Thermal Properties of Aliphatic Polypeptoids

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Methods


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | Tg | |
|---|---|---|
| PSar | 71,000 K·g/mol | 428 K |
| P(N-EtGly) | 52,000 K·g/mol | 396 K |
| P(N-PrGly) | 69,000 K·g/mol | 373 K |
| P(N-PenGly) | 35,000 K·g/mol | 283 K |
2.3. Synthetic Procedures
2.3.1. Monomer Synthesis
2.3.2. Preparation of Polymers
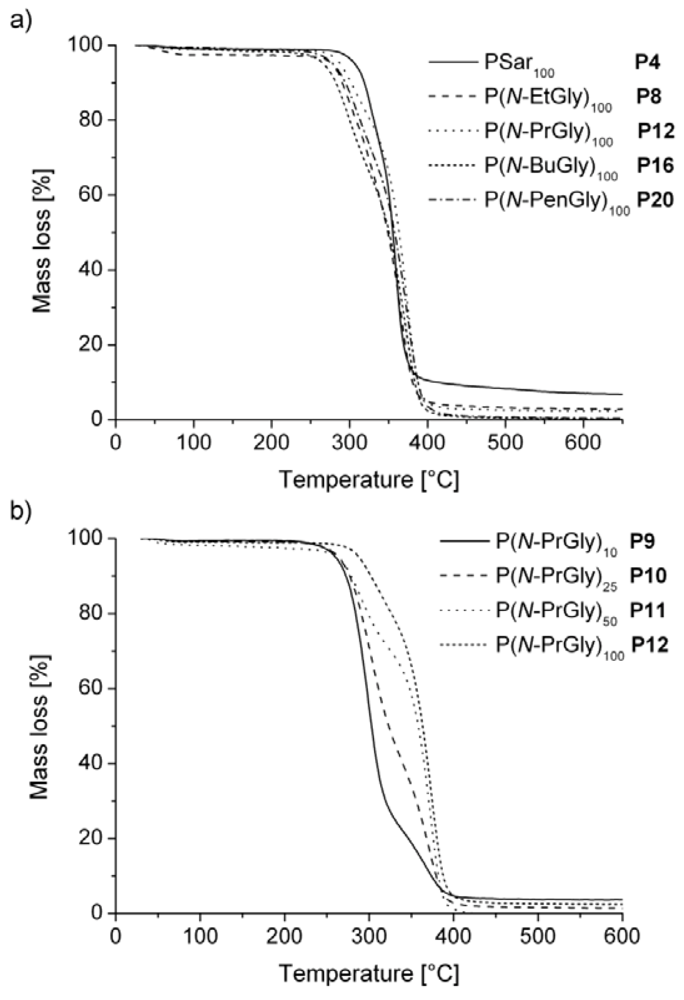
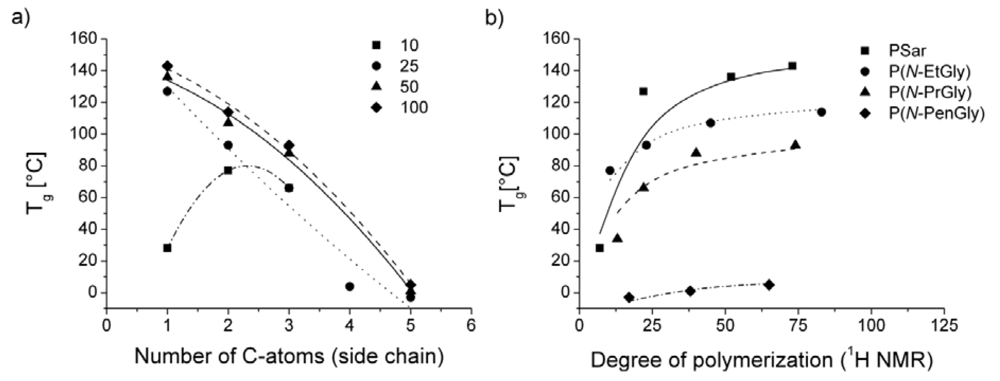
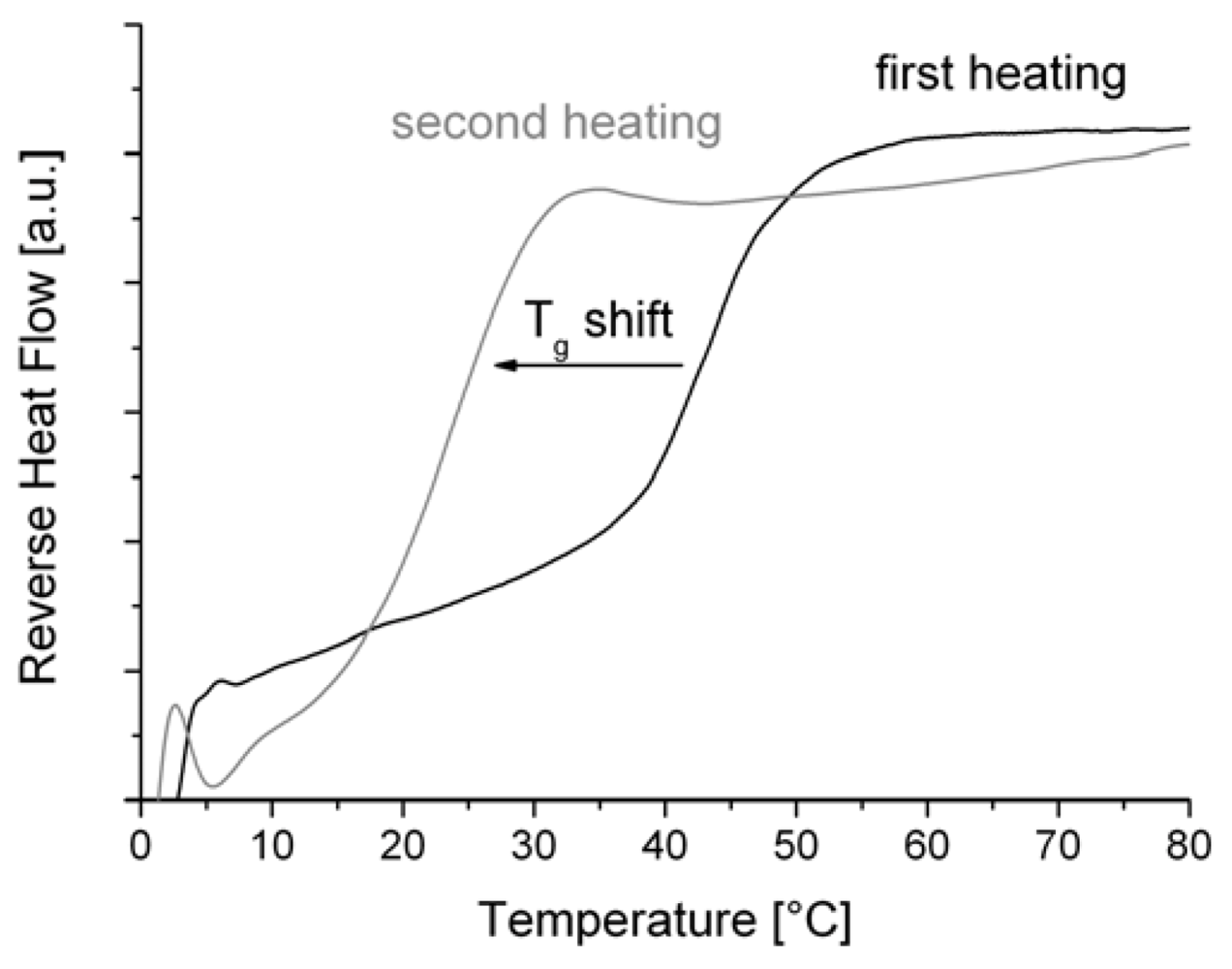
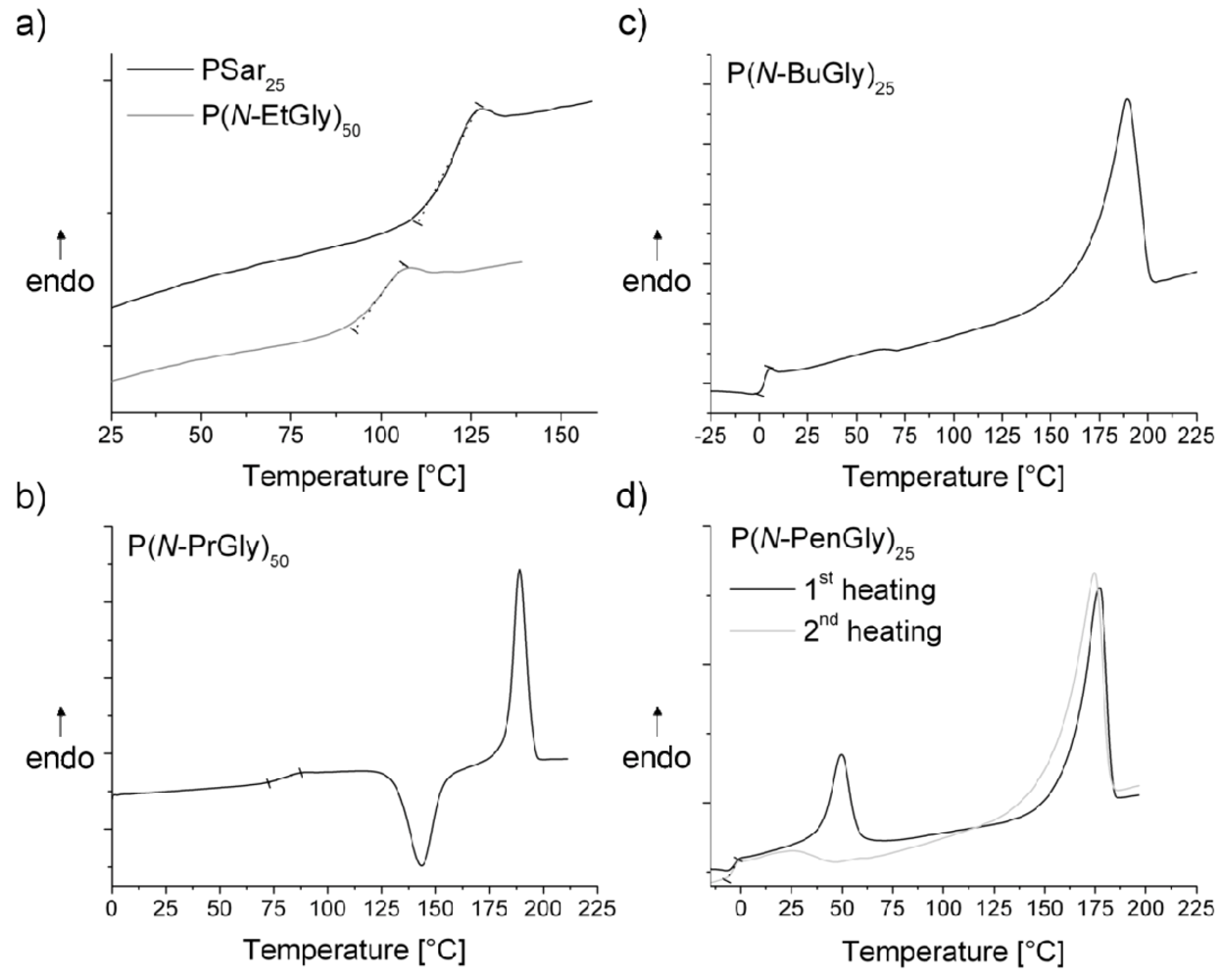
3. Results and Discussion

| Polymer a | Mn b [kg/mol] | ÐM b | Mn c [kg/mol] | ÐM c | Mn d [kg/mol] | DP d | yield [%] | |
|---|---|---|---|---|---|---|---|---|
| P1 | PSar10 | - | - | 0.7 | 1.06 | 0.6 e | 7 e | 65 |
| P2 | PSar25 | 1.3 | 1.31 | 1.6 | 1.08 | 1.7 e | 22 e | 83 |
| P3 | PSar50 | 4.0 | 1.09 | 3.5 | 1.01 | 3.8 e | 52 e | 95 |
| P4 | PSar100 | 8.9 | 1.05 | 7.2 | 1.01 | 5.3 e | 73 e | >99 i |
| P5 | P(N-EtGly)10 | - | - | 1.0 | 1.06 | 1.0 e | 10.5 e | - |
| P6 | P(N-EtGly)25 | 1.0 | 1.65 | 2.0 | 1.05 | 2.1 e | 23 e | 62 |
| P7 | P(N-EtGly)50 | 3.3 | 1.25 | 3.3 | 1.06 | 3.9 e | 45 e | 92 |
| P8 | P(N-EtGly)100 | 6.8 | 1.22 | 6.6 | 1.01 | 7.2 e | 83 e | >99 i |
| P9 | P(N-PrGly)10 | 0.6 | 1.58 | 1.4 | 1.03 | 1.4 f | 13 f | 28 |
| P10 | P(N-PrGly)25 | 1.6 | 1.32 | 2.0 | 1.04 | 2.3 f | 22 f | 68 |
| P11 | P(N-PrGly)50 | 5.0 | 1.22 | 4.2 | 1.04 | 4.1 g | 40 g | 83 |
| P12 | P(N-PrGly)100 | 7.5 | 1.15 | 6.0 | 1.03 | 7.4 f | 74 f | 64 |
