1. Introduction
Functional polymeric nanoparticles (NPs) are of considerable interest in the emerging fields of nanomedicine, nanolithography and nanoelectronic, among others [
1,
2,
3,
4]. As a result, the synthetic methodologies for the functional polymeric NPs have attracted a wide range of attention. Perhaps the most universal technique for the preparation of functional polymeric NPs having controlled morphologies is the self-assembly of amphiphilic block copolymers [
5]. However, these self-assembled nanostructures tend to spontaneously dissociate under high dilution or high ionic strength, which are typically encountered in the blood circulation system [
6,
7]. Thus, the self-assembled polymeric NPs are frequently cross-linked to prevent disintegration at low concentrations or upon environmental changes [
8,
9].
“Click chemistry”, especially the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction [
10], is a powerful tool to introduce cross-linking points to a polymer system [
11]. It has been extended to design hydrogels [
12,
13,
14], core or shell cross-linked self-assembled micelles [
15,
16], layer-by-layer cross-linked films [
17,
18,
19], hollow spheres [
20,
21] and intermolecular cross-linked polymeric NPs [
22,
23], based on low molecular weight or polymeric precursors. Very recently, it also became a popular tool to synthesize single-chain cross-linked polymeric NPs via intramolecular “click” reactions [
24,
25]. Atom transfer radical polymerization (ATRP) [
26], a well-known controlled radical polymerization (CRP) technique, not only shares a number of attractive features with “click chemistry”, such as a high tolerance toward a wide range of functional groups and protic solvents, but also uses the same copper catalyst system [
27]. The combination of these two techniques in one-pot synthesis provides a versatile approach for the preparation of block copolymers [
28,
29,
30], functional macromolecules [
31,
32,
33,
34] and interpenetrating network hydrogels [
13], among others. However, the combination of two chemistries has rarely been utilized to prepare the functional polymeric NPs in one-pot synthesis [
35]. Thus, it is very interesting to prepare the polymeric NPs with suitable surface functionalization via the combination of ATRP and “click chemistry” in a one-pot approach.
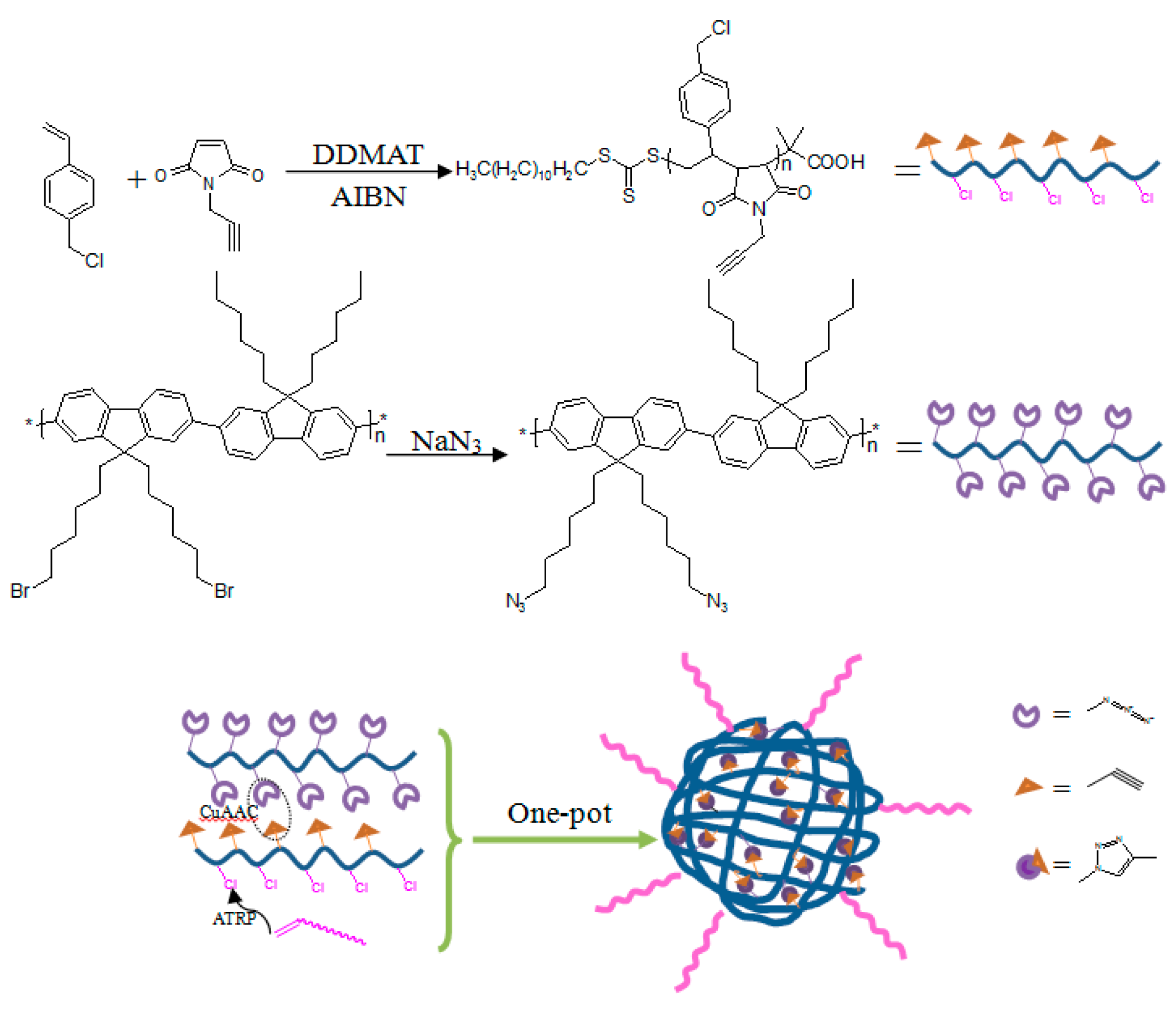
Herein, we reported a general synthetic route to PEGylated fluorescent NPs based on simultaneous intermolecular chain “click” cross-linking reaction and ATRP. Alkynyl groups and ATRP initiating sites were introduced on poly(
p-chloromethyl styrene-
alt-
N-propargylmaleimide (P(CMS-
alt-NPM)) copolymers via reversible addition-fragmentation chain transfer (RAFT) polymerization of
N-propargylmaleimide (NPM) and
p-chloromethyl styrene (CMS). As one of the most widely-used conjugated polymers, a fluorene-based polymer with pendent azido functionalities (poly[(9,9-dihexylfluorene)-
alt-(9,9-bis-(6-azidohexyl)fluorene)] (PFC6N
3)) was selected as the cross-linker. Intermolecular “click” cross-linking between P(CMS-
alt-NPM) and PFC6N
3 with a 1:1 alkynyl/azido ratio and ATRP of poly(ethylene glycol) methyl ether methacrylate (PEGMMA) from the initiating sites of P(CMS-
