Ring-Opening Polymerization—An Introductory Review

Abstract
:1. Introduction

| Name | Structure | Ring size | Mechanism |
|---|---|---|---|
| Olefin |  | 4,5,8 | Metathesis |
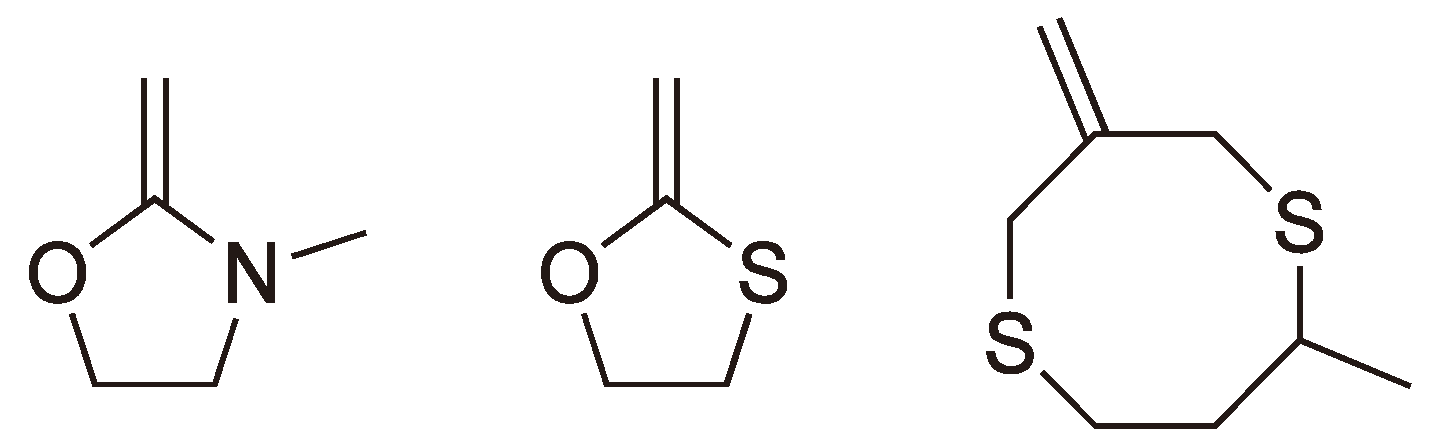
| Ether |  | 3–5,7 | Cationic, anionic |
| Thioether |  | 3,4 | Cationic, anionic |
| Amine |  | 3,4,7 | Cationic |
| Lactone |  | 4,6–8 | Anionic, cationic |
| Thiolactone |  | 4–8 | Anionic, cationic |
| Lactam |  | ≥4 | Anionic, cationic |
| Disulfide |  | ≥8 | Radical |
| Anhydride |  | 5 and ≥7 | Anionic |
| Carbonate |  | 6–8 and ≥20 | Anionic |
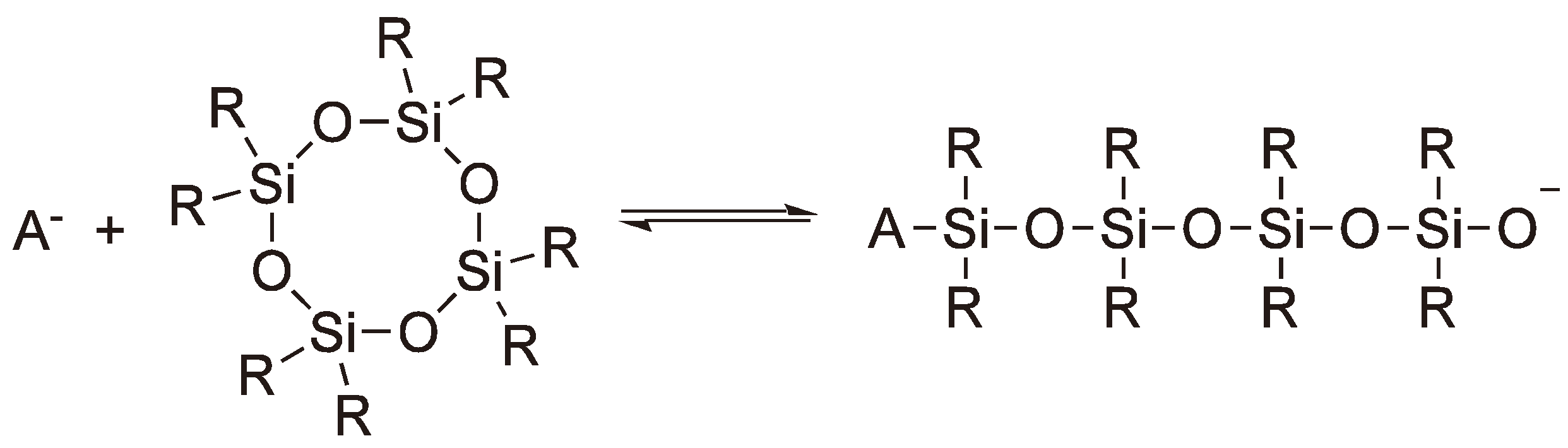
| Silicone |  | 6,8 and ≥10 | Anionic, cationic |
| Phosphazene |  | 6 | Cationic |
| Phosphonite |  | 3,5–7 | Anionic |
2. Radical Ring-Opening Polymerization

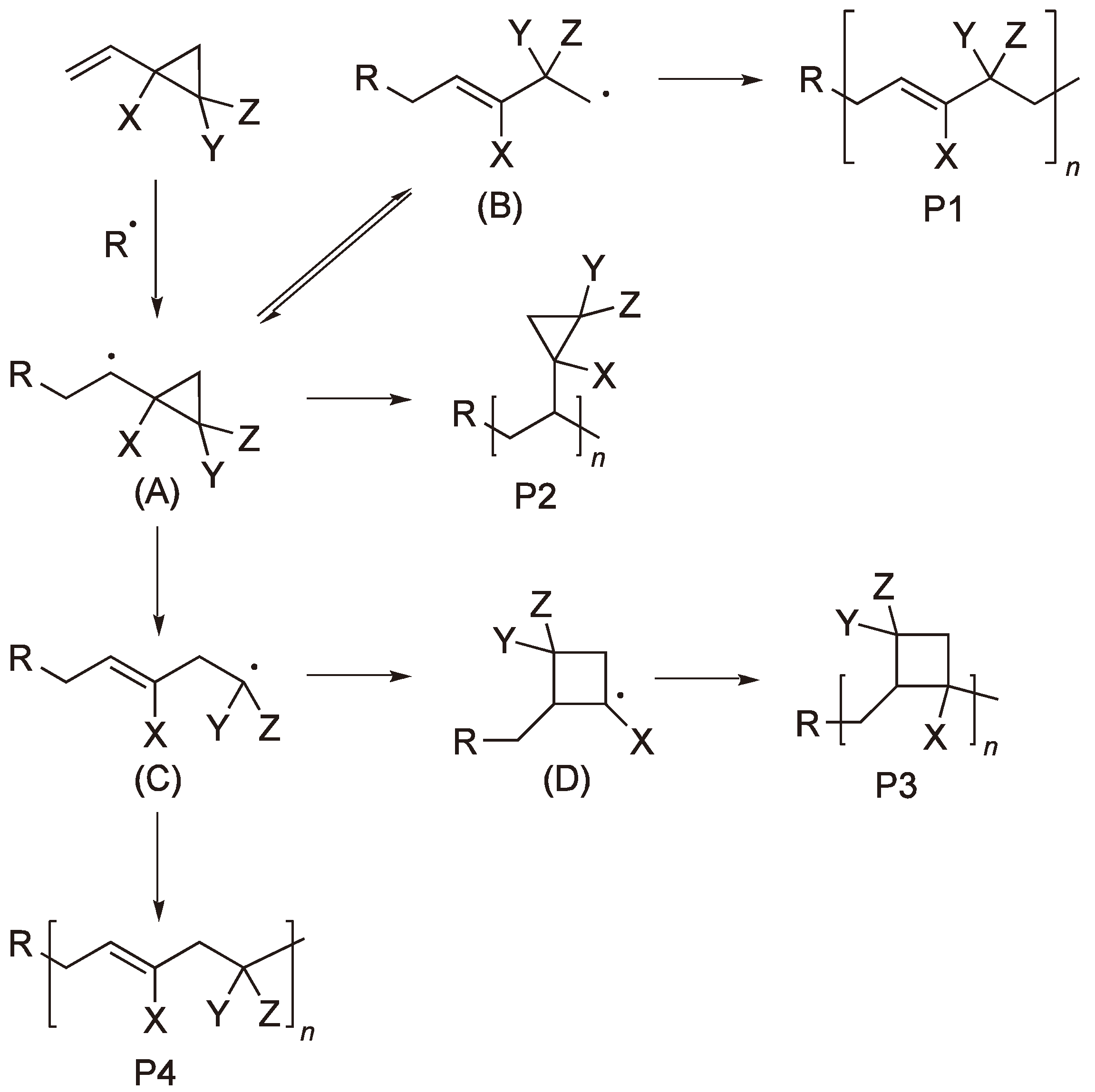
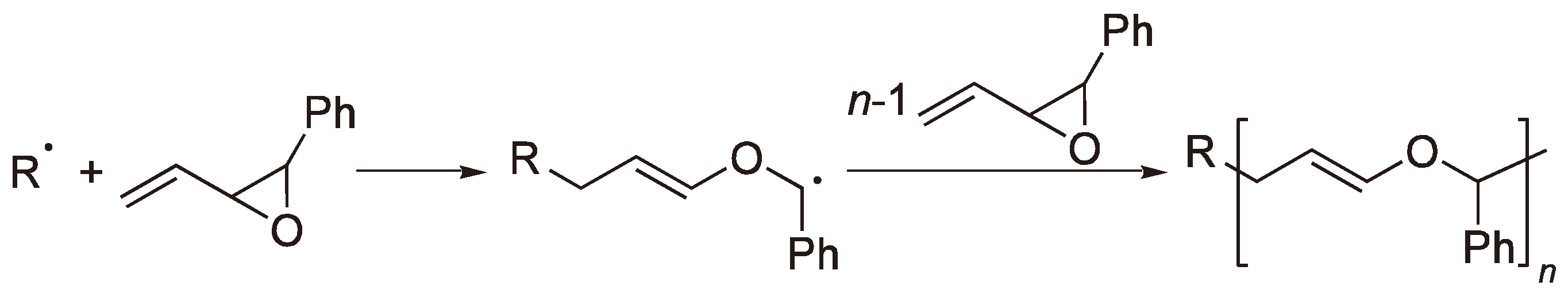
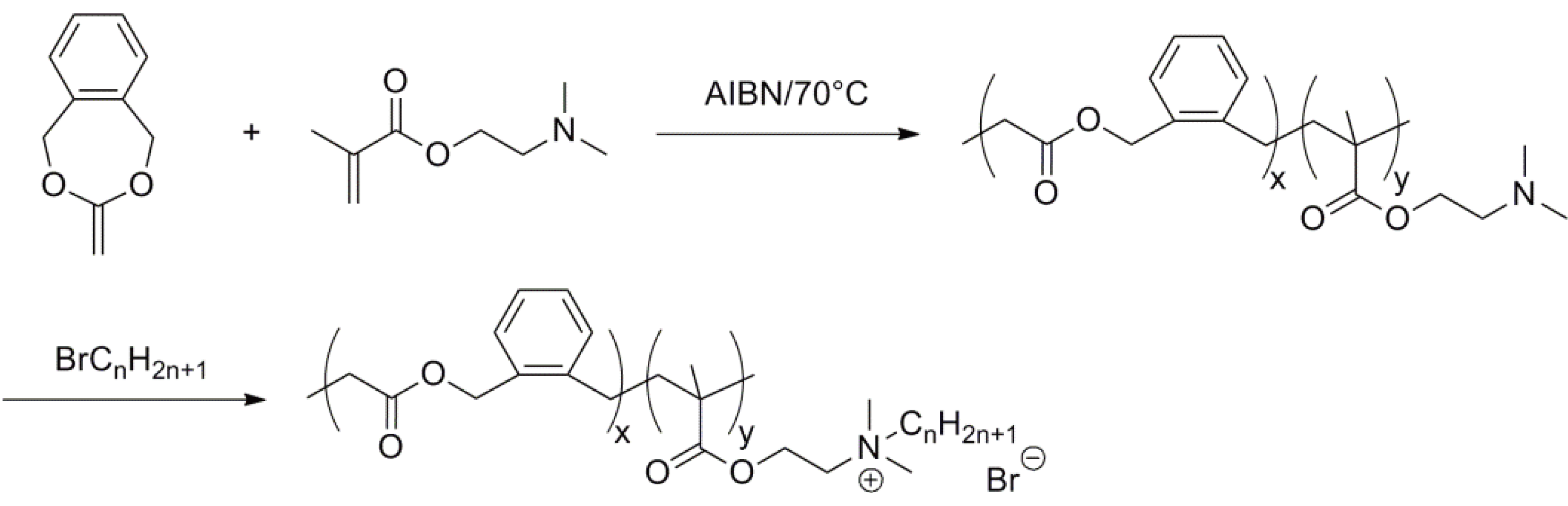
2.1. Vinyl Substituted Cyclic Monomers



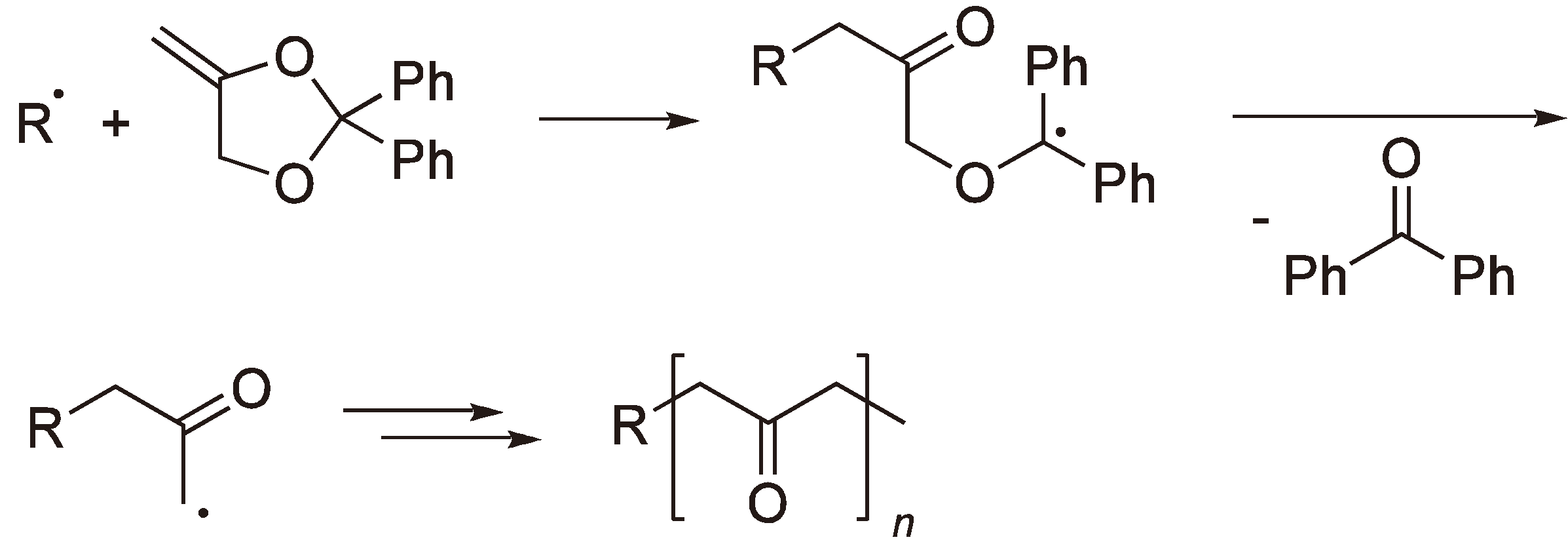
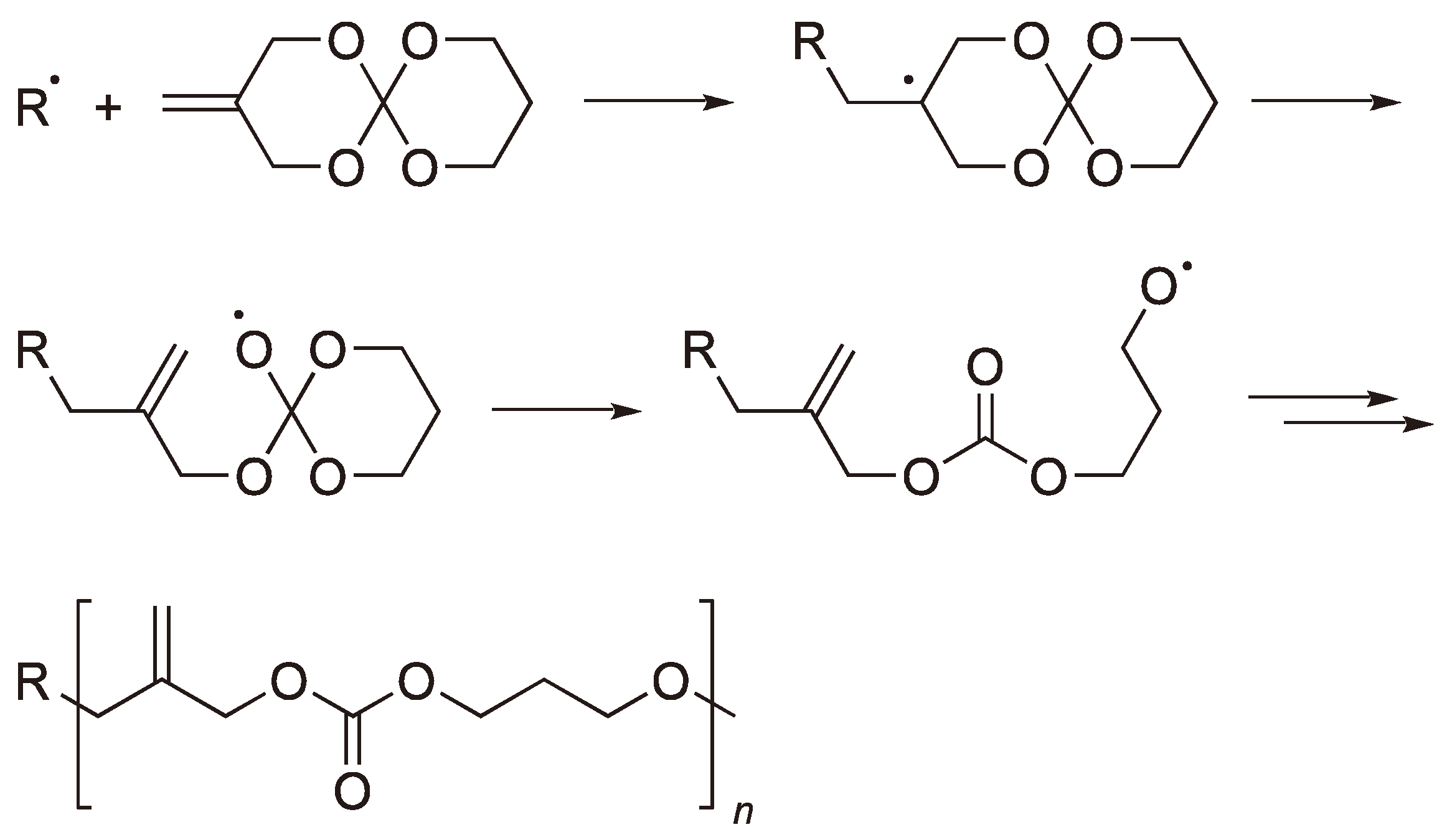
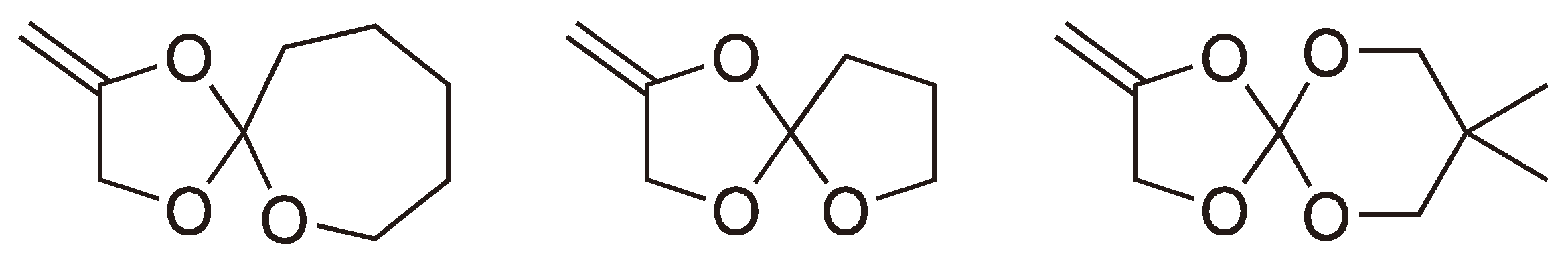
2.2. Methylene Substituted Cyclic Monomers