| P13 | P(N-BuGly)10 | 0.9 | 1.55 | 1.4 | 1.06 | 1.4 h | 11 h | 89 |
| P14 | P(N-BuGly)25 | 1.9 | 1.27 | 1.9 | 1.16 | 2.2 h | 18.5 h | 93 |
| P15 | P(N-BuGly)50 | 3.2 | 1.40 | 4.4 | 1.04 | 4.4 h | 38 h | 98 |
| P16 | P(N-BuGly)100 | 4.2 | 1.16 | - | - | 6.8 h | 59 h | >99 i |
| P17 | P(N-PenGly)10 | 0.9 | 1.38 | 1.4 | 1.03 | 1.5 h | 11 h | 64 |
| P18 | P(N-PenGly)25 | 2.1 | 1.2 | 1.9 | 1.16 | 2.3 h | 17 h | - |
| P19 | P(N-PenGly)50 | - | - | - | - | 4.9 h | 38 h | >99 i |
| P20 | P(N-PenGly)100 | - | - | - | - | 8.4 h | 65 h | >99 i |

| polymera | Tg [°C] | Tm [°C] | Tc [°C]b | Td [°C]c | |
|---|---|---|---|---|---|
| P1 | PSar10 | 28 | - | - | 175 |
| P2 | PSar25 | 127 | - | - | 210 |
| P3 | PSar50 | 136 | - | - | 215 |
| P4 | PSar100 | 143 | - | - | 250 |
| P5 | P(N-EtGly)10 | 77 | - | - | n.d. |
| P6 | P(N-EtGly)25 | 93 | - | - | 220 |
| P7 | P(N-EtGly)50 | 107 | - | - | 215 |
| P8 | P(N-EtGly)100 | 114 | - | - | 240 |
| P9 | P(N-PrGly)10 | 34 | - | - | 205 |
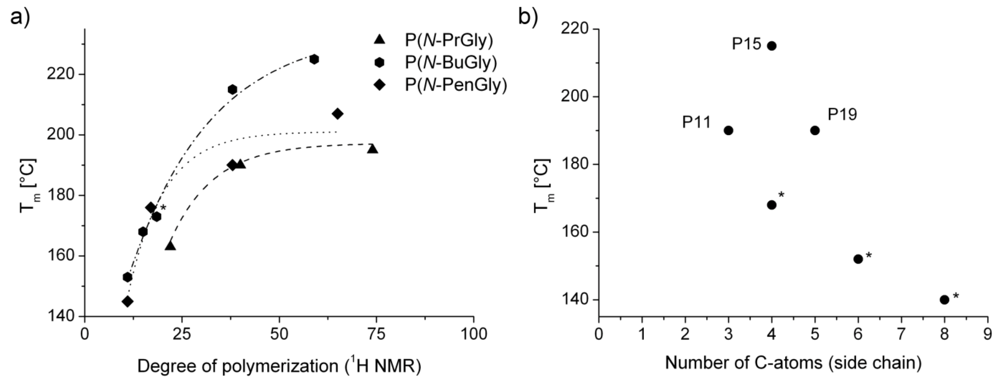
| P10 | P(N-PrGly)25 | 66 | 163 | - | 210 |
| P11 | P(N-PrGly)50 | 88 | 190 | 150 | 205 |
| P12 | P(N-PrGly)100 | 93 | 198 | 166 | 233 |
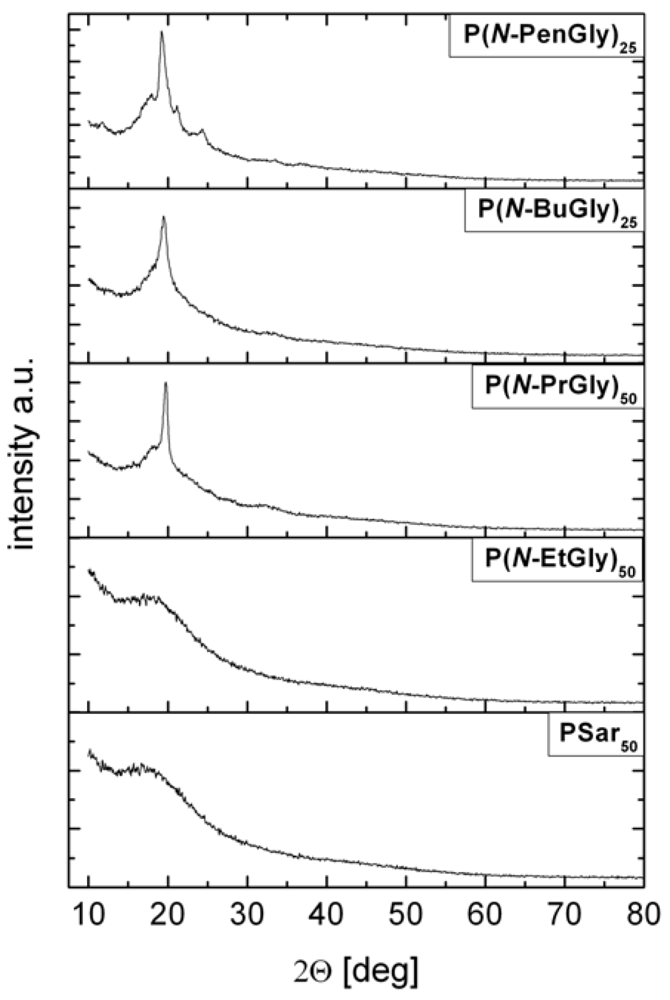
| P13 | P(N-BuGly)10 | - | 153 | - | 210 |
| P14 | P(N-BuGly)25 | 4 | 173 | - | n.d. |
| P15 | P(N-BuGly)50 | - | 215 (2nd Tm = 63 °C) | - | 215 |
| P16 | P(N-BuGly)100 | - | 225 (2nd Tm = 70 °C) | - | 213 |
| P17 | P(N-PenGly)10 | - | 145 (2nd Tm = 56 °C) | - | 205 |
| P18 | P(N-PenGly)25 | −3 | 176 (2nd Tm = 49 °C) | - | n.d. |
| P19 | P(N-PenGly)50 | 1 | 190 (2nd Tm = 70 °C) | - | 220 |
