, W.; Keßler, B. Preparation and life science applications of polyelectrolyte complex nanoparticles. Curr. Trends Polym. Sci. 2009, 13, 1–10. [Google Scholar]
- Feng, X.; Leduc, M.; Pelton, R.H. Polyelectrolyte complex characterization with isothermal titration calorimetry and colloid titration. Coll. Surf. A 2008, 317, 535–542. [Google Scholar]
- Müller, M.; Rieser, T.; Lunkwitz, K.; Berwald, S.; Meier-Haack, J.; Jehnichen, D. An in-situ ATR-FTIR study on polyelectrolyte multilayer assemblies on solid surfaces and their susceptibiliy to fouling. Macromol. Rapid Commun. 1998, 19, 333. [Google Scholar]
- Müller, M.; Rieser, T.; Dubin, P.; Lunkwitz, K. Selective interaction between proteins and the outermost surface of polyelectrolyte multilayers: Influence of the polyanion type, pH and salt. Macromol. Rapid Commun. 2001, 22, 390–395. [Google Scholar]
- Müller, M.; Keßler, B.; Houbenov, N.; Bohata, K.; Pientka, Z.; Brynda, E. pH Dependence and protein selectivity of poly(ethyleneimine)/poly(acrylic acid) multilayers studied by in-situ ATR-FTIR spectroscopy. Biomacromolecules 2006, 7, 1285–1294. [Google Scholar]
- Dick, C.R.; Ham, G.E. Characterization of polyethyleneimine. J. Macromol. Sci. Chem. 1970, A4, 130. [Google Scholar]
- Horn, D. Polyethyleneimine-Physicochemical properties and applications. In Polymeric Amines and Ammonium Salts; Goethals, E.J., Ed.; Pergamon Press: Oxford, UK, 1980; p. 333. [Google Scholar]
- Freudenberg, U.; Zschoche, S.; Simon, F.; Janke, A.; Schmidt, K.; Behrens, S.H.; Auweter, H.; Werner, C. Covalent immobilization of cellulose layers onto maleic anhydride copolymer thin films. Biomacromolecules 2005, 6, 1628–1634. [Google Scholar]
- Fröhlich, J.; Poeschla, S.; Keβler, B.; Müller, M. Adsorption of polyelectrolyte systems at cellulose model films. Unpublished work, 2011.
- Fringeli, U.P. Internal Reflection Spectroscopy, Theory and Applications; Mirabella, F.M., Ed.; Dekker: New York, NY, USA, 1992; p. 255. [Google Scholar]
- Schatz, C.; Domard, A.; Viton, C.; Pichot, C.; Delair, T. Versatile and efficient formation of colloids of biopolymer-based polyelectrolyte complexes. Biomacromolecules 2004, 5, 1882–1892. [Google Scholar]
- Schatz, C.; Lucas, J.M.; Viton, C.; Domard, A.; Pichot, C.; Delair, T. Formation and properties of positively charged colloids based on polyelectrolyte complexes of biopolymers. Langmuir 2004, 20, 7766–7778. [Google Scholar]
- Drogoz, A.; David, L.; Rochas, C.; Domard, A.; Delair, T. Polyelectrolyte complexes from polysaccharides: Formation and stoichiometry monitoring. Langmuir 2007, 23, 10950–10958. [Google Scholar]
- Wandrey, C.; Hunkeler, D.; Wendler, U.; Jaeger, W. Counterion activity of highly charged strong polyelectrolytes. Macromolecules 2000, 33, 7136. [Google Scholar]
- Wagberg, L.; Hägglund, R. Kinetics of Poyelectrolyte Adsorption on Cellulosic Fibers. Langmuir 2001, 17, 1096–1103. [Google Scholar]
- Wagberg, L. Polyelectrolyte complexes for surface modification of wood fibres, II. Influence of complexes on wet and dry strength of paper. Coll. Surf. A 2003, 218, 137–149. [Google Scholar]
- Feng, X.; Pouw, K.; Leung, V.; Pelton, R.H. Adhesion of colloidal polyelectrolyte complexes to wet cellulose. Biomacromolecules 2007, 8, 2161–2166. [Google Scholar]
- Müller, M.; Werner, C.; Grundke, K.; Eichhorn, K.J.; Jacobasch, H.J. Thermodynamic and spectroscopic characterization of the adsorption of plasma proteins onto cellulosic substrates. Macromol. Symp. 1996, 103, 55–72. [Google Scholar]
- Müller, M. ATR-FTIR spectroscopy at polyelectrolyte multilayer systems. In Handbook of Polyelectrolytes and Their Applications; Tripathy, S.K., Kumar, J., Nalwa, H.S., Eds.; American Scientific Publishers (ASP): Valencia, CA, USA, 2002; Volume 1, pp. 293–312. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Müller, M.; Keßler, B.; Fröhlich, J.; Poeschla, S.; Torger, B. Polyelectrolyte Complex Nanoparticles of Poly(ethyleneimine) and Poly(acrylic acid): Preparation and Applications. Polymers 2011, 3, 762-778. https://doi.org/10.3390/polym3020762
Müller M, Keßler B, Fröhlich J, Poeschla S, Torger B. Polyelectrolyte Complex Nanoparticles of Poly(ethyleneimine) and Poly(acrylic acid): Preparation and Applications. Polymers. 2011; 3(2):762-778. https://doi.org/10.3390/polym3020762
Chicago/Turabian StyleMüller, Martin, Bernd Keßler, Johanna Fröhlich, Sebastian Poeschla, and Bernhard Torger. 2011. "Polyelectrolyte Complex Nanoparticles of Poly(ethyleneimine) and Poly(acrylic acid): Preparation and Applications" Polymers 3, no. 2: 762-778. https://doi.org/10.3390/polym3020762
APA StyleMüller, M., Keßler, B., Fröhlich, J., Poeschla, S., & Torger, B. (2011). Polyelectrolyte Complex Nanoparticles of Poly(ethyleneimine) and Poly(acrylic acid): Preparation and Applications. Polymers, 3(2), 762-778. https://doi.org/10.3390/polym3020762