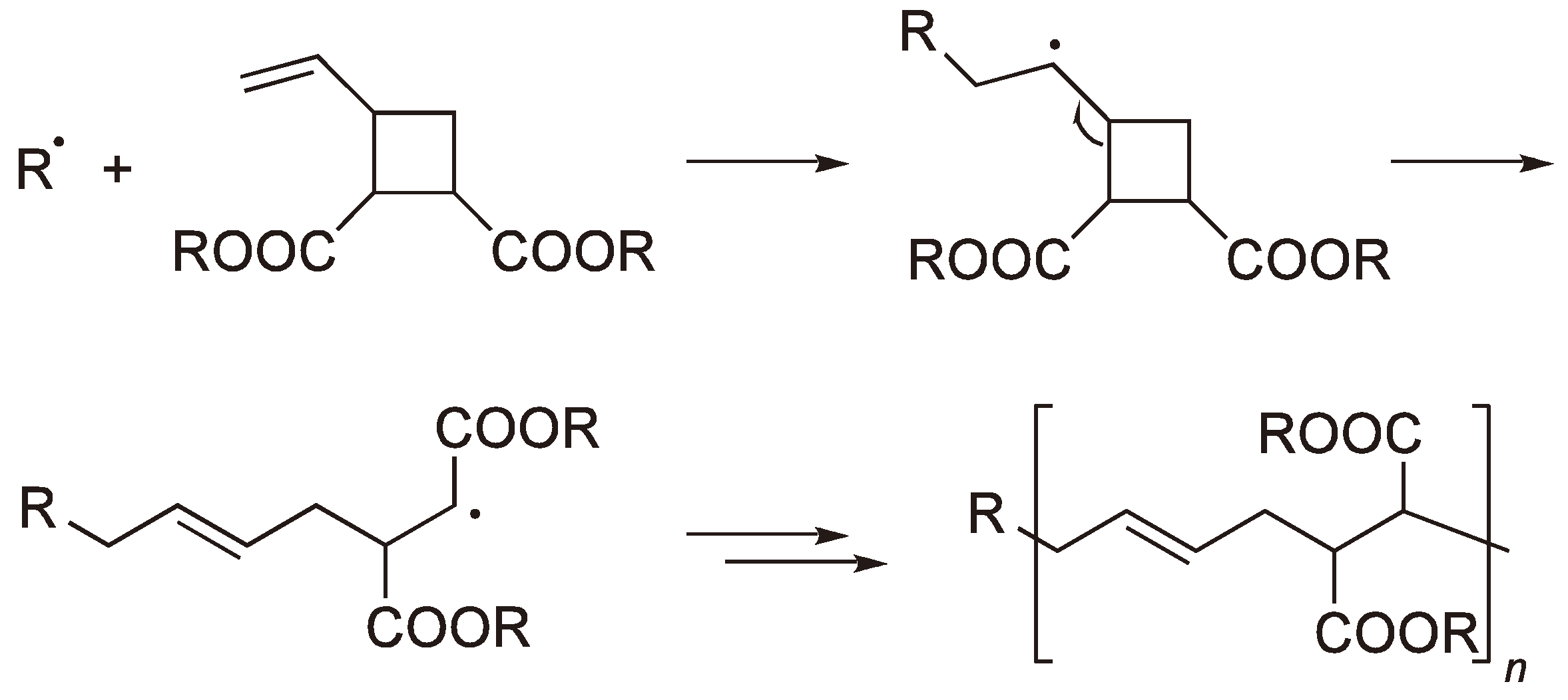
2.3. Double Ring-Opening


2.4. Degradable Polyesters via Radical Ring-Opening Homo- and Copolymerization

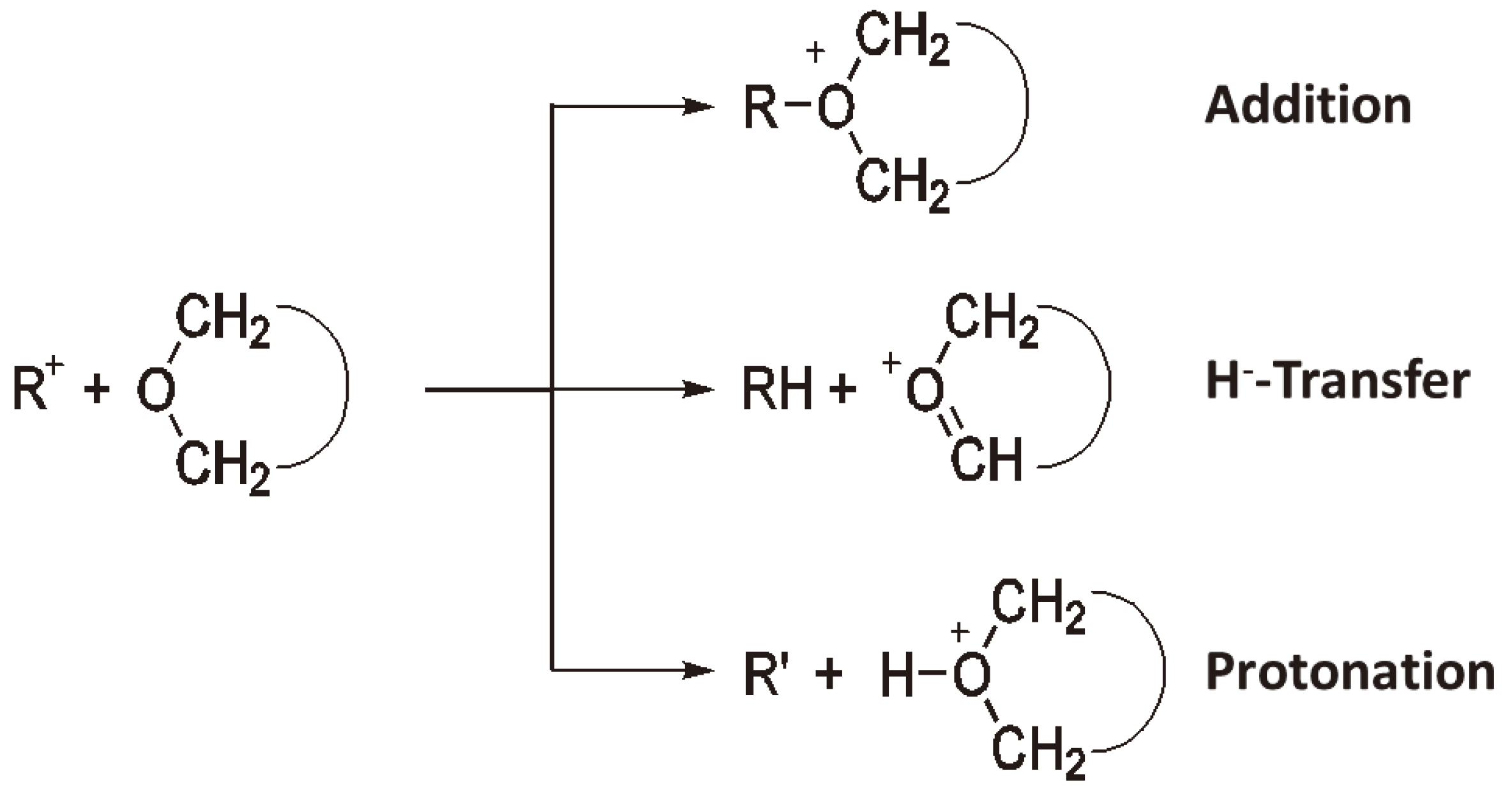
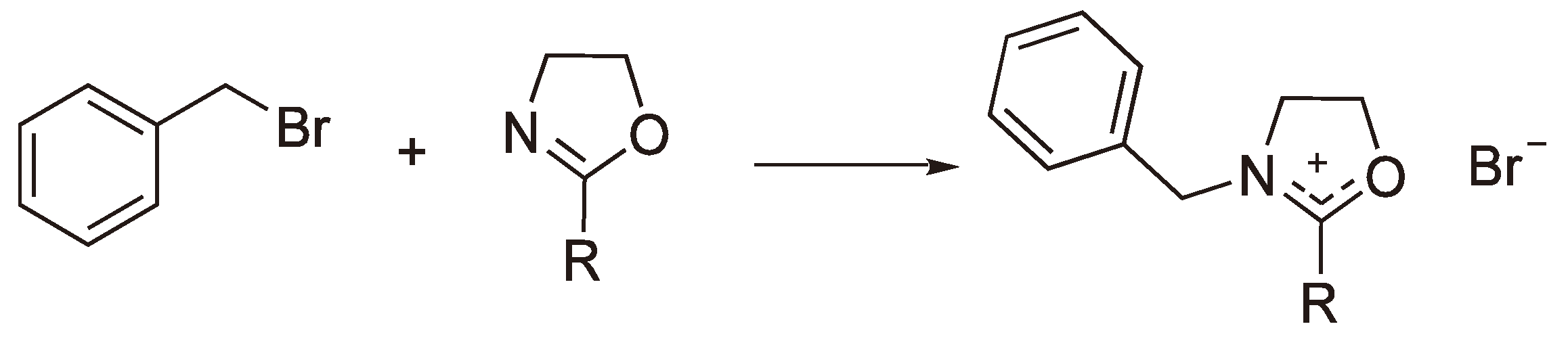
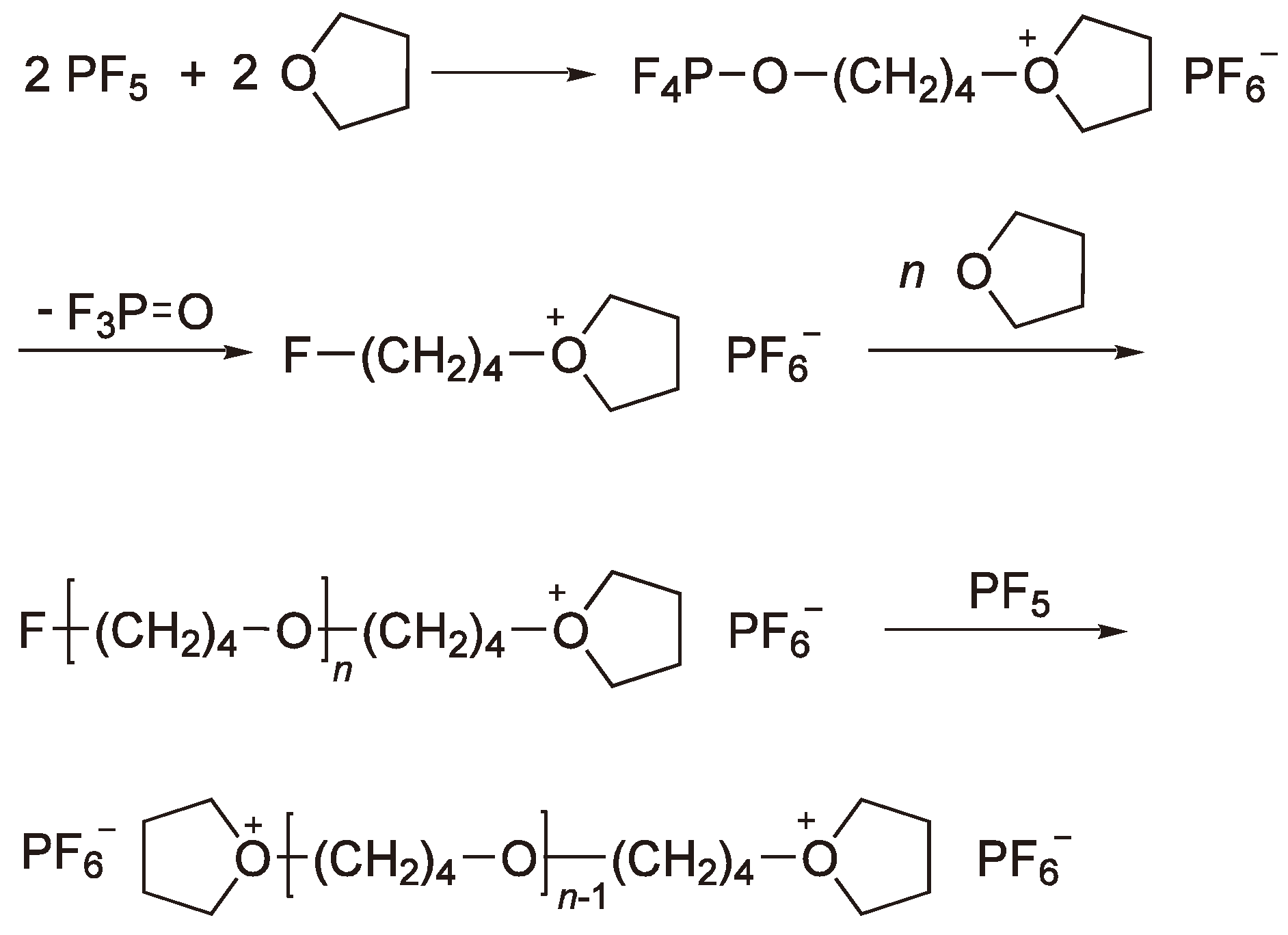
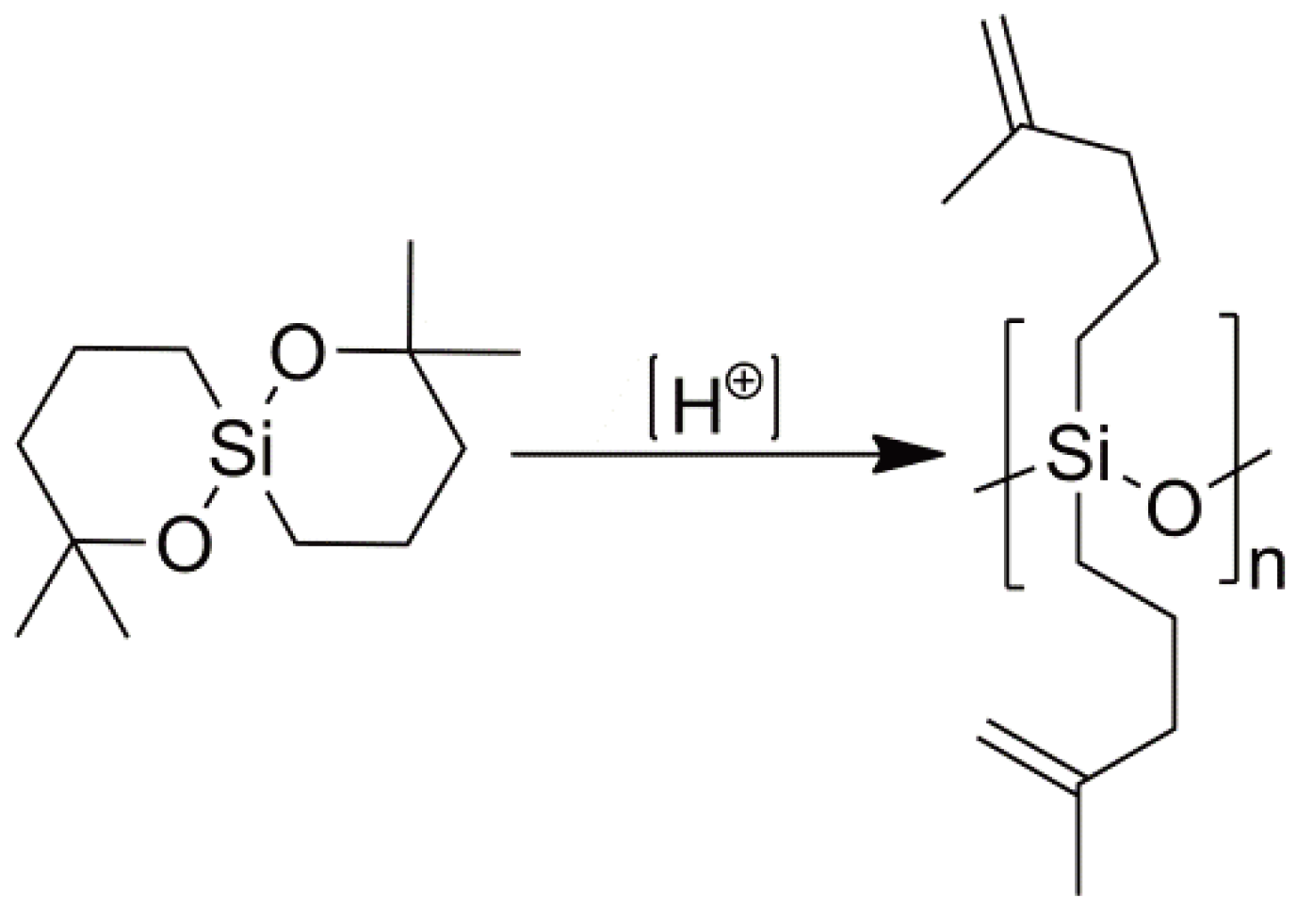
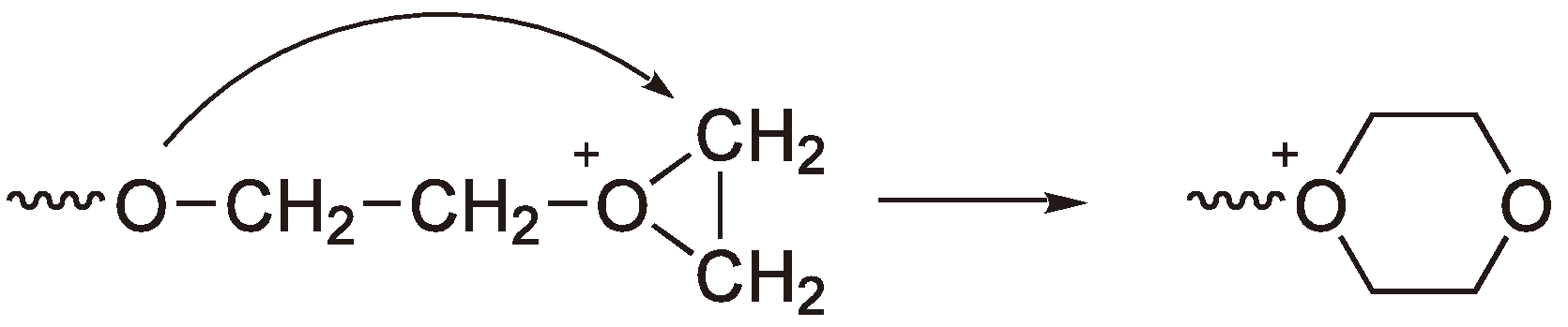
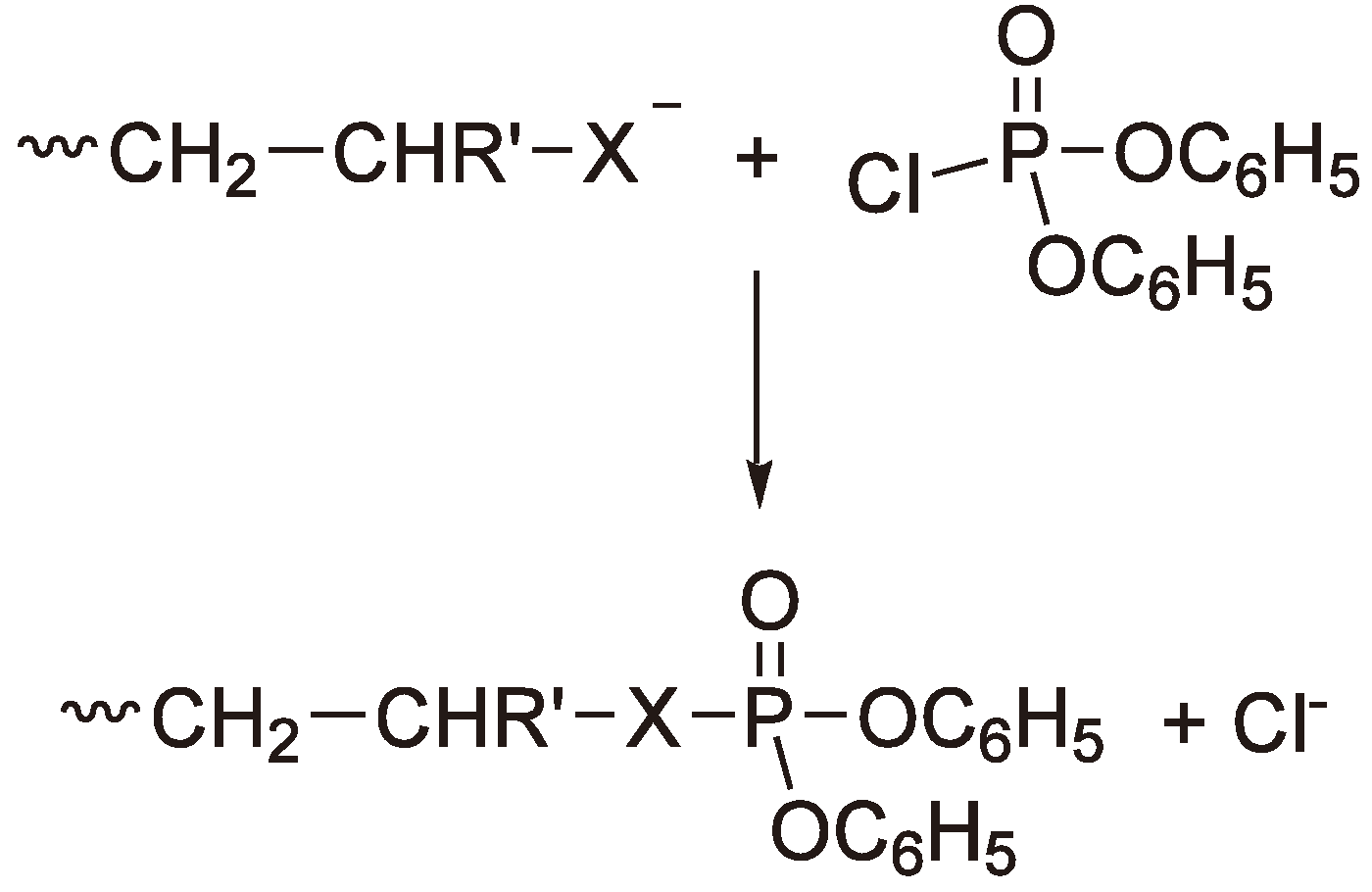
3. Cationic Ring-Opening Polymerization