| P20 | P(N-PenGly)100 | 5 | 207 (2nd Tm = 67 °C) | - | 220 |






4. Conclusions
Acknowledgments
References
- Leuchs, H.; Geiger, W. Über die Anhydride von α-Amino-N-Carbonsäuren und die von α-Aminosäuren. Chem. Ber. 1908, 41, 1721–1726. [Google Scholar] [CrossRef]
- Leuchs, H.; Manasse, W. Über die Isomerie der Carbäthoxyl-glycyl glycinester. Chem. Ber. 1907, 40, 3235–3249. [Google Scholar]
- Leuchs, H. Über die Glycin-Carbonsäure. Chem. Ber. 1906, 39, 857–861. [Google Scholar] [CrossRef]
- Sigmund, F.; Wessely, F. Untersuchungen über α-Amino-N-Carbonsäureanhydride. II. H-S Z. Physiol. Chem. 1926, 157, 91–105. [Google Scholar] [CrossRef]
- Wessely, F.; Riedl, K.; Tuppy, H. Untersuchungen über alpha-Amino-N-Carbonsäureanhydride VI. Monatsh. Chem. 1950, 81, 861–872. [Google Scholar] [CrossRef]
- Waley, S.G.; Watson, J. The Kinetics of the Polymerization of Sarcosine Carbonic Anhydride. P. Roy. Soc. Lond. A Mat. 1949, 199, 499–517. [Google Scholar] [CrossRef]
- Bamford, C.H.; Block, H.; Pugh, A.C. P. The polymerization of 3-substituted oxazolidine-2,5-diones. J. Chem. Soc. 1961, 1961, 2057–2063. [Google Scholar]
- Hanby, W.E.; Waley, S.G.; Watson, J. Synthetic Polypeptides. Part I. J. Chem. Soc. 1950, 3009–3013. [Google Scholar]
- Ballard, D.G.; Bamford, C.H. Kinetics of the Formation of Polypeptides from N-Carboxy-α-amino-acid Anhydrides. Nature 1953, 4365, 907–908. [Google Scholar]
- Ballard, D.G.H.; Bamford, C.H. Studies in polymerization. X. “The chain-effect”. P. Roy. Soc. Lond. A Mat. 1956, 236, 384–396. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pitsikalis, M.; Sakellariou, G. Synthesis of well-defined polypeptide-based materials via the ring-opening polymerization of alpha-amino acid N-carboxyanhydrides. Chem. Rev. 2009, 109, 5528–5578. [Google Scholar]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of DL-beta-Phenylalanine N-Carboxyanhydride by Poly(N-n-Propylglycine) Diethylamide. Polym. J. 1970, 1, 198–203. [Google Scholar]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of amino acid derivatives by polymer catalysts. V. Polymerization of DL-β-phenylalanine N-carboxyanhydride by several poly(N-alkylamino acid) diethylamides. Biopolymers 1970, 9, 791–797. [Google Scholar] [CrossRef]
- Sisido, M.; Imanishi, Y.; Okamura, S. Polymerization of amino acid derivatives by polymer catalysts. III. Chain effect polymerization induced by poly(N-ethylglycine) diethylamide. Biopolymers 1969, 7, 937–947. [Google Scholar] [CrossRef]
- Sisido, M.; Imanishi, Y.; Higashimura, T. Molecular weight distribution of polysarcosine obtained by NCA polymerization. Makromol. Chem. 1977, 178, 3107–3114. [Google Scholar] [CrossRef]
- Aoi, K.; Hatanaka, T.; Tsutsumiuchi, K.; Okada, M.; Imae, T. Synthesis of a novel star-shaped dendrimer by radial-growth polymerization of sarcosine N-carboxyanhydride initiated with poly (trimethyleneimine) dendrimer. Macromol. Rapid Commun. 1999, 20, 378–382. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; von Lossow, C.; Schwarz, G. Primary Amine-Initiated Polymerizations of Alanine-NCA and Sarcosine-NCA. Macromol. Chem. Phys. 2004, 205, 918–924. [Google Scholar] [CrossRef]