3.1. Initiation
- Brönsted acids:


- Carbenium Ions:

- Onium-Ions:

- Covalent Initiators:

- Lewis acids:

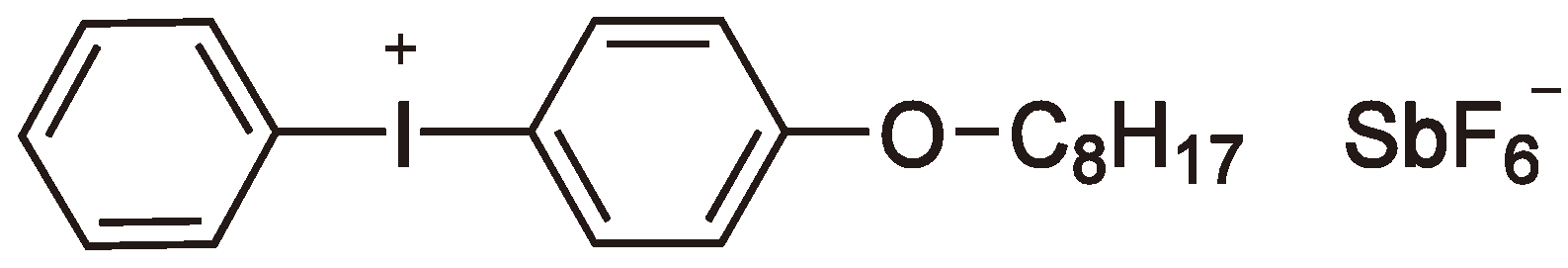
- Photoinitiators





3.2. Chain Growth

| Monomer | Structure of the Growing Chain End [51,52,53,56] |
|---|---|
 | |





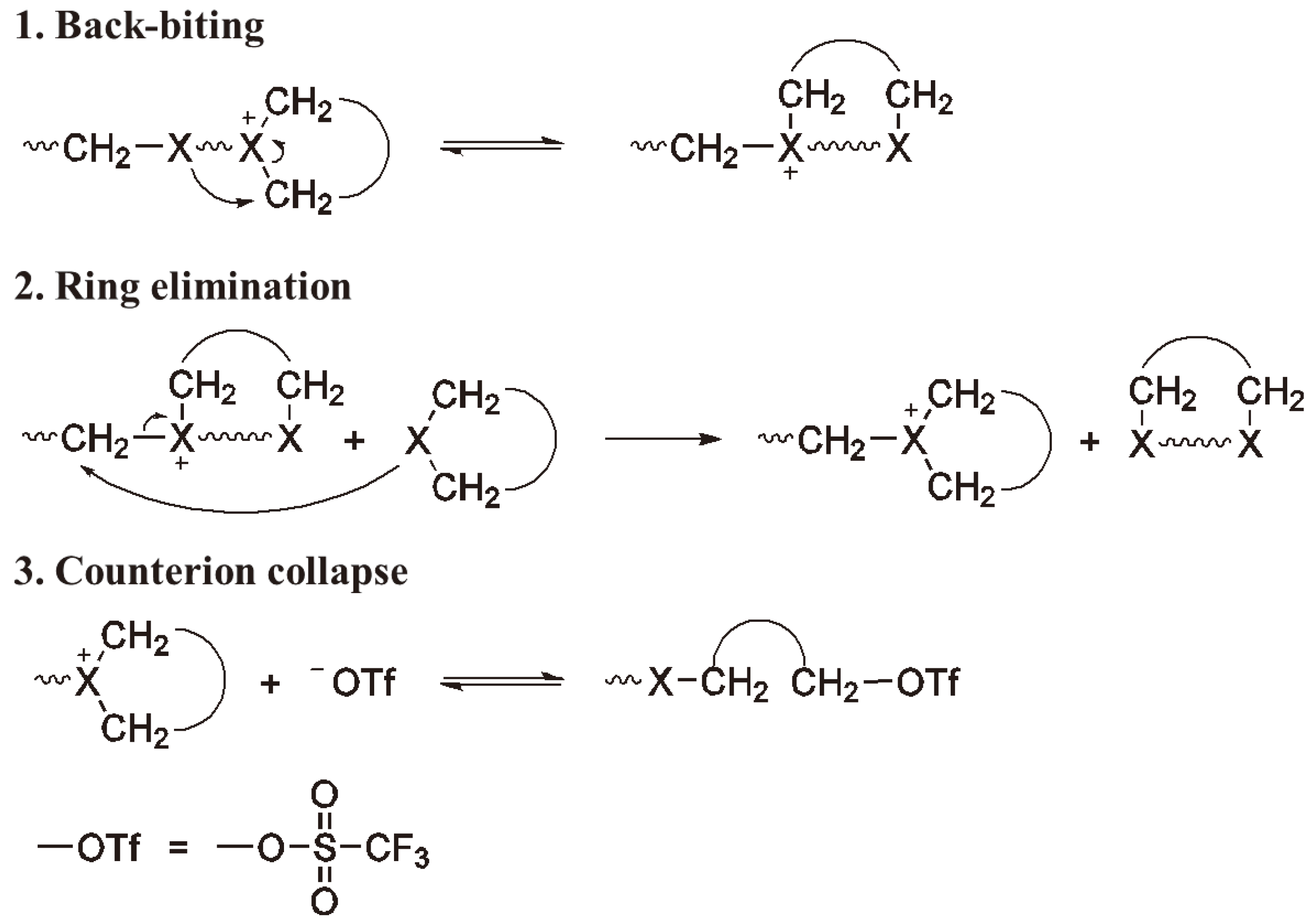
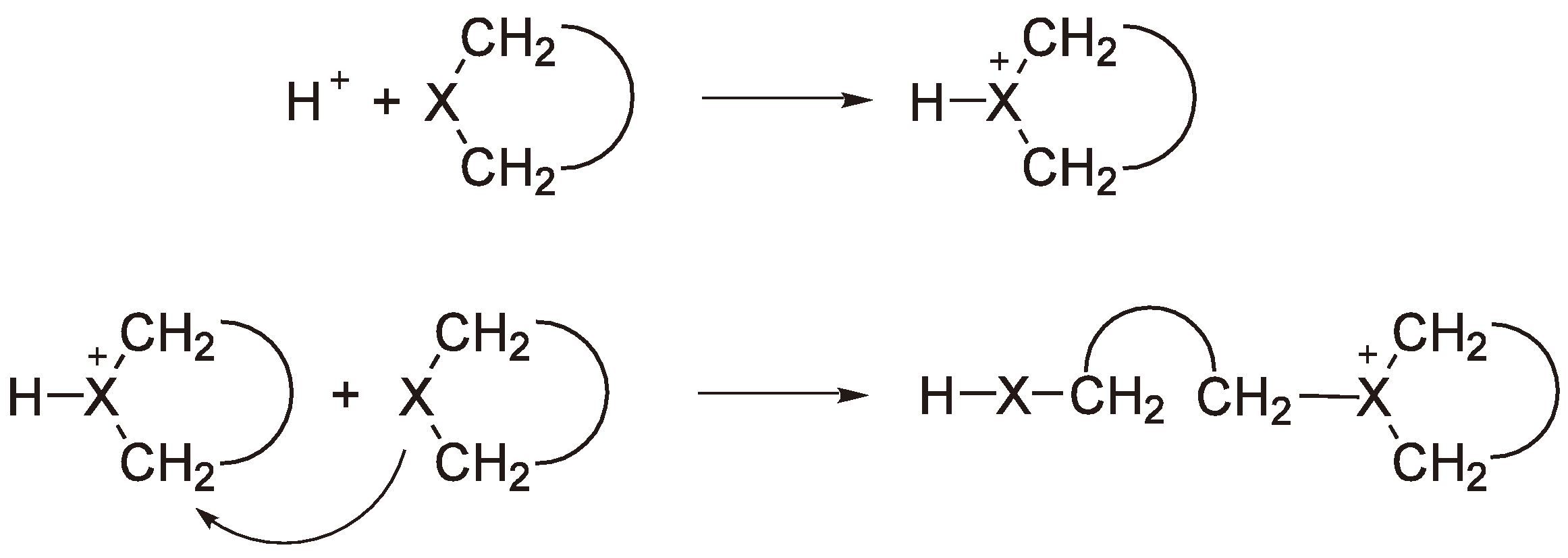
3.3. Termination





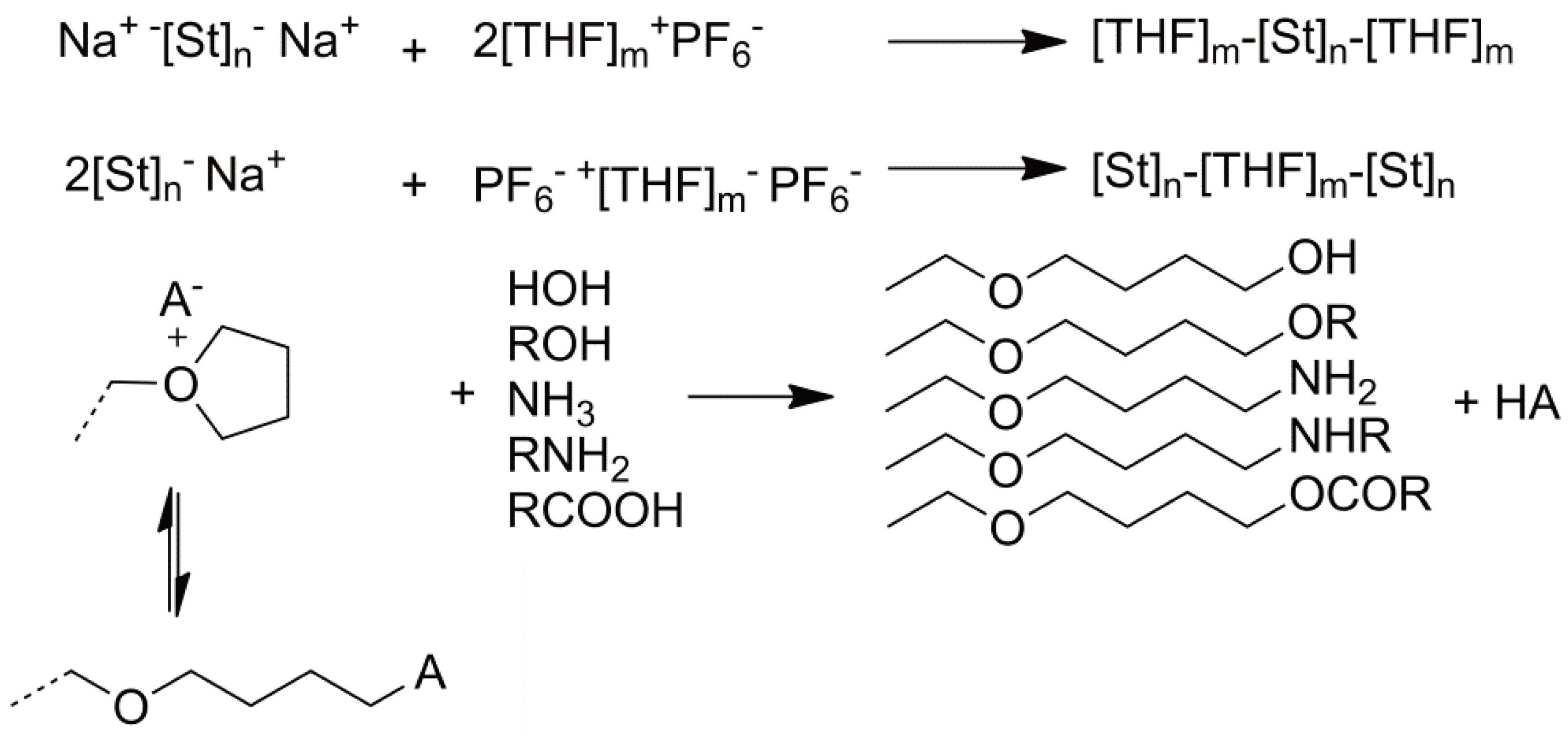
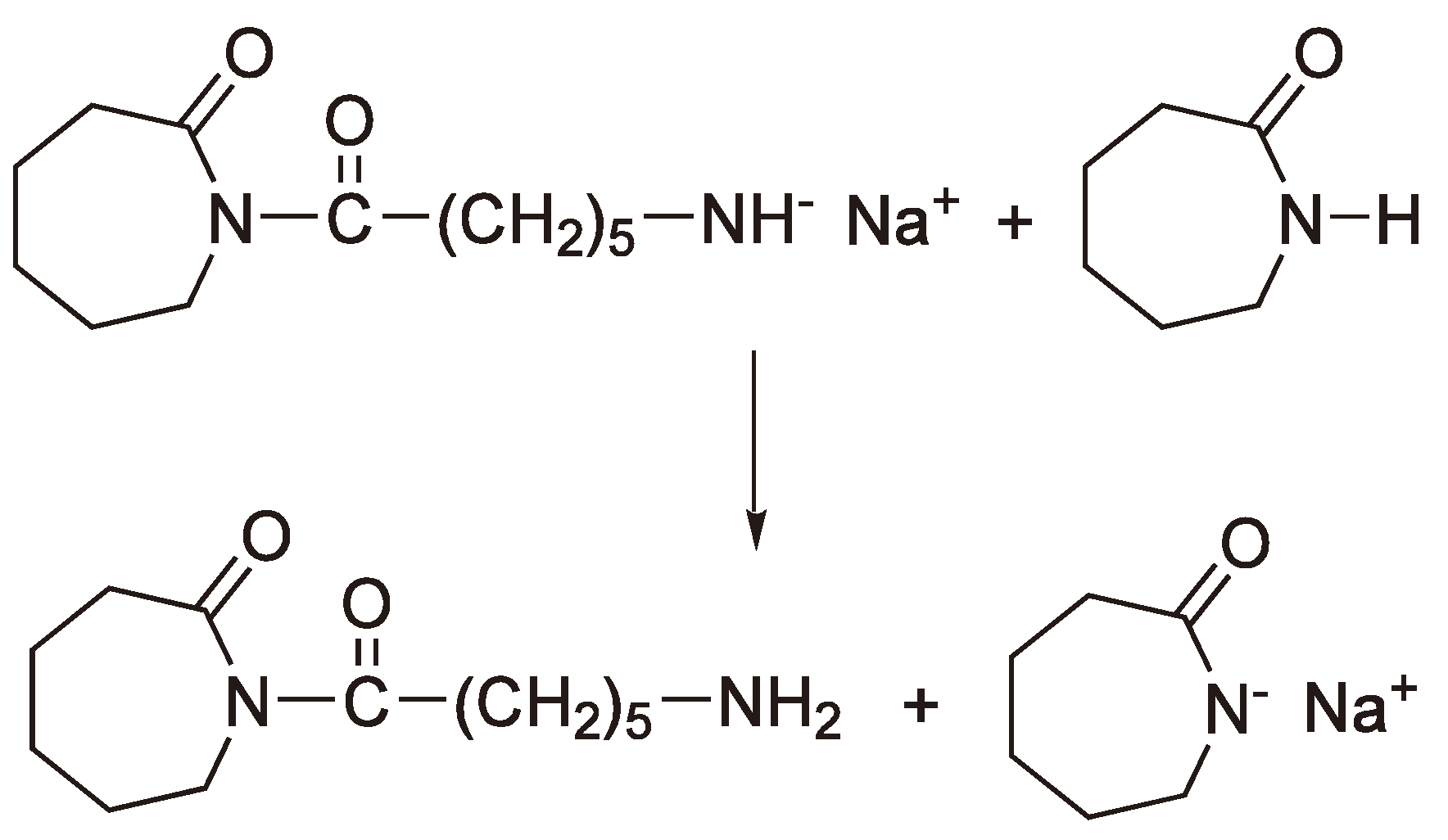
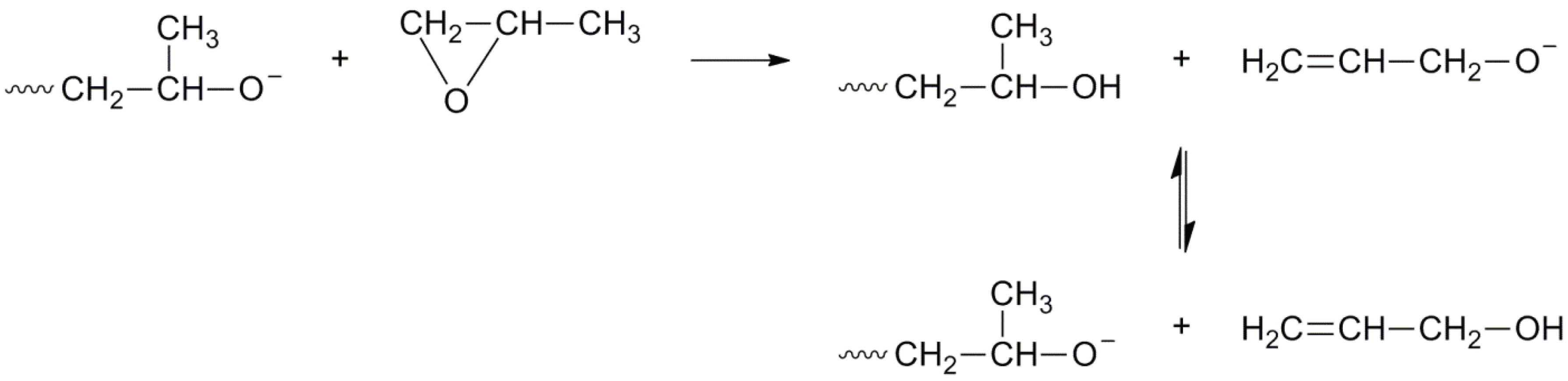
4. Anionic Ring-Opening Polymerization