- Tanisaka, H.; Kizaka-Kondoh, S.; Makino, A.; Tanaka, S.; Hiraoka, M.; Kimura, S. Near-infrared fluorescent labeled peptosome for application to cancer imaging. Bioconjug. Chem. 2008, 19, 109–117. [Google Scholar] [CrossRef]
- Makino, A.; Kizaka-Kondoh, S.; Yamahara, R.; Hara, I.; Kanzaki, T.; Ozeki, E.; Hiraoka, M.; Kimura, S. Near-infrared fluorescence tumor imaging using nanocarrier composed of poly(L-lactic acid)-block-poly(sarcosine) amphiphilic polydepsipeptide. Biomaterials 2009, 30, 5156–5160. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, D. Cyclic poly(alpha-peptoid)s and their block copolymers from N-heterocyclic carbene-mediated ring-opening polymerizations of N-substituted N-carboxylanhydrides. J. Am. Chem. Soc. 2009, 131, 18072–18074. [Google Scholar]
- Guo, L.; Li, J.; Brown, Z.; Ghale, K.; Zhang, D. Synthesis and Characterization of Cyclic and Linear Helical Poly(alpha-peptoid)s by N-Heterocyclic Carbene-Mediated Ring-Opening Polymerizations of N-Substituted N-Carboxyanhydrides. Biopolymers 2011, 96, 596–603. [Google Scholar] [CrossRef]
- Fetsch, C.; Grossmann, A.; Holz, L.; Nawroth, J.F.; Luxenhofer, R. Polypeptoids from N-Substituted Glycine N-Carboxyanhydrides: Hydrophilic, Hydrophobic, and Amphiphilic Polymers with Poisson Distribution. Macromolecules 2011, 44, 6746–6758. [Google Scholar] [CrossRef]
- Fetsch, C.; Luxenhofer, R. Highly Defined Polypeptoids via Multiple Chain Extension and Macroinitiators. Macromol. Rapid. Commun. 2012, 33, 1708–1713. [Google Scholar] [CrossRef]