| Monomer | Structure of the Growing Chain | Reference | ||
|---|---|---|---|---|
 |  | [94] | ||
 |  | [95] | ||
 |  | [96,99] | ||
 |  | [97] | ||
 |  | [98] | ||
4.1. Initiation
| Description | Structure | Monomer | Reference |
|---|---|---|---|
| Radical anion |  | Ethylene oxide | [100] |
| Propylene sulfide | [101] | ||
| Carbanion | C2H5–Li+ | Thietane | [99] |
| n-C4H9–Li+ | Propylene sulfide Hexamethylcyclo-trisiloxane | [102] [102 ] | |
 | Ethylene oxide | [103] | |
| Alcoholate | CH3O−K+ | Styrene oxide | [104] |
| β-Propiolactone | |||
| Silanolate | (CH3)3SiO−K+ | β-Propiolactone | [105] |
| ε-Caprolactone | [106] | ||
| Carboxylate | CH3COO−K+ | β-Propiolactone | [106] |
| Ethylene oxide | [106] | ||
| Propylene oxide | [107] | ||
| Thiolate | C2H5S−K+ | Propylene sulfide | [107] |
| Lactam anion |  | ε-Caprolactam | [108] |
| Amine | (C2H5)3N | Propylene sulfide | [95] |
| Al-trialkoxide | Al(O-t-C3H7)3 | ε-Caprolactone | [109,110] |
| Al-Dialkyl monoalkoxide | (C2H5)2AlOCH3 | Lactide ε-Caprolactone | [111,112] |
4.2. Propagation











4.3. Transfer and Termination
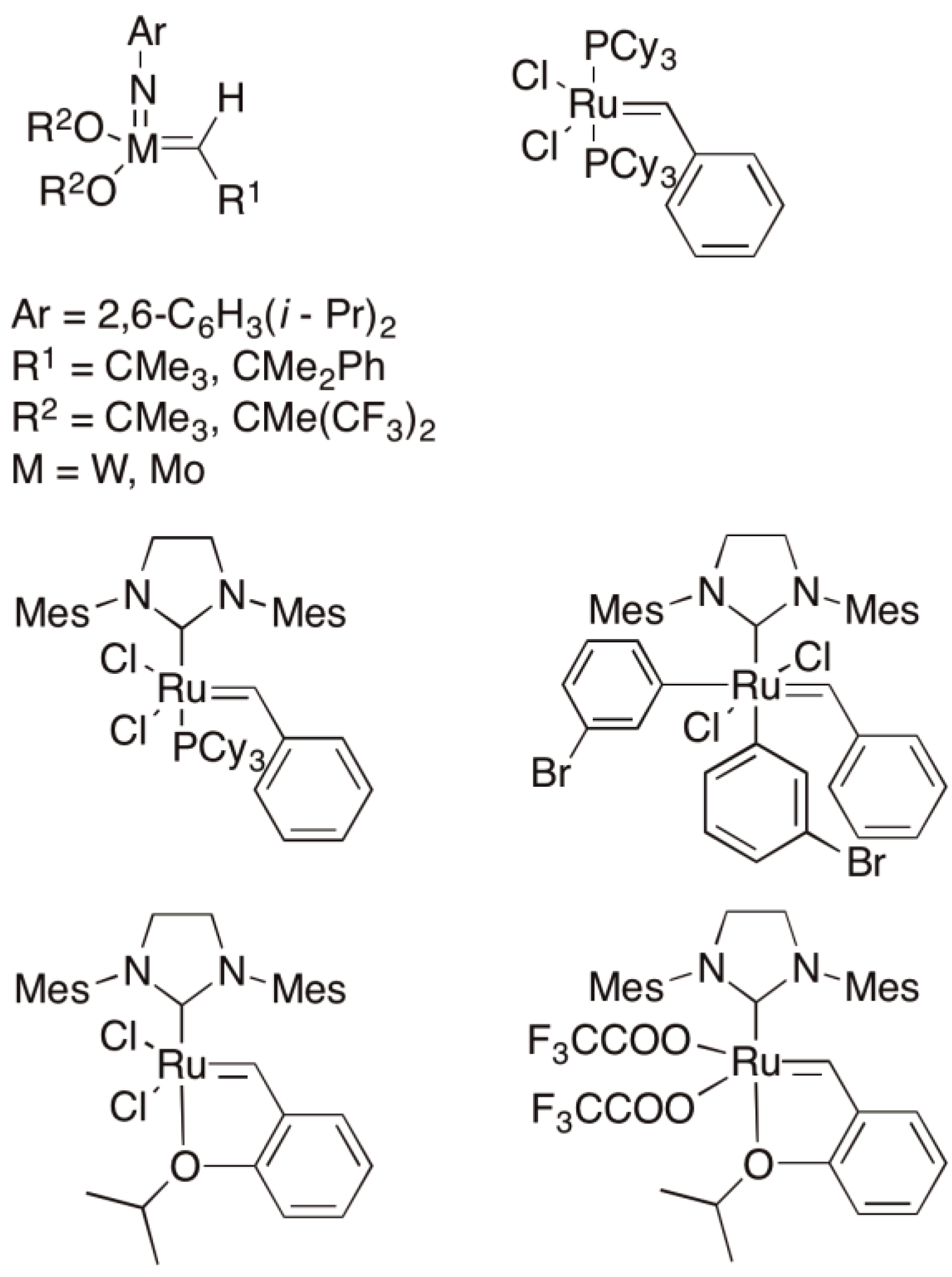
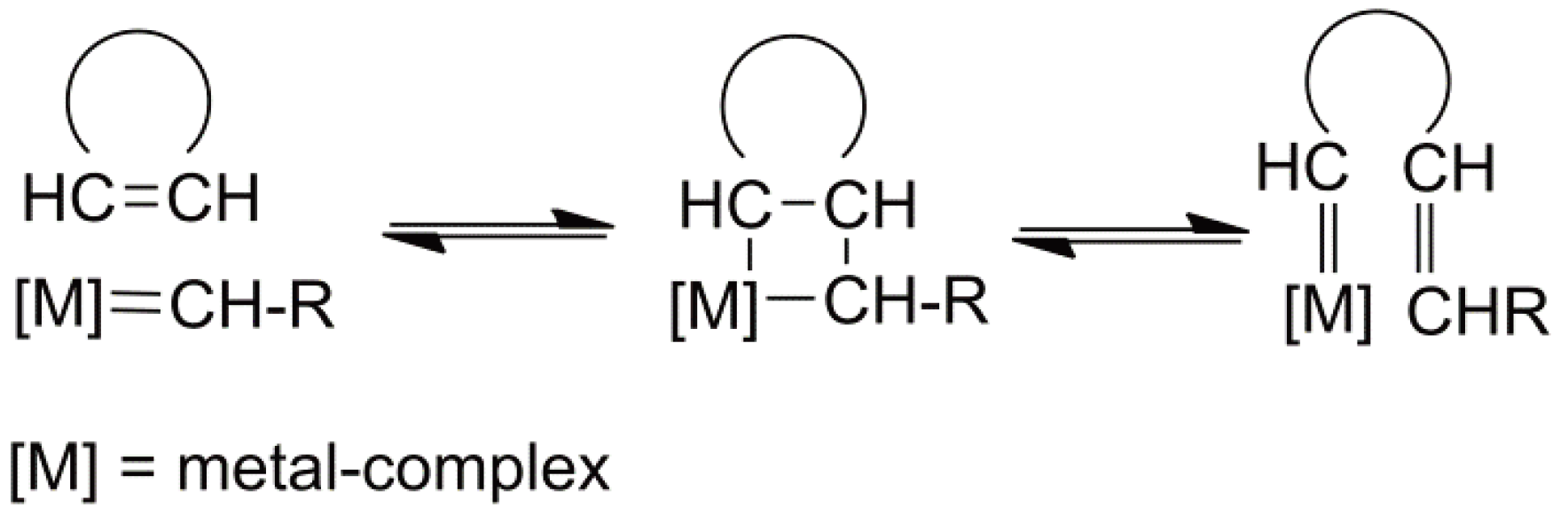
5. Ring-Opening Metathesis Polymerization (ROMP)


| Monomer | Configuration of the double bond in the polymer | ΔH0/(kJ/mol) | ΔS0/(J/(mol K)) | ΔG0/(kJ/mol) |
|---|---|---|---|---|
| Cyclopentene | cis | −15.4 | −52 | −0.3 |
| trans | −18 | −52 | −2.3 | |
| Cyclohexene | cis | −1–3 | −31 | 6.2 |
| trans | 1–3 | −28 | 7.3 | |
| Cycloheptene | cis | −16 | −20 | −10.0 |
| trans | −20 | −17 | −15.0 | |
| Cyclooctene | cis | −20 | −2 | −19.4 |
| trans | −22 | −2 | −21.5 | |
| 1,5-Cyclooctadiene | cis | −25 | −5 | −23.5 |
| trans | −33 | −5 | −31.5 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
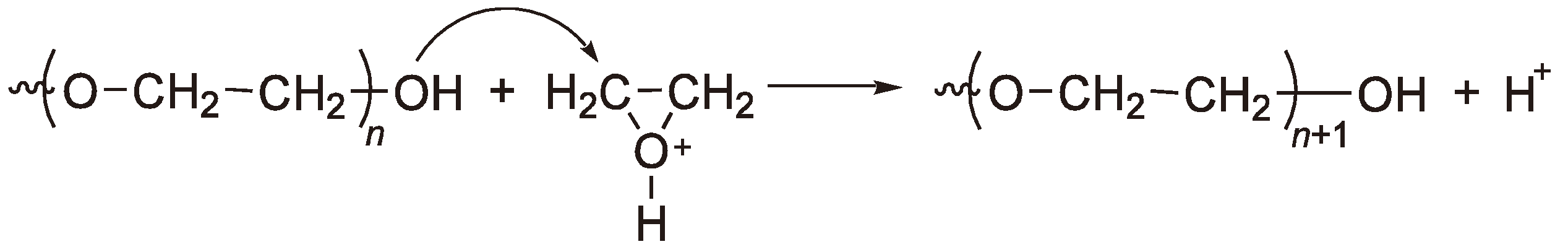{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
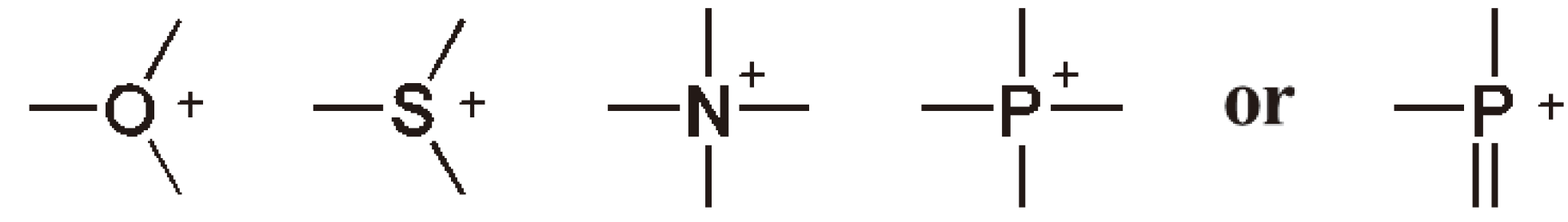{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
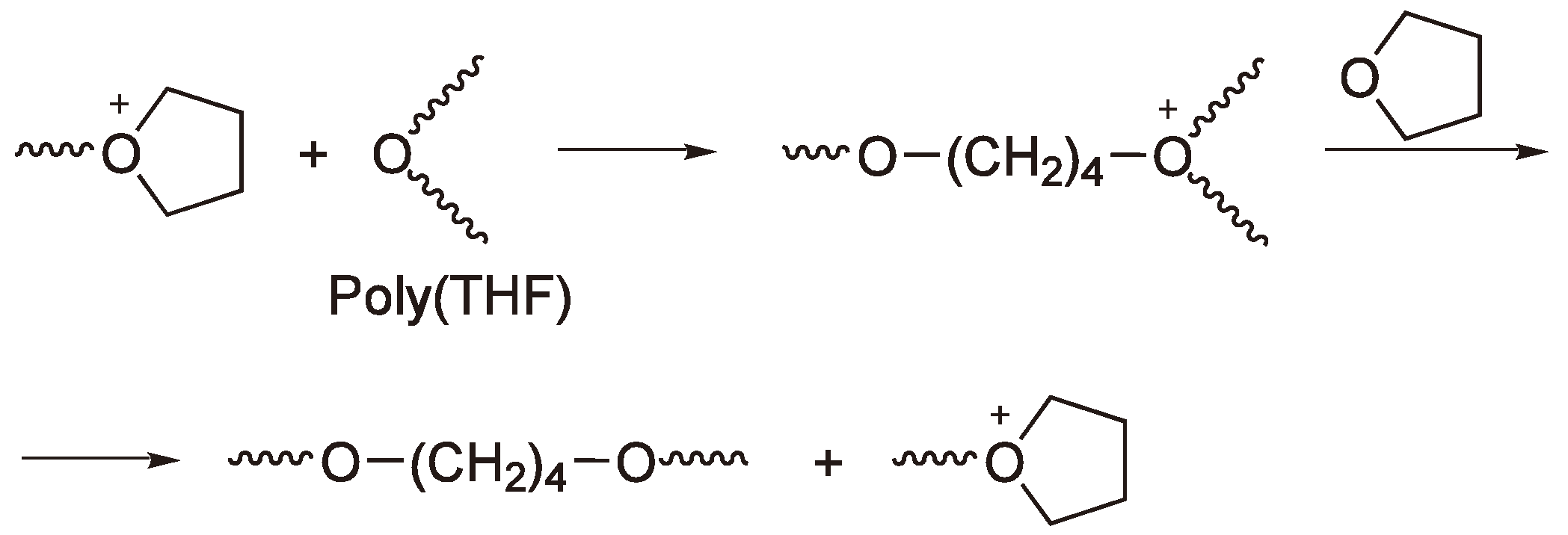{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
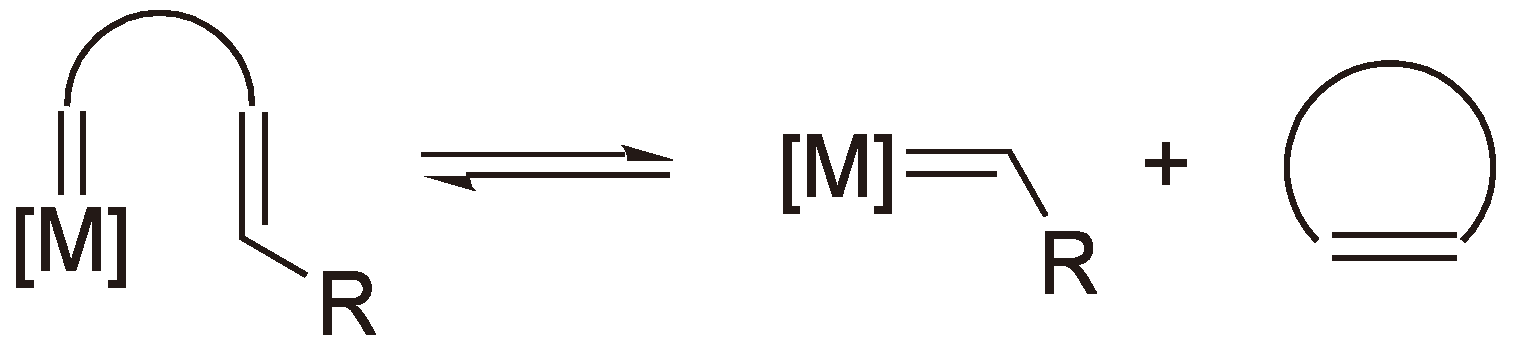{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






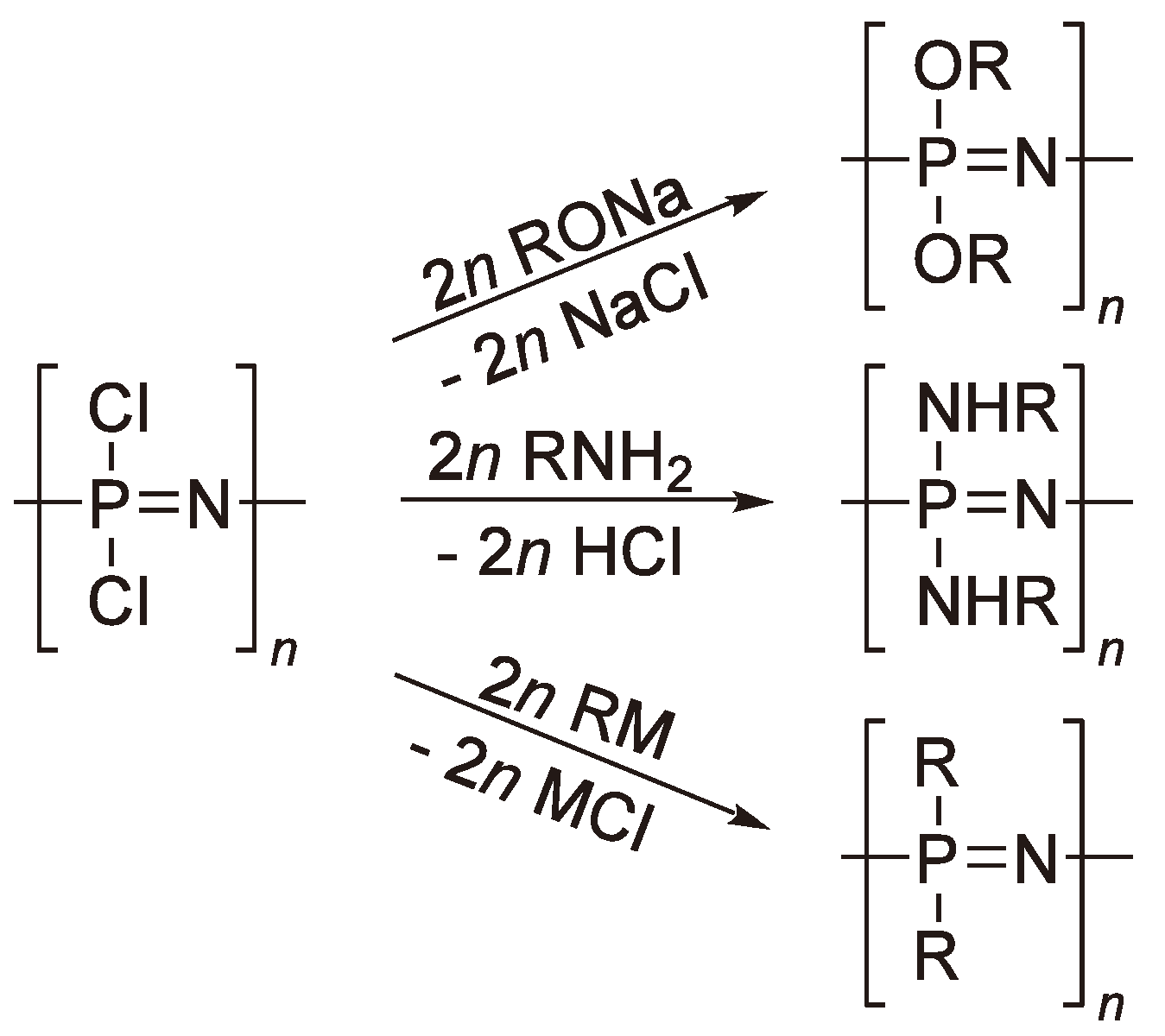
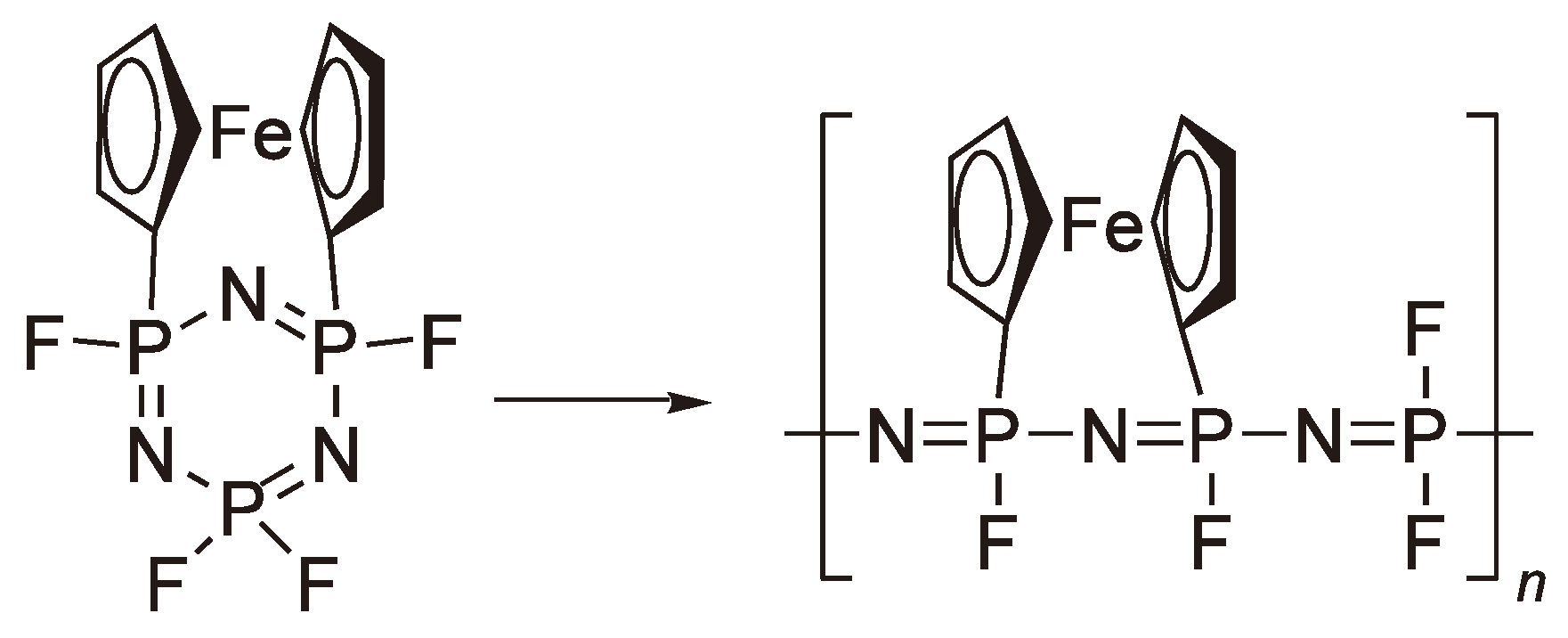
6. Other Ring-Opening Polymerizations