- Lahasky, S.H.; Serem, W.K.; Guo, L.; Garno, J.C.; Zhang, D. Synthesis and Characterization of Cyclic Brush-Like Polymers by N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Propargyl N-Carboxyanhydride and the Grafting-to Approach. Macromolecules 2011, 44, 9063–9074. [Google Scholar] [CrossRef]
- Robinson, J.W.; Schlaad, H. A versatile polypeptoid platform based on N-allyl glycine. Chem. Commun. 2012, 48, 7835–7837. [Google Scholar] [CrossRef]
- Lahasky, S.H.; Hu, X.; Zhang, D. Thermoresponsive Poly(α-peptoid)s: Tuning the Cloud Point Temperatures by Composition and Architecture. ACS Macro Lett. 2012, 1, 580–584. [Google Scholar] [CrossRef]
- Robinson, J.W.; Secker, C.; Weidner, S.; Schlaad, H. Thermo-responsive Poly(N-C3 glycine)s. Macromolecules 2013, 47. in print. [Google Scholar]
- Zhang, D.; Lahasky, S.H.; Guo, L.; Lee, C.-U.; Lavan, M. Polypeptoid Materials: Current Status and Future Perspectives. Macromolecules 2012, 45, 5833–5841. [Google Scholar] [CrossRef]
- Rosales, A.M.; Murnen, H.K.; Zuckermann, R.N.; Segalman, R.A. Control of Crystallization and Melting Behavior in Sequence Specific Polypeptoids. Macromolecules 2010, 43, 5627–5636. [Google Scholar]
- Aoi, K.; Nakamura, R.; Okada, M. Polypeptide-synthetic polymer hybrids, 2. Miscibility of poly(vinyl alcohol) with polysarcosine. Macromol. Chem. Phys. 2000, 201, 1059–1066. [Google Scholar] [CrossRef]
- Rettler, E.F.J.; Kranenburg, J.M.; Lambermont-Thijs, H.M.L.; Hoogenboom, R.; Schubert, U.S. Thermal, Mechanical, and Surface Properties of Poly(2-N-alkyl-2-oxazoline)s. Macromol. Chem. Phys. 2010, 211, 2443–2448. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Claudy, P.; Létoffe, J.M.; Camberlain, Y.; Pascault, J.P. Glass transition of polystyrene versus molecular weight. Polym. Bull. 1983, 9, 208–215. [Google Scholar] [CrossRef]