7. Ring-Opening Polymerization of Phosphazenes





8. Summary and Outlook
References
- Duda, A.; Kowalski, A. Thermodynamics and Kinetics of Ring Opening Polymerisation. In Handbook of Ring Opening Polymerisation; Dubois, P., Coulembier, O., Raquez, J-M., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2009; pp. 1–51. [Google Scholar]
- Kubisa, P. Cationic Polymerization of Heterocyclics. In Cationic Polymerizations; Matyjaszewski, K., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 437–553. [Google Scholar]
- Ivin, K.J. Polymer Handbook, 2nd; Brandrup, J., Immergut, E.H., Eds.; John Wiley & Sons: New York, NY, USA, 1975; p. 8. [Google Scholar]
- Brunelle, D.J. Introduction. In Ring-Opening Polymerization; Hanser Publishers: Munich, Germany, 1993; pp. 2–4. [Google Scholar]
- Kubisa, P.; Vairon, J.P. Cationic Ring Opening Polymerization of Cyclic Acetals. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier BV: Amsterdam, the Netherlands, 2012; Volume 4, pp. 183–211. [Google Scholar]
- Endo, T. General Mechanism in Ring-Opening Polymerization. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulembier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; pp. 53–63. [Google Scholar]
- Hall, H.K.; Ykman, P. Addition polymerization of cyclobutene and bicyclobutane monomers. J. Polym. Sci. Macromol. Rev. 1976, 11, 1–45. [Google Scholar] [CrossRef]
- Sarker, P.; Ebdon, J.R.; Rimmer, S. Branched oligovinylcyclopropane by transfer to allylic carbonate oligomers via radical ring-opening polymerization. Macromol. Rapid Commun. 2006, 27, 2007–2013. [Google Scholar]
- Singha, N.K.; Kavitha, A.; Sarker, P.; Rimmer, S. Copper Mediated Controlled Radical Ring-Opening Polymerization (RROP) of Vinylcycloalkane. Chem. Commun. 2008, 3049–3051. [Google Scholar]
- Sanda, F.; Endo, T. Radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 2001, 39, 265–276. [Google Scholar] [CrossRef]
- Moszner, N.; Zeuner, F.; Völkel, T.; Rheinberger, V. synthesis and polymerization of vinylcyclopropanes. Macromol. Chem. Phys. 1999, 200, 2173–2187. [Google Scholar]
- Cho, I.; Kim, J.-B. Exploratory ring-opening polymerization. XIII, Radical ring-opening polymerization of 2-phenyl-3-vinyloxirane: A C–C-Bond scission polymerization of the epoxide ring. J. Polym. Sci. Polym. Lett. 1983, 21, 433–436. [Google Scholar]
- Ochiai, B.; Endo, T. Computational evaluation of radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 2007, 45, 2827–2834. [Google Scholar] [CrossRef]
- Endo, T.; Kanda, N. Syntheses of 2-phenyl-3-vinyloxirane derivatives that undergo radical ring-opening polymerization. J. Polym. Sci. Polym. Chem. 1985, 23, 1931–1938. [Google Scholar] [CrossRef]
- Hiraguri, Y.; Endo, T. Synthesis and radical ring-opening polymerization of 1,2-dicarbomethoxy-3-vinylcyclobutane. J. Polym. Sci. Polym. Lett. 1989, 27, 333–337. [Google Scholar]
- Bailey, W.J. Ring-Opening Polymerization. In Comprehensive Polymer Science; Allen, G., Bevington, A.G., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 283–320. [Google Scholar]
- Hiraguri, Y.; Endo, T. Synthesis and radical ring-opening polymerization of 2,2-diphenyl-4-methylene-5-methyl-l,3-dioxolane, alternating copolymer of propylene, and carbon monoxide. J. Polym. Sci. Polym. Chem. 1992, 30, 689–690. [Google Scholar] [CrossRef]
- Evans, R.A.; Rizzardo, E. Free-Radical ring-opening polymerization of cyclic allylic sulfides. Macromolecules 1996, 29, 6983–6989. [Google Scholar] [CrossRef]
- Angermann, J.; Burtscher, P.; Fischer, U.K.; Moszner, N.; Rheinberger, V. Dental Materials Based on Multicyclic Allyl Sulfides. Eur. Patent 1825843 Al, 29 August 2007. [Google Scholar]
- Luck, R.M.; Sadhir, R.K. Monomers that Expand During Polymerization. In Expanding Monomers; Sadhir, R.K., Luck, R.M., Eds.; CRC-Press: Boca Raton, FL, USA, 1992; pp. 21–61. [Google Scholar]
- Endo, T.; Bailey, W.J. Synthesis and radical ring-opening polymerization of spiro orthocarbonates. J. Polym. Sci. Polym. Chem. 1975, 13, 2525–2530. [Google Scholar] [CrossRef]
- Tagoshi, H.; Endo, T. Syntheses of polymers that undergo no shrinkage on crosslinking. J. Polym. Sci. Polym. Lett. 1988, 26, 77–81. [Google Scholar] [CrossRef]
- Bailey, W.J.; Zheng, Z.-F. Synthesis and double ring-opening polymerization of cyclic ketals of γ-methylenelactones. J. Polym. Sci. Polym. Chem. 1991, 29, 437–446. [Google Scholar] [CrossRef]
- Endo, T.; Bailey, W.J. Synthesis and radical ring-opening polymerization of 2-Methylene-l,4,6-trioxaspiro[4,4]nonane. J. Polym. Sci. Polym. Lett. 1980, 18, 25–27. [Google Scholar] [CrossRef]
- Agarwal, S. Chemistry, chances and limitations of the radical ring-opening polymerization of cyclic ketene acetals for the synthesis of degradable polyesters. Polym. Chem. 2010, 1, 953–964. [Google Scholar] [CrossRef]
- Ren, L.; Agarwal, S. Synthesis, characterization, and properties evaluation of poly[(N-isopropylacrylamide)-co-ester]s. Macromol. Chem. Phys. 2007, 208, 245–253. [Google Scholar] [CrossRef]
- Agarwal, S.; Ren, L.; Kissel, T.; Bege, N. Synthetic route and characterization of main chain ester-containing hydrolytically degradable poly(N,N-dimethylaminoethylmethacrylate)-Based polycations. Macromol. Chem. Phys. 2010, 211, 905–915. [Google Scholar] [CrossRef]
- Agarwal, S.; Ren, L. Polycaprolactone-based novel degradable ionomers by radical ring-opening polymerization of 2-methylene-1,3-dioxepane. Macromolecules 2009, 42, 1574–1579. [Google Scholar] [CrossRef]
- Grabe, N.; Zhang, Y.; Agarwal, S. Degradable elastomeric block copolymers based on polycaprolactone by free-radical chemistry. Macromol. Chem. Phys. 2011, 212, 1327–1334. [Google Scholar] [CrossRef]
- Zhang, Y.; Dafeng, C.; Zheng, M.; Kissel, T.; Agarwal, S. Biocompatible and degradable poly(2-hydroxyethyl methacrylate) based polymers for biomedical applications. Polym. Chem. 2012, 3, 2752–2759. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, M.; Kissel, T.; Agarwal, S. Design and biophysical characterization of bioresponsive degradable poly(dimethylaminoethyl methacrylate) based polymers for in vitro dna transfection. Biomacromolecules 2012, 13, 313–322. [Google Scholar] [CrossRef]
- Jin, Q.; Maji, S.; Agarwal, S. Novel amphiphilic, biodegradable, biocompatible, crosslinkable copolymers: Synthesis, characterization and drug delivery applications. Polym. Chem. 2012, 3, 2785–2793. [Google Scholar] [CrossRef]
- Undin, J.; Illanes, T.; Finne-Wistrand, A.; Albertsson, A.-C. Random introduction of degradable linkages into functional vinyl polymers by radical ring-opening polymerization, tailored for soft tissue engineering. Polym. Chem. 2012, 3, 1260–1266. [Google Scholar] [CrossRef]
- Plikk, P.; Tyson, T.; Finne-Winstrand, A.; Albertsson, A.-C. Mapping the characteristics of radical ring-opening polymerization of a cyclic ketene acetal towards the creation of a functional polyester. J. Polym. Sci. A Polym. Chem. 2009, 47, 4587–4601. [Google Scholar] [CrossRef]
- Wei, Y.; Connors, E.J.; Jia, X.; Wang, J. Controlled free radical ring-opening polymerization and chain extension of the living polymer. J. Polym. Sci. A Polym. Chem. 1998, 36, 761–771. [Google Scholar] [CrossRef]
- He, T.; Zou, Y.-F.; Pan, C.-Y. Controlled/“Living” radikal ring-opening polymerization of 5,6-benzo-2-methylene-1,3-dioxepane based on reversible addition fragmentation chain transfer mechanism. Polym. J. 2002, 34, 138–143. [Google Scholar] [CrossRef]
- Yuan, G.-Y.; Pan, C.-Y.; Tang, B.-Z. “Living” free radical ring-opening polymerization of 5,6-benzo-2-methylene-1,3-dioxopane using the atom transfer radical polymerization method. Macromolecules 2001, 34, 211–214. [Google Scholar] [CrossRef]
- Penczek, S.; Kubisa, P. Cationic Ring-Opening Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 13–86. [Google Scholar]
- Vairon, J.-P.; Spassky, N. Industrial Cationic Polymerization: An Overview. In Cationic Polymerizations; Matyjaszewski, K., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 683–750. [Google Scholar]
- Motokucho, S.; Sudo, A.; Endo, T. Living cationic-ring-opening polymerization of five-membered cyclic dithiocarbonate controlled by neighboring group participation of carbamate group. J. Polym. Sci. Polym. Chem. 2007, 45, 4459–4464. [Google Scholar] [CrossRef]
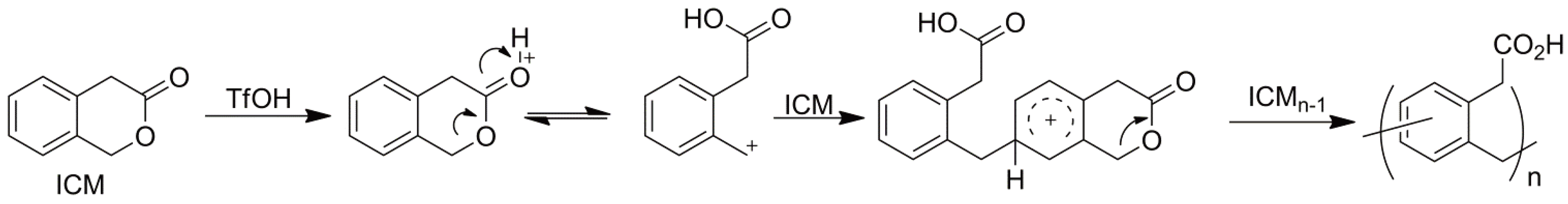
- Suzuki, A.; Sudo, A.; Endo, T. Cationic ring-opening-polymerization of 3-isochromanone through the formation of benzyl cationic intermediate and its friedel-crafts reaction. J. Polym. Sci. Polym. Chem. 2009, 47, 2214–2218. [Google Scholar] [CrossRef]
- Firat, Y.; Jones, F.R.; Plesch, P.H.; Westermann, P.H. The propagating species in the polymerization of 1,3-dioxacycloalkanes by percholoric acid. Makromol. Chem. Suppl. 1975, 1, 203–216. [Google Scholar] [CrossRef]
- Jaacks, V.; Boehlke, K.; Eberius, E. A method of determining the active centers in cationic polymerization of dioxolane, trioxane and formaldehyde. Makromol. Chem. 1968, 118, 354–360. [Google Scholar] [CrossRef]
- Brzezinska, K.; Chwialkowska, W.; Kubisa, P.; Matyjaszewski, K.; Penczek, S. Ion trapping in cationic polymerizations. Makromol. Chem. 1977, 178, 2491–2494. [Google Scholar] [CrossRef]
- Penczek, S. Cationic Ring-Opening Polymerization(CROP) major mechanistic phenomena. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 1919–1933. [Google Scholar]
- Bednarek, M.; Kubisa, P.; Penczek, S. Coexistence of activated monomer and active chain end mechanisms in cationic copolymerization of tetrahydrofuran with ethylene oxide. Macromolecules 1999, 32, 5257–5263. [Google Scholar] [CrossRef]
- Bednarek, M.; Brzezinska, K.; Stasinski, J.; Kubisa, P.; Penczek, S. Heteropolyacids—New efficient initiators of cationic polymerization. Makromol. Chem. 1989, 190, 929–938. [Google Scholar] [CrossRef]
- Ryu, C.Y.; Spencer, M.J.; Crivello, J.V. Involvement of supramolecular complexes in the capture and release of protonic acids during the cationic ring-opening polymerization of epoxides. Macromolecules 2012, 45, 2233–2241. [Google Scholar] [CrossRef]
- Slomkowski, S.; Penczek, S. Kinetics of hydride transfer from 1,3-dioxolane to the free triphenymethyl cation preceeding the true initiation. Chem. Commun. l970, 1347–1348. [Google Scholar]
- Afshar-Taromi, F.; Scheer, M.; Rempp, P.; Franta, E. A new efficient cationic initiator for the polymerization of tetrahydrofuran. Makromol. Chem. 1978, 179, 849–853. [Google Scholar] [CrossRef]
- Szymanski, R.; Penczek, S. Dynamic NMR studies of equilibria involving active species in the polymerization of cyclic acetals. Reaction of methoxycarbenium ion (CH3OCH2)+ with 5, 6 and 7 membered cyclic acetals. Makromol. Chem. 1982, 183, 1587–1602. [Google Scholar]
- Matyjaszewski, K.; Penczek, S. The macroester-macroion equilibrium in the cationic polymerization of THF observed directly by 300 MHz H NMR. J. Polym. Sci. Polym. Chem. 1974, 12, 1905–1912. [Google Scholar] [CrossRef]
- Goethals, E.J.; Drijvers, W. Cationic polymerization of cyclic sulfides. IV. Determination of the structure and concentration of the propagating species during the polymerization of 3,3-dimethoxythietane by 300 MHz NMR spectroscopy. Makromol. Chem. 1973, 165, 329–333. [Google Scholar]
- Goethals, E.J.; Schacht, E.H. Cationic polymerization of cyclic imines. II. 300 MHz 1H NMR of the structure and concentration of propagating species during the polymerization of 1,3,3-trimethylazetidine. J. Polym. Sci. Polym. Lett. Ed. l973, 11, 497–501. [Google Scholar]
- Li, Y.; Yu, B. Glycolization initiated cationic ring-opening polymerization of tetrahydrofuran to prepare neoglycolpolymers. Chem. Commun. 2010, 46, 6060–6062. [Google Scholar]
- Saegusa, T.; Kobayashi, S.; Yamada, A. Kinetics of the isomerization polymerization of 2-methyl-2-oxazoline by benzyl chloride and bromide initiators. Makromol. Chem. 1976, 177, 2271–2283. [Google Scholar]
- Smith, S.; Hubin, A.J. Preparation and chemistry of dicationically active polymers of tetrahydrofuran. J. Macromol. Sci. Chem. 1973, A7, 1399–1413. [Google Scholar]
- Kobayashi, S.; Danda, H.; Saegusa, T. Superacids and their derivatives. IV. Kinetic studies on the ring-opening polymerization of tetrahydrofuran initiated with ethyl trifluoromethanesulfonate by means of 19F and 1H nuclear magnetic resonance spectroscopy. Evidence for the oxonium-ester equilibrium of the propagating species. Macromolecules 1974, 7, 415–420. [Google Scholar]
- Matyjaszewski, K.; Kubisa, P.; Penczek, S. Ion ⇄ Ester equilibria in the living cationic polymerization of tetrahydrofuran. J. Polym. Sci. Polym. Chem. 1974, 12, 1333–1336. [Google Scholar]
- Kennedy, J.P.; Marechal, E. Carbocationic. Polymerization; John Wiley & Sons: New York, NY, USA, 1982; pp. 95–97. [Google Scholar]
- Hoene, R.; Reichert, K.-H. Elucidation of the initation step in the cationic polymerization of tetrahydrofuran with phospherous pentafluoride. Makromol. Chem. 1976, 177, 3545–3570. [Google Scholar] [CrossRef]
- Crivello, J.V.; Lam, J.H.W. Diaryliodonium salts. A new class of photoinitiators for cationic polymerization. Macromolecules 1977, 10, 1307–1315. [Google Scholar]
- Sangermano, M.; Crivello, J.V. Visible and long-wavelength cationic photopolymerization. ACS Symp. Ser. 2003, 847, 242–252. [Google Scholar]
- Crivello, J.V. Cationic photopolymerization of alkyl glycidyl ethers. J. Polym. Sci. Polym. Chem. 2006, 44, 3036–3052. [Google Scholar] [CrossRef]
- Putzien, S.; Louis, E.; Nuyken, O.; Crivello, J.V.; Kühn, F.E. UV curing of epoxy functional hybrid silicones. J. Appl. Polym. Sci. 2012, 126, 1188–1197. [Google Scholar]
- Anger, C.A.; Hinelang, K.; Helbich, T.; Halbach, T.; Stohrer, J.; Rieger, B. Photoinduced polysiloxane architechtures from spirosiloxane precursors via intramolecular hydrosilylation. ACS Macro Lett. 2012, 1, 1204–1207. [Google Scholar]
- Hoefler, T.; Griesser, M.; Gruber, G.; Jakopic, G.; Trimmel, G.; Kern, W. Photo fries rearrangement in polymer media: An investigation on fully aromatic esters containing the naphthyl chromophore. Macromol. Chem. Phys. 2008, 209, 488–498. [Google Scholar]
- Buijsen, P.F.A.; Hacker, N.P. Photochemical reaction of onium salts with N,N-Dimethylaniline; evidence for photo induced electron transfer. Tetrahedron Lett. 1993, 34, 1557–1560. [Google Scholar]
- Jungermann, S.; Riegel, N.; Mueller, D.; Meerholz, K.; Nuyken, O. Novel photo-cross-linkable hole transporting polymers: synthesis, characterization and application in organic light emitting diodes. Macromolecules 2006, 39, 8911–8919. [Google Scholar]
- Schelter, J.; Mielke, G.F.; Koehnen, A.; Wies, J.; Koeber, S.; Nuyken, O.; Meerholz, K. Novel non-conjugated main-chain hole-transporting polymers for organic electronics application. Macromol. Rapid. Commun. 2010, 31, 1560–1567. [Google Scholar]
- Müller, C.D.; Falcon, A.; Reckefuss, N.; Rojahn, M.; Wiederhirn, V.; Rudati, P.; Frohne, H.; Nuyken, O.; Becker, H.; Meerholz, K. Multi-colour organic light emitting displays by solution processing. Nature 2003, 421, 829–833. [Google Scholar]
- Feser, S.; Meerholz, K. Investigation of the photocrosslinking mechanism in oxetane functionalized semicunductors. Chem. Mater. 2011, 23, 5001–5005. [Google Scholar] [CrossRef]
- Makino, A.; Kobayashi, S. Chemistry of 2-oxazolines: A crossing of the cationic ring-opening polymerization and enzymatic ring-opening polyaddition. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 1251–1270. [Google Scholar] [CrossRef]
- Novel polyoxazolines polymer drug delivery platform. Available online: http://www.baypat.de/en/industry/technology-offers.html?tech_ang=1684 (accessed on 17 April 2013).
- Kourti, M.-E.; Vougioukalakis, G.C.; Hadjichristidis, N.; Pitsikalis, M. Metallocene-mediated cationic-ring-opening polymerization of 2-methyl and 2-phenyl-oxazoline. J. Polym. Sci. Polym. Chem. 2011, 49, 2520–2527. [Google Scholar]
- Saegusa, T.; Matsumoto, S. Determination of concentration of propagating species in cationic polymerization of tetrahydrofuran. J. Polym. Sci. A 1968, 6, 1559–1565. [Google Scholar]
- Saegusa, T.; Kobayashi, S. Cationic ring-opening polymerization of cyclic ethers. Progr. Polym. Sci. Jpn. 1973, 4, 106–151. [Google Scholar]
- Matyjaszewski, K.; Penczek, S. Ion trapping in cationic polymerization. II, Relative rates of trapping and relative chemical shifts of structural differing phosphines as trapping agents. Makromol. Chem. 1981, 182, 1735–1742. [Google Scholar]
- Brzezinska, R.; Szymanski, P.; Kubisa, P.; Penczek, S. Activated monomer mechanism in cationic polymerization. Ethylenoxide, formulation of mechanism. Makromol. Chem. Rapid Commun. 1986, 7, 1–4. [Google Scholar]
- Penczek, S.; Kubisa, P.; Szymanski, R. Activated monomer propagation in cationic polymerization. Makromol. Chem. Macromol. Symp. 1986, 3, 203–220. [Google Scholar] [CrossRef]
- Qayouh, H.; Lahcini, M.; Six, J.-L.; Kricheldorf, H.R. Polymerizations of hexamethylcyclosiloxane catalyzed by metal sulfonate/acid chloride combinations. J. Appl. Polym. Sci. 2012, 124, 4114. [Google Scholar]
- Sauvet, G.; Lebrun, J.J.; Sigwalt, P. Cationic Polymerization of Cyclosilanes: The Various Processes Involved. In Cationic Polymerization and Related Processes; Goethals, E.J., Ed.; Academic Press: London, UK, 1984; pp. 237–251. [Google Scholar]
- Wilczek, L.; Chojnowski, J. Acidolytic ring-opening of cyclic siloxane and acetal monomers. Role of hydrogen bonding in cationic polymerization initiated with protonic acids. Macromolecules 1981, 14, 9–17. [Google Scholar]
- Richards, D.H.; Kingston, S.B.; Souel, T. Block copolymer synthesis by reaction of living polystyrene with living polytetrahydrofuran. Polymer 1978, 19, 68–72. [Google Scholar]
- Nagai, D.; Sato, M.; Ochai, B.; Endo, T. Synthesis and properties of the polythiourethanes obtained by the cationic ring-opening polymerization of cyclic thiourethanes. J. Polym. Sci. Polym. Chem. 2006, 44, 4795–4803. [Google Scholar]
- Krieg, A.; Weber, C.; Hoogenboom, R.; Becer, C.R.; Schubert, U.S. Block Copolymers of Poly (2-Oxazoline)s and Poly (Methyl) Acrylates; A Crossover between Cationic Ring-Opening Polymerization (CROP) and Reversible Addition-Fragmentation Chain Transfer (RAFT). ACS Macro Lett. 2012, 1, 776–779. [Google Scholar]
- Lefevre, N.; Fustin, C.-A.; Hoogenboom, R.; Schubert, U.S.; Gohy, J.-F. Nanostructured surfaces from block copoly(2-oxazoline)s prepared by microwave-assisted cationic ring-opening polymerization. Polym. Prepr. 2008, 49, 949–950. [Google Scholar]
- Paulus, R.M.; Erdmenger, T.; Becer, C.R.; Hoogenboom, R.; Schubert, U.S. Scale-Up of microwave-assisted polymerizations in continuous flow mode: Cationic ring-opening polymerization of 2-Ethyl-2-oxazoline. Macromol. Rapid Commun. 2007, 28, 484–491. [Google Scholar]
- Hoogenboom, R. A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar]
- Krause, J.O.; Zarka, M.T.; Anders, U.; Weberskirch, R.; Nuyken, O.; Buchmeiser, M.R. Simple synthesis of Poly(acetylene) latex particles in aqueous media. Angew. Chem. Int. Ed. 2003, 42, 5965–5969. [Google Scholar]
- Gall, B.; Bortenschlager, M.; Nuyken, O.; Weberskirch, R. Cascade reaction in polymeric nanoreactors. Mono(Rh) and Bimetallic(Rh, Ir) micellar catalysis in the hydroformylation of octene. Macromol. Chem. Phys. 2008, 209, 1152–1159. [Google Scholar]
- Schönfelder, D.; Fischer, K.; Schmidt, M.; Nuyken, O.; Weberskirch, R. Poly(2-oxazoline) functionalized with palladium carbene complexes: soluble amphiphilic polymer supports for C–C coupling reactions in water. Macromolecules 2005, 8, 254–262. [Google Scholar]
- Slomkowski, S.; Duda, A. Anionic Ring-Opening Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 88–128. [Google Scholar]
- Ostrovskii, V.E.; Khodzhemirov, V.A.; Barkova, A.P. Heat of ethylene oxide polymerization and kinetics of polymerization on solid potassium hydroxide. Dokl. Akad. Nauk. SSSR 1970, 191, 1095–1098. [Google Scholar]
- Nicco, A.; Boucheron, R.G. Anionic polymerization of thiirane. Eur. Polym. J. 1970, 6, 1477–1490. [Google Scholar]
- Richards, D.H.; Eastmond, G.C.; Stewart, M.J. Anionically Prepared Telechelic Polymers. In Telechelic Polymers: Synthesis and Applications; Goethals, E.J., Ed.; CRC Press: Boca Rotan, FL, USA, 1989; p. 43. [Google Scholar]
- Duda, A.; Penczek, S. Thermodynamics of L-Lactide polymerization. Equilibium monomer concentration. Macromolecules 1990, 23, 1636–1639. [Google Scholar]
- Lebedev, B.V.; Mukhina, N.N.; Kulagina, T.G. Thermodynamics of Poly(dimethyldisiloxane) in the Range of 0–350 K. Vysokomol. Soed. Ser. A 1978, 20, 1297–1303. [Google Scholar]
- Morton, M.; Kammereck, R.F. Nucleophilic substitution at bivalent sulfur, reaction of alkyllithium with cyclic sulfides. J. Am. Chem. Soc. 1970, 92, 3217–3218. [Google Scholar]
- Richards, D.N.; Szwarc, M. Block polymers of ethylene oxide and its analogs with styrene. Trans. Faraday Soc. 1959, 55, 1644–1650. [Google Scholar]
- Boileau, S.; Champetier, G.; Sigwalt, P. Initiation mechanism for the polymerization of episulfides by sodium naphthalene. J. Polym. Sci. Polym. Symp. 1967, 16, 3021–3031. [Google Scholar]
- Boileau, S. Use of cryptates in anionic polymerization of heterocyclic compounds. ACS Symp. Ser. 1981, 166, 283–305. [Google Scholar]
- Deffieux, A.; Boileau, S. Anionic polymerization of heterocyclic compounds. Polymer 1977, 18, 1047–1050. [Google Scholar]
- Hofman, A.; Slomkowski, S.; Penczek, S. Structure and active centers and mechanism of the anionic polymerization of lactones. Makromol. Chem. 1984, 185, 91–101. [Google Scholar]
- Koinuma, H.; Naito, K.; Hirai, H. Anionic polymerization of oxiranes and cyclic siloxanes initiated with potassium salt-crown ether systems. Makromol. Chem. 1982, 183, 1383–1392. [Google Scholar]
- Sosnowski, S.; Slomkowski, S.; Penczek, S. Kinetics of ε-caprolactone polymerization and formation of cyclic monomers. Makromol. Chem. 1983, 184, 2159–2171. [Google Scholar]
- Morton, M.; Kammereck, R.F.; Fetters, L.J. Polymerization and block copolymerization of cyclic sulfides. Br. Polym. J. 1971, 3, 120–128. [Google Scholar]
- Stehlicek, J.; Labsky, J.; Sebenda, J. Alkaline polymerization of 6-caprolactam XXV. The effect of structure of the acyl on polymerization activated by acylcaprolactams or diacrylamines. Collect. Czech. Chem. Commun. 1987, 32, 545–557. [Google Scholar]
- Hamitou, A.; Ouhadi, T.; Jerome, R.; Teyssie, P. Soluble bimetallic μ-oxoalkoxides: VII. Characteristics and mechanism of ring-opening polymerization of lactones. J. Polym. Sci. Polym. Chem. 1977, 15, 865–873. [Google Scholar]
- Vion, J.M.; Jerome, R.; Teyssie, P.; Aubin, H.; Prud’homme, R. Synthesis and characterization and miscibility of caprolactone copolymers. Macromolecules 1986, 19, 1828–1838. [Google Scholar]
- Hofman, A.; Slomkowski, S.; Penczek, S. Polymerization of ε-caprolactone with kinetic suppression of macrocycles. Makromol. Chem. Rapid Commun. 1987, 8, 387–391. [Google Scholar]
- Duda, A.; Florjanczyk, Z.; Hofman, A.; Slomkowski, S.; Penczek, S. Living pseudoanionic polymerization of ε-caprolactone. Poly(caprolactone) free of cyclics and with controlled end groups. Macromolecules 1990, 23, 1640–1646. [Google Scholar]
- Inoue, S.; Tsukuma, I.; Kawaguchi, M.; Tsurata, T. Synthesis of optically active polymers by asymmetric catalysts VI. Behavior of organozinc catalyst systems in the stereoselective polymerization of propylene oxide. Makromol. Chem. 1967, 103, 151–163. [Google Scholar]
- Sosnowski, S.; Duda, A.; Slomkowski, S.; Penczek, S. Determination of the structure of active centers in the anionic polymerization by phosphorus-31 NMR, introducing a phosphorus containing end group. Makromol. Chem. Rapid. Commun. 1984, 5, 551–557. [Google Scholar] [CrossRef]
- Michalski, J.; Skowronska, A.; Bodalski, R. Mechanisms of Reactions of Phosphorus Compounds. In Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; Wiley VCH: New York, NY, USA, 1987; p. 255. [Google Scholar]
- Sebenda, J. Anionic Ring-Opening Polymerization: Lactams. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 511–530. [Google Scholar]
- Yu, G.-E.; Heatley, F.; Booth, C.; Blease, T.G. Anionic polymerization of propylene oxide; isomerization of allylic ether to propenyl ether end groups. J. Polym. Sci. A Polym. Chem. 1994, 32, 1131–1135. [Google Scholar]
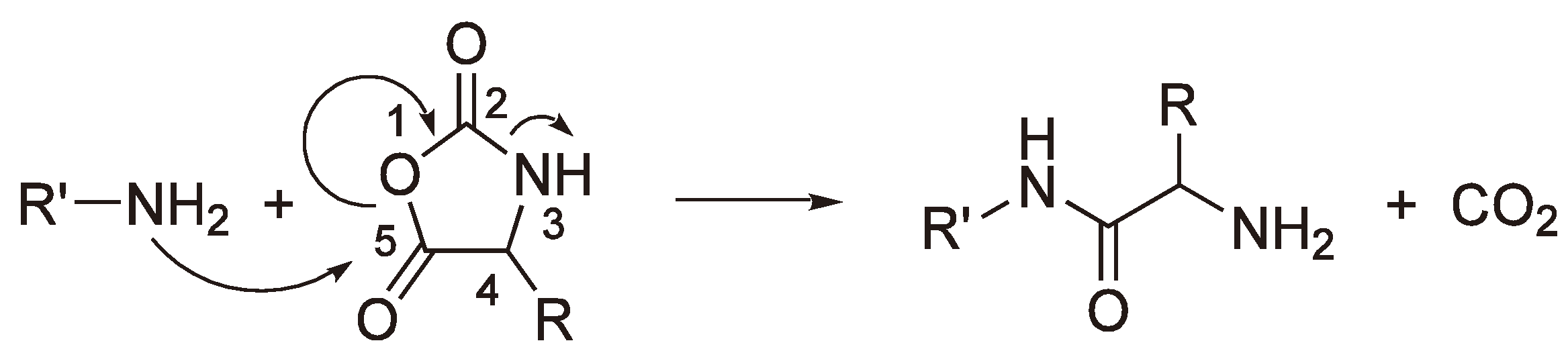
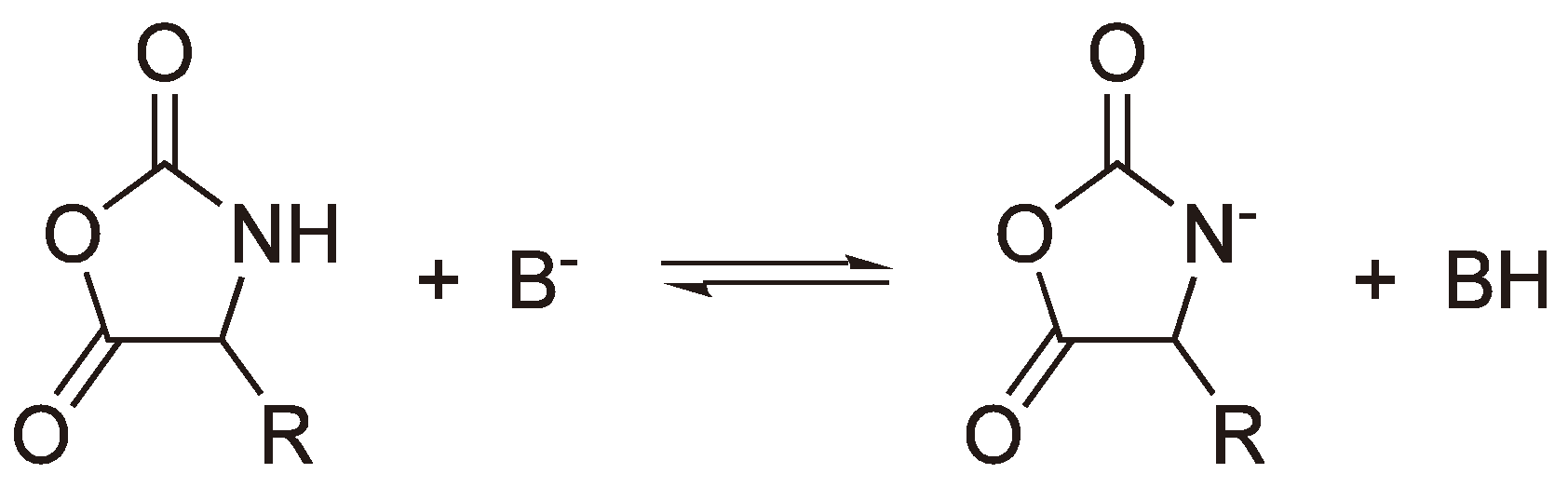
- Kricheldorf, H.R. Anionic Ring-Opening Polymerization: N-Carboxyanhydrides. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 531–551. [Google Scholar]
- Duda, A.; Kowalski, A. Thermodynamics and Kinetics of Ring-Opening Polymerization. In Handbook of Ring-Opening Polymerization; Dubois, P., Coulombier, O., Raquez, J.-M., Eds.; Wiley-VCH: Weinheim, Germany, 2009; p. 8. [Google Scholar]
- Ackermann, J.; Damroth, V. Chemie und Technologie der Silikone II. Herstellung und Verwendung von Siliconpolymeren. Chemie in unserer Zeit 1989, 23, 86–99. [Google Scholar] [CrossRef]
- Choijnowski, J. Kinetically controlled ring-opening polymerization. J. Inorg. Organomet. Polym. 1991, 1, 299–323. [Google Scholar] [CrossRef]
- Boileau, S. Anionic Ring-Opening Polymerization: Epoxides and Episulfides. In Comprehensive Polymer Science; Allen, G., Bevington, J.C., Eds.; Pergamon Press: Oxford, UK, 1989; Volume 3, pp. 467–487. [Google Scholar]
- Anderson, A.N.; Merckling, N.G. Polymeric Bicyclo[2.2.1]hept-2-ene. U.S. Patent 2,721,189, 18 October 1955. [Google Scholar]
- Eleuterio, H.S. Polymerization of Cyclic Olefins. U.S. Patent 3,074,918, 22 January 1967. [Google Scholar]
- Ivin, K.J.; Mol, J.C. Metathesis and Metathesis Polymerization; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Buchmeiser, M.R. Homogeneous metathesis polymerization by well-defined group VI and Group VII transition metal alkylidenes: Fundamentals and applications in the preparation of advanced materials. Chem. Rev. 2000, 100, 1565–1604. [Google Scholar]
- Frenzel, U.; Müller, B.K.M.; Nuyken, O. Metathesis Polymerization of Cycloolefins. In Handbook of Polymer Synthesis, 2nd; Kricheldorf, H.R., Nuyken, O., Swift, G., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 381–426. [Google Scholar]
- Nuyken, O.; Schneider, M.; Frenzel, U. Metathesis Polymerization. In Encyclopedia of Polymer Science and Technology; Wiley: New York, NY, USA, 2012. [Google Scholar]