- berreiter, K.; Kanig, G. Self-plasticization of polymers. J. Colloid Sci. 1952, 7, 569–583. [Google Scholar] [CrossRef]
- Lee, C.U.; Smart, T.P.; Guo, L.; Epps, T.H.; Zhang, D. Synthesis and Characterization of Amphiphilic Cyclic Diblock Copolypeptoids from N-Heterocyclic Carbene-Mediated Zwitterionic Polymerization of N-Substituted N-carboxyanhydride. Macromolecules 2011, 44, 9574–9585. [Google Scholar] [CrossRef]
- Nam, K.T.; Shelby, S.A.; Choi, P.H.; Marciel, A.B.; Chen, R.; Tan, L.; Chu, T.K.; Mesch, R.A.; Lee, B.C.; Connolly, M.D.; Kisielowski, C.; Zuckermann, R.N. Free-floating ultrathin two-dimensional crystals from sequence-specific peptoid polymers. Nat. Mater. 2010, 9, 454–460. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fetsch, C.; Luxenhofer, R. Thermal Properties of Aliphatic Polypeptoids. Polymers 2013, 5, 112-127. https://doi.org/10.3390/polym5010112
Fetsch C, Luxenhofer R. Thermal Properties of Aliphatic Polypeptoids. Polymers. 2013; 5(1):112-127. https://doi.org/10.3390/polym5010112
Chicago/Turabian StyleFetsch, Corinna, and Robert Luxenhofer. 2013. "Thermal Properties of Aliphatic Polypeptoids" Polymers 5, no. 1: 112-127. https://doi.org/10.3390/polym5010112
APA StyleFetsch, C., & Luxenhofer, R. (2013). Thermal Properties of Aliphatic Polypeptoids. Polymers, 5(1), 112-127. https://doi.org/10.3390/polym5010112