- Lebedev, B.; Smirnova, N. Thermodynamics of cycloalkenes, of their bulk polymerization in the presence of metathesis catalysts and of polyalkenes. Macromol. Chem. Phys. 1994, 195, 35–63. [Google Scholar] [CrossRef]
- Cheridnichenko, V.M. The equilibrium polymerization of cyclic olefins. Polym. Sci. USSR 1978, 20, 1225–1233. [Google Scholar] [CrossRef]
- Andrews, J.M.; Jones, F.R.; Semlyen, J.A. Equilibrium ring concentration and the statistical conformations of polymeric chains. 12. Cycles in molten and solid Nylon-6. Polymer 1974, 15, 420–424. [Google Scholar] [CrossRef]
- Schrock, R.R. Multiple metal-carbon bonds for catalytic metathesis reactions (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3748–3759. [Google Scholar] [CrossRef]
- Grubbs, R.H. Olefin metathesis catalysts for the preparation of molecules and materials (Nobel lecture). Angew. Chem. Int. Ed. 2006, 45, 3760–3765. [Google Scholar] [CrossRef]
- Chauvin, Y. Olefin metathesis: The early days (Nobel Lecture). Angew. Chem. Int. Ed. 2006, 45, 3741–3747. [Google Scholar] [CrossRef]
- Schrock, R.H. On the trail of metathesis catalysts. Organomet. Chem. 1986, 300, 249–262. [Google Scholar]
- Schwab, P.; France, M.B.; Ziller, J.W.; Grubbs, R.H. A series of well-defined metathesis catalysts of [RuCl2(=CHR′)(PR3)2] and their reactions. Angew. Chem. Int. Ed. 1995, 34, 2039–2041. [Google Scholar]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Love, J.A.; Morgan, J.P.; Tmka, T.M.; Grubbs, R.H. A practical and highly active ruthenium-based catalysts that effects the cross metathesis of acrylonitrile. Angew. Chem. Int. Ed. 2002, 41, 4035–4037. [Google Scholar] [CrossRef]
- Kingsbury, J.; Harrity, J.; Bonnitatebus, P.; Hoveyda, A.H. A recycable Ru-based metathesis catalysts. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Halbach, T.S.; Mix, S.; Fischer, D.; Maechling, S.; Krause, J.O.; Sievers, C.; Blechert, S.; Nuyken, O.; Buchmeiser, M.R. Novel ruthenium-based metathesis catalysts containing electron withdrawing ligands, synthesis, immobilization and reactivity. J. Org. Chem. 2005, 70, 4687–4694. [Google Scholar] [CrossRef]
- Schrock, R.R. Ring-Opening Metathesis Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; p. 141. [Google Scholar]
- Trimmel, G.; Riegler, S.; Fuchs, G.; Slugovc, C.; Stelzer, F. Liquid crystalline polymers by metathesis polymerization. Adv. Polym. Sci. 2003, 176, 43–87. [Google Scholar]
- Herisson, J.L.; Chauvin, Y. Catalysis of olefin transformation by tungsten complexes, telomerization of cyclic olefins in the presence of acyclic olefins. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Grubbs, R.H. The development of functional group tolerant ROMP catalysts. J. Macromol. Sci. Chem. 1994, A31, 1829–1833. [Google Scholar]
- Krause, J.O.; Nuyken, O.; Wurst, K.; Buchmeiser, M.R. Synthesis and reactivity of homogeneous and heterogoneous ruthenium-based metathesis catalysts containing electron withdrawing ligands. Chem. Eur. J. 2004, 10, 777–784. [Google Scholar] [CrossRef]
- Hilf, S.; Kilbinger, A.F.M. Functional endgroups for polymers prepared using ring-opening metathesis polymerization. Nat. Chem. 2009, 1, 537–546. [Google Scholar] [CrossRef]
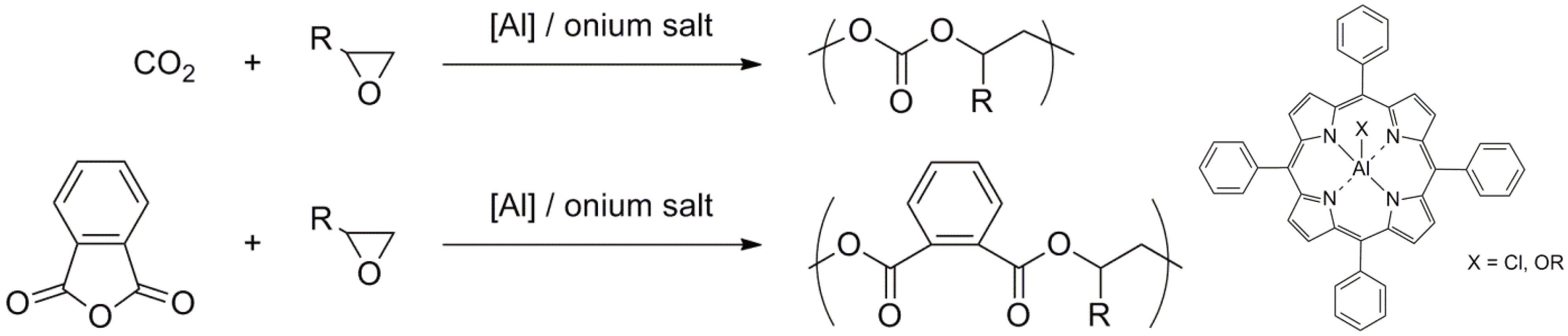
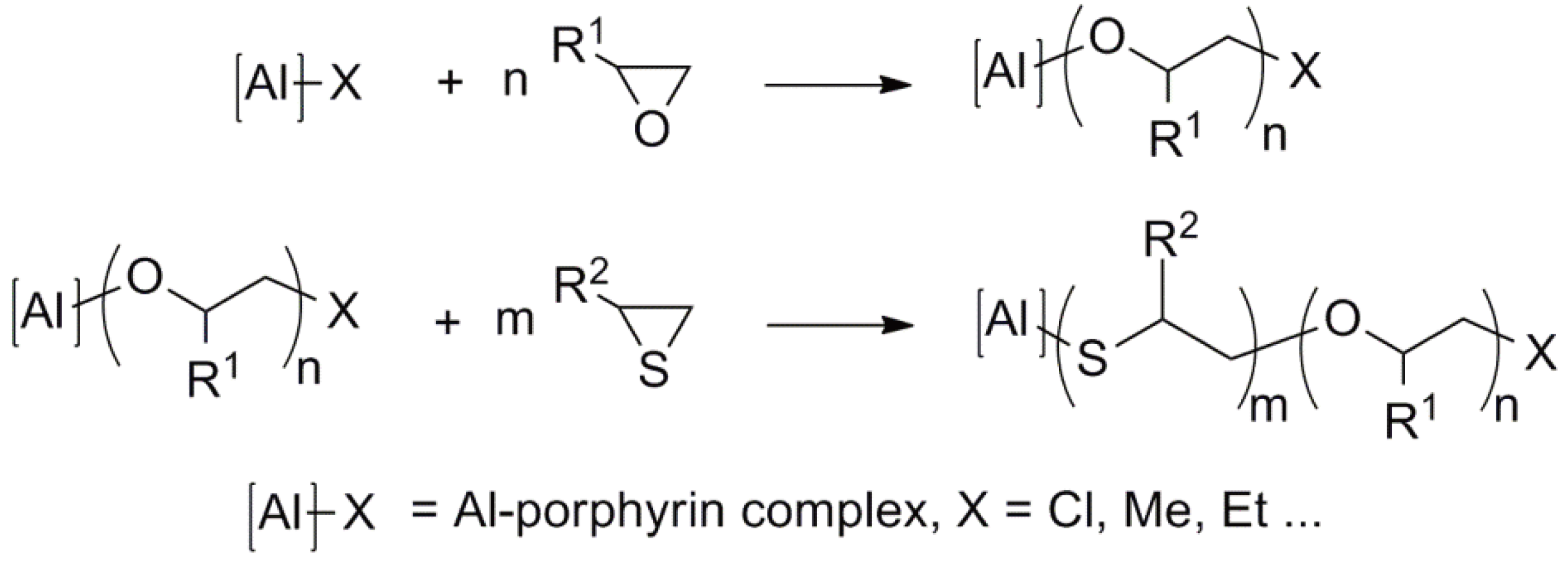
- Inoue, S.; Aida, T. Catalysts for Living and Immortal Polymerization. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 197–215. [Google Scholar]
- Kobayashi, S. Ring-Opening Polymerization Involving Oxidation-Reduction-Processes. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 337–350. [Google Scholar]
- Inoue, S. Immortal polymerization: The outset, development, and application. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 2861–2871. [Google Scholar] [CrossRef]
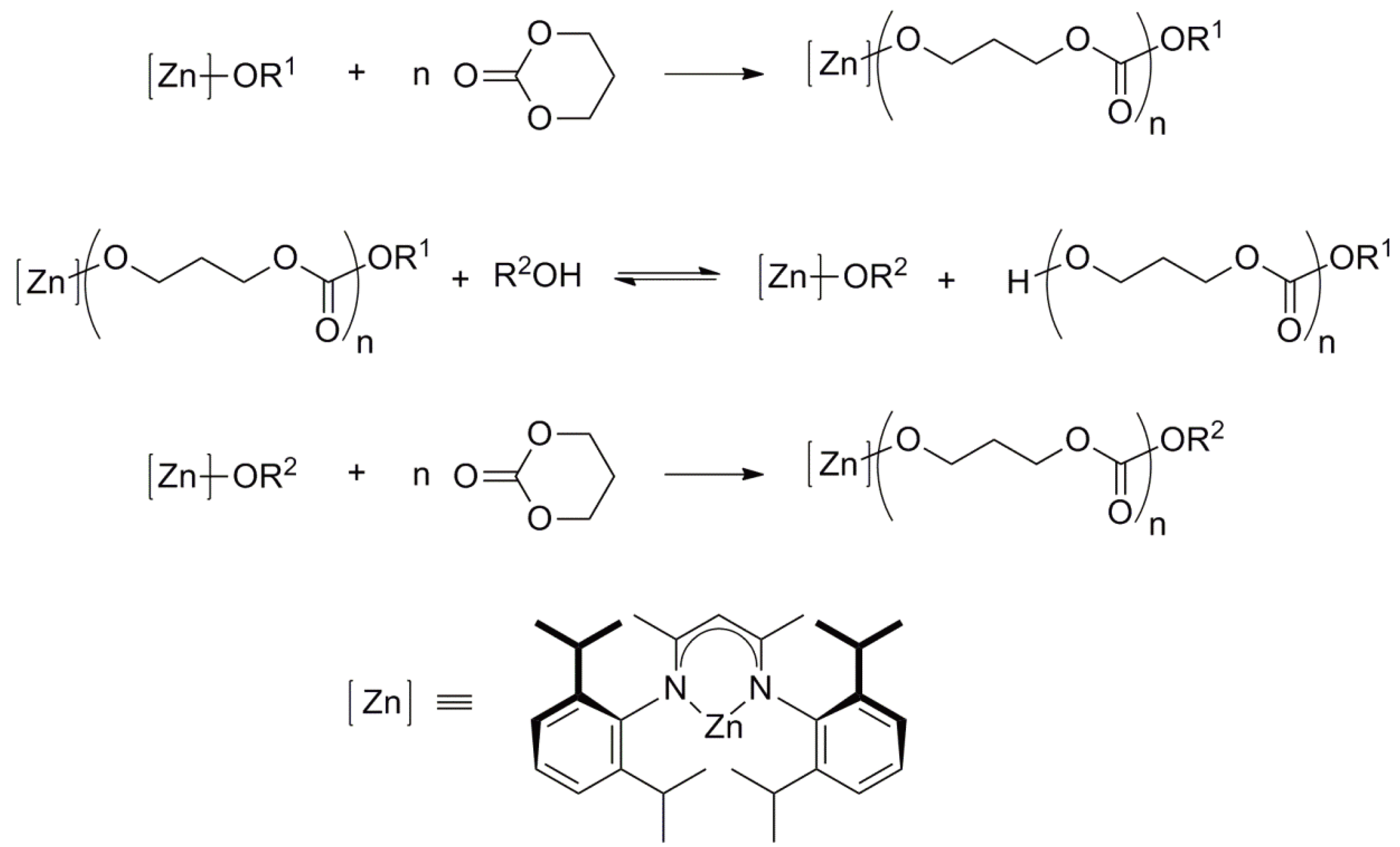
- Helou, M.; Miserque, O.; Brusson, J.-M.; Carpentier, J.-F.; Guillaume, S.M. Ultraproductive, Zinc-Mediated, Immortal Ring-Opening Polymerization of Trimethylene Carbonate. Chem. Eur. J. 2008, 14, 8772–8775. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Liu, X.; Cui, D. Facile synthesis of pendant- and α,ω-chain-end- functionalized polycarbonates via immortal polymerization by using salan lutetium alkyl precursor. Chem. Commun. 2012, 48, 4588–4590. [Google Scholar] [CrossRef]
- Ajellal, N.; Carpentier, J.-F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trionov, A. Metal-catalyzed immortal ring-opening of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef]
- Guillaume, S.M.; Carpentier, J.-F. Recent advances in metallo/organo-catalyzed immortal ring-opening polymerization of cyclic carbonates. Catal. Sci. Technol. 2012, 2, 898–906. [Google Scholar] [CrossRef]
- Amgoune, A.; Thomas, C.M.; Carpentier, J.-F. Yttrium complexes as catalysts for living and immortal polymerization of lactide to highly heterotactic PLA. Macromol. Rapid Commun. 2007, 28, 693–697. [Google Scholar] [CrossRef]
- Endo, M.; Aida, T.; Inoue, S. Immortal Polymerization of epsilon-caprolactone, initiated by aluminum porphyrin in the presence of alcohol. Macromolecules 2002, 20, 2982–2988. [Google Scholar] [CrossRef]
- Xu, C.; Yu, I.; Mehrkhodavandi, P. Highly controlled immortal polymerization of β-butyrolactone by a dinuclear indium catalyst. Chem. Commun. 2012, 48, 6806–6808. [Google Scholar] [CrossRef]
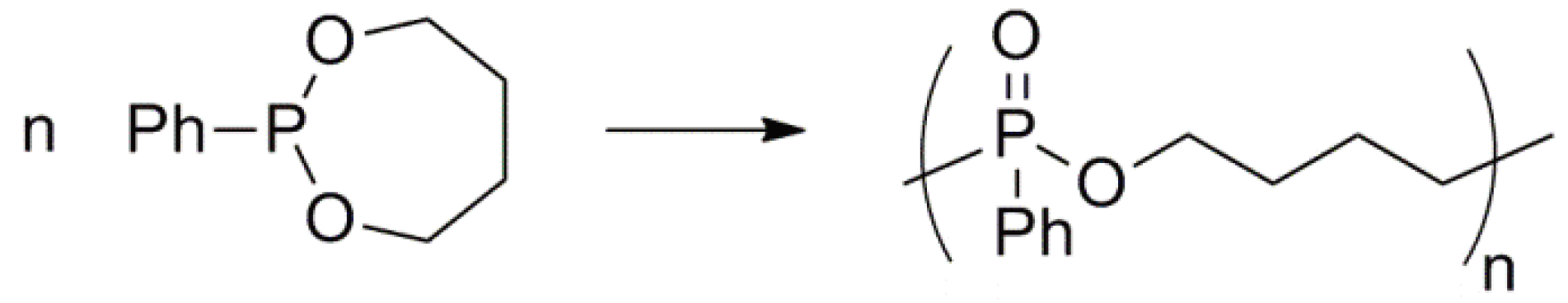
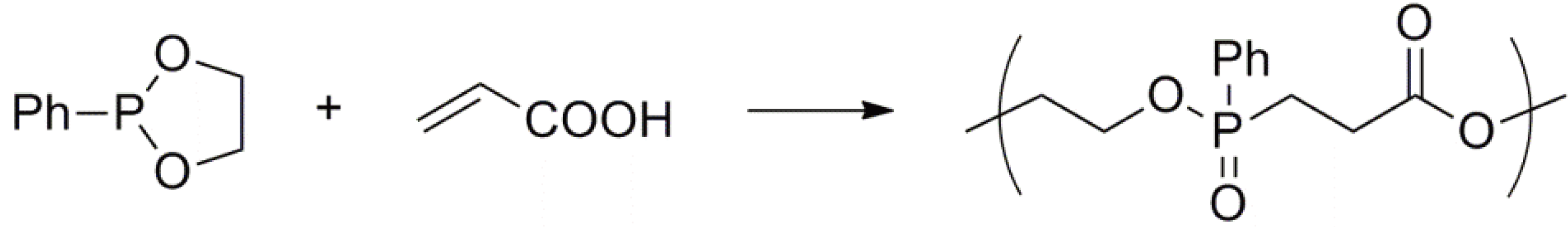
- Kobayashi, S.; Tokunoh, M.; Saegusa, T. Cationic ring-opening polymerization of 2-phenyl-1,3,2-dioxaphosphepane, a seven-membered cyclic phosphonite. Macromolecules 1986, 19, 466–469. [Google Scholar] [CrossRef]
- Kobayashi, S.; Saegusa, T. Alternating Copolymerization Involving Zwitterions. In Alternating Copolymers; Cowie, J.M.G., Ed.; Plenum: New York, NY, USA, 1985; pp. 189–239. [Google Scholar]
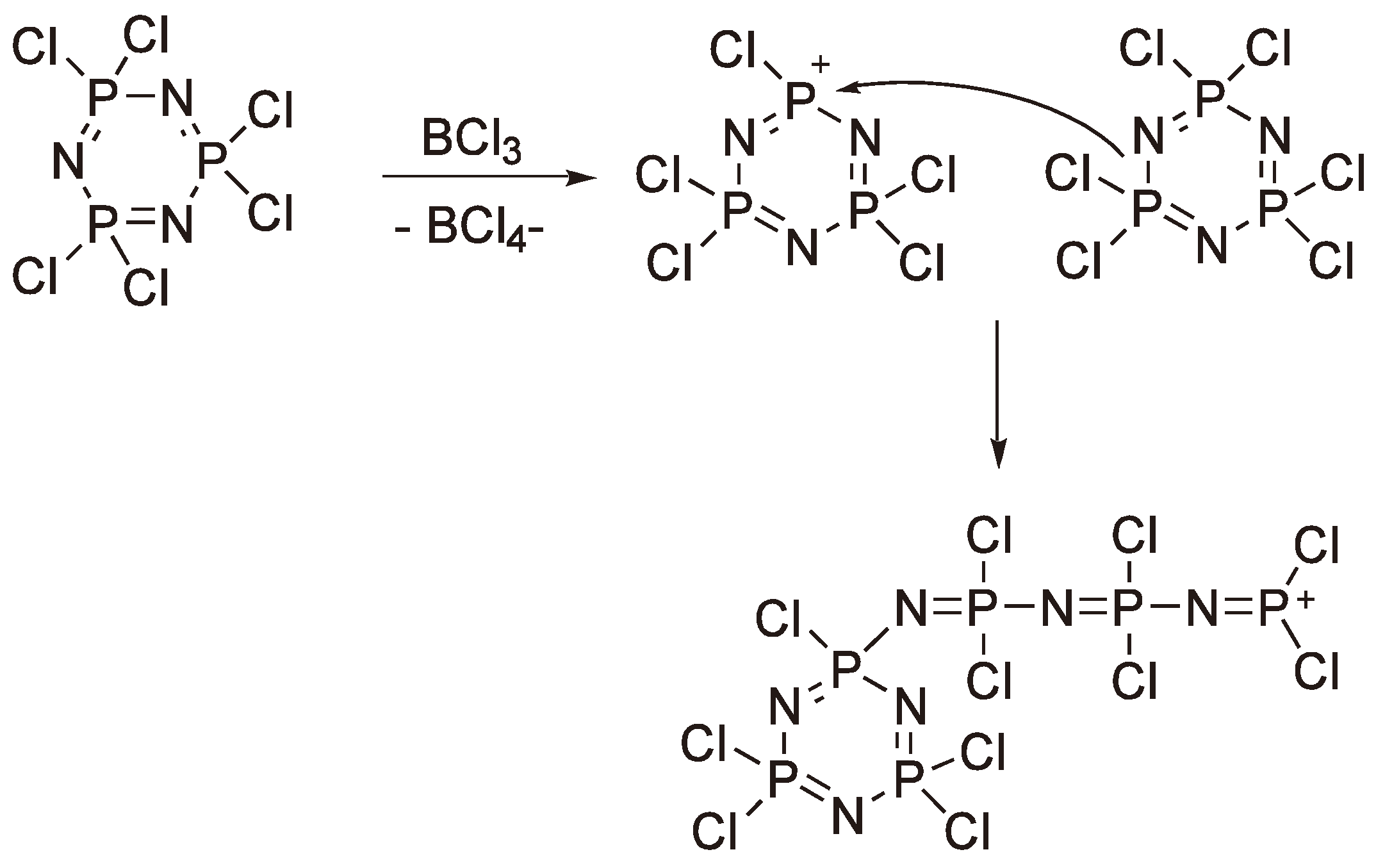
- Allcock, H.R. Ring-Opening Polymerization in Phosphazene Chemistry. In Ring-Opening Polymerization; Brunelle, D.J., Ed.; Hanser Publishers: Munich, Germany, 1993; pp. 217–237. [Google Scholar]
- Allcock, H.R. Current status of polyphosphazene chemistry. ACS Symp. Ser. 1988, 360, 250–267. [Google Scholar] [CrossRef]
- Allcock, H.R. Phosphorus-Nitrogen-Compounds; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- v.Liebig, J. Über Phosphorstickstoff. Ann. Chem. 1834, 11, 139. [Google Scholar]
- Stokes, H.N. Chloronitrides of phosphorus. Am. Chem. J. 1897, 19, 782–796. [Google Scholar] [CrossRef]
- D’Halluin, G.; de Jaeger, R.; Potin, P. Polydichlorophosphazenes: Synthèse à Partir De Cl3Pnp(O)Cl2. Bull. Soc. Chim. Belg. 1989, 98, 653–666. [Google Scholar]
- Matyjaszewski, K.; Cypryk, M.; Dauth, J.; Montague, R.; White, M. New synthetic routes towards polyphosphazenes. Makromol. Chem. Macromol. Sym. 1992, 54–55, 13–30. [Google Scholar]
- Matyjaszewski, K.; Franz, U.; Montague, R.A.; White, M.L. Synthesis of polyphosphazenes from phosphoranimines and phosphine azides. Polymer 1994, 35, 5005–5011. [Google Scholar] [CrossRef]
- Franz, U.; Nuyken, O.; Matyjaszewski, K. Synthesis and characterization of poly(phenyl-p-tolylphosphazene), prepared via in situ polymerization of phenyl-p-tolylphosphine azide. Macromol. Rapid Comm. 1994, 15, 169–174. [Google Scholar] [CrossRef]
- Allcock, H.R. Chemistry and Applications of Polyphosphazenes; Wiley-Interscience: New York, NY, USA, 2003. [Google Scholar]
- Neilson, R.H.; Wisian-Neilson, P. Poly(alkyl/arylphosphazenes) and their precursors. Chem. Rev. 1988, 88, 541–562. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kugel, R.L.; Valan, K.J. High molecular weight Poly(alkoxy and aryloxy-phosphazenes). Inorg. Chem. 1966, 5, 1709–1715. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kugel, R.L. High molecular weight Poly(diaminophosphazenes). Inorg. Chem. 1966, 5, 1716–1718. [Google Scholar] [CrossRef]
- Rose, S.H. Synthesis of phosphonitrilic fluoroelastomers. J. Polym. Sci. 1968, 6, 837–839. [Google Scholar] [CrossRef]
- Singler, R.E.; Schneider, N.S.; Hagnauer, G.L. Polyphosphazenes: Synthesis—Properties—Applications. Polym. Eng. Sci. 1975, 15, 321–338. [Google Scholar] [CrossRef]
- Kolich, C.H.; Klobucar, W.D.; Books, J.T. Process for surface treating phosphonitrilic fluoroelastomers. U.S. Patent 4,945,139 A, 31 July 1990. [Google Scholar]
- Tate, D.P. Polyphosphazene elastomers. J. Polym. Sci. 1974, 48, 33–45. [Google Scholar]
- Gettleman, L.; Farris, C.L.; Rawls, H.R.; LeBouef, R.J. Soft and firm denture liner for a composite denture and method of fabricating. U.S. Patent 4,432,730, 21 February 1984. [Google Scholar]
- Blonsky, P.M.; Shriver, D.F.; Austin, P.E.; Allcock, H.R. Polyphosphazene solid electrolytes. J. Am. Chem. Soc. 1984, 106, 6854–6855. [Google Scholar] [CrossRef]
- Allcock, H.R.; O’Connor, S.J.M.; Olmeijer, D.L.; Napierala, M.E.; Cameron, C.G. Cation complexation and conductivity in crown ether bearing polyphosphazenes. Macromolecules 1996, 23, 7544–7552. [Google Scholar]
- Fei, S.-T.; Allcock, H.R. Methoxyethoxyethoxyphosphazenes as Ionic Conductive Fire Retardant/Additives for Lithium Battery Systems. J. Power Source 2010, 19, 2082–2088. [Google Scholar]
- Tang, H.; Pintauro, P.N. Polyphosphazene membranes. IV. Polymer morphology and proton conductivity in sulfonated poly[bis(3-methylphenoxy)phosphazene] films. J. Appl. Polym. Sci. 2001, 79, 49–59. [Google Scholar] [CrossRef]
- Allcock, H.R.; Kwon, S.; Riding, G.H.; Fitzpatrick, R.J.; Bennett, J.L. Hydrophilic Polyphosphazenes as Hydrogels: Radiation Crosslinking and Hydrogel Characteristics of Poly[bis(methoxyethoxyethoxy)phosphazene]. Biomaterials 1988, 9, 509–513. [Google Scholar] [CrossRef]
- Kim, J.; Chun, C.; Kim, B.; Hong, J.M.; Cho, J.-K.; Lee, S.H.; Song, S.-C. Thermosensitive/magnetic poly(organophosphazene) hydrogel as a long-term magnetic resonance contrast platform. Biomaterials 2012, 33, 218–224. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Turner, M.L.; Fitzpatrick, R.J. Poly(organophosphazenes) with Poly(alkyl ether) Side Groups: A Study of Their Water Solubility and the Swelling Characteristics of Their Hydrogels. Macromolecules 1992, 25, 5573–5577. [Google Scholar] [CrossRef]
- Allcock, H.R.; Fitzpatrick, R.J.; Visscher, K.B. Thin layer grafts of Poly[bis(methoxyethoxy-ethoxy)-phosphazene] on organic polymer surfaces. Chem. Mater. 1992, 4, 775–780. [Google Scholar] [CrossRef]
- Allcock, H.R.; Ambrosio, A.M. A. synthesis and characterization of pH-senstitive poly(organophosphazene) hydrogels. Biomaterials 1996, 17, 2295–2302. [Google Scholar] [CrossRef]
- Allcock, H.R.; Pucher, S.R.; Scopelianos, A.G. Poly[amino acid ester)phosphazenes] as substrates for the controlled release of small molecules. Macromolecules 1994, 6, 516–524. [Google Scholar]
- Deng, M.; Kumbar, S.G.; Wan, Y.; Toti, U.S.; Allcock, H.R.; Laurencin, C.T. Polyphosphazene polymers for tissue engineering: An analysis of material synthesis, characterization, and applications. Soft Matter 2010, 6, 3119–3132. [Google Scholar] [CrossRef]
- Deng, M.; Kumbar, S.G.; Nair, L.S.; Arlin, L.; Weikel, A.L.; Allcock, H.R.; Laurencin, C.T. Biomimetic structures: Biological implications of dipeptide-substituted polyphosphazene–polyester blend nanofiber matrices for load-bearing bone regeneration. Adv. Funct. Mater. 2011, 21, 2641–2651. [Google Scholar] [CrossRef]
- Allcock, H.R.; Morozowich, N. Bioerodible polyphosphazenes and their medical potential. Polym. Chem. 2012, 3, 578–590. [Google Scholar] [CrossRef]
- Manners, I.; Renner, G.; Nuyken, O.; Allcock, H.R. Poly(carbophosphazenes): A new class of inorganic-organic macromolecules. J. Am. Chem. Soc. 1989, 111, 5478–5480. [Google Scholar] [CrossRef]
- Allcock, H.R.; Coley, S.M.; Manners, I.; Renner, G.; Nuyken, O. Poly[(aryloxy)carbophosphazenes]: Synthesis, properties, and thermal transition behavior. Macromolecules 1991, 24, 2024–2028. [Google Scholar] [CrossRef]
- Dodge, J.A.; Manners, I.; Allcock, H.R.; Renner, G.; Nuyken, O. Poly(thiophospazenes): New inorganic macromolecules with backbone composed of phosphorus, nitrogen and sulfur atoms. J. Am. Chem. Soc. 1990, 112, 1268–1269. [Google Scholar] [CrossRef]
- Manners, I.; Riding, G.H.; Dodge, J.A.; Allcock, H.R. Role of ring strain and steric hindrance in a new method of the synthesis of macrocycles and high polymer phosphazenes. J. Am. Chem. Soc. 1989, 111, 3067–3069. [Google Scholar] [CrossRef]
- Voit, B. New developments in hyperbranched polymers. J. Polym. Sci. Polym. Chem. 2000, 38, 2505–2525. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nuyken, O.; Pask, S.D. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 361-403. https://doi.org/10.3390/polym5020361
Nuyken O, Pask SD. Ring-Opening Polymerization—An Introductory Review. Polymers. 2013; 5(2):361-403. https://doi.org/10.3390/polym5020361
Chicago/Turabian StyleNuyken, Oskar, and Stephen D. Pask. 2013. "Ring-Opening Polymerization—An Introductory Review" Polymers 5, no. 2: 361-403. https://doi.org/10.3390/polym5020361
APA StyleNuyken, O., & Pask, S. D. (2013). Ring-Opening Polymerization—An Introductory Review. Polymers, 5(2), 361-403. https://doi.org/10.3390/polym5020361