alt-NPM) proceeded simultaneously, leading to the formation of PEGylated fluorescent NPs (
Scheme 1).
Scheme 1.
Synthesis of alkynyl and ATRP-initiating sites-containing alternate copolymer poly(p-chloromethyl styrene-alt-N-propargylmaleimide (P(CMS-alt-NPM)) and the azido-bearing conjugated fluorene copolymer poly[(9,9-dihexylfluorene)-alt-(9,9-bis-(6-azidohexyl)fluorene)] (PFC6N3) and the formation of PEGylated fluorescent NPs via simultaneous ATRP and “click chemistry”. DDMAT, 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid; AIBN, 2,2’-azoisobutyronitrile.
Scheme 1.
Synthesis of alkynyl and ATRP-initiating sites-containing alternate copolymer poly(p-chloromethyl styrene-alt-N-propargylmaleimide (P(CMS-alt-NPM)) and the azido-bearing conjugated fluorene copolymer poly[(9,9-dihexylfluorene)-alt-(9,9-bis-(6-azidohexyl)fluorene)] (PFC6N3) and the formation of PEGylated fluorescent NPs via simultaneous ATRP and “click chemistry”. DDMAT, 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid; AIBN, 2,2’-azoisobutyronitrile.
2. Experimental Section
2.1. Materials
p-Chloromethyl styrene (CMS, 90%), CuCl (99%), anhydrous sodium acetate (99%), maleic anhydride (95%), propargylamine (98%), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT, 98%), poly(ethylene glycol) methyl ether methacrylate (PEGMMA,
Mn = 300),
N,N,N',N'',N''-pentamethyldiethylenetriamine (PMDETA, 98%) and 2,2’-azoisobutyronitrile (AIBN, 98%) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). The monomer was used after removal of the inhibitors in a ready-to-use disposable inhibitor removal column. CuCl was washed with acetic acid followed by methanol to remove impurities. All other regents were purchased from Sigma-Aldrich Chem. Co. (St. Louis, MO, USA) or Merck Chem. Co. (Darmstadt, Germany), and were used without further purification. Poly[(9,9-dihexylfluorene)-
alt-(9,9-bis(6-bromohexyl)fluorene)] (PFC6Br) was synthesized according to the methods reported in the literature [
36].
2.2. Synthesis of N-Propargylmaleimide (NPM)
Maleic anhydride (9.81 g, 0.1 mol) was dissolved in acetone (50 mL), and the solution was cooled in an ice bath. Then, propargylamine (6.40 mL, 0.1 mol) in acetone (25 mL) was added in portions over 20 min under vigorous stirring. The mixture was refluxed for 1 h under nitrogen atmosphere, followed by a removal of the solvent using the rotary evaporator. After recrystallization in acetone, the beige crystal (N-propargyl-substituted maleamic acid) was filtered and dried under reduced pressure. N-propargyl-substituted maleamic acid (7.65 g, 0.05 mol) was suspended in acetic anhydride (25 mL) in the presence of anhydrous sodium acetate (2.5 g, 0.03 mol) at 65 °C for 2 h. After cooling down to room temperature, the mixture was poured into ice water (150 mL) and neutralized with K2CO3 under vigorous stirring. After extraction with diethyl ether (3 × 50 mL), the organic phase was dried over anhydrous Na2SO4, filtered and concentrated using the rotary evaporator. The residue was further purified by distillation in vacuum, resulting in a light yellow crystal (2.36 g, 35% yield). 1H NMR (CDCl3): 6.78 (2H, CH=CH), 4.31 (2H, N–CH2), 2.23 (1H, ≡CH). FTIR (cm-1): ν(C≡CH) = 3271 cm−1.
2.3. Preparation of Poly(p-chloromethyl styrene-alt-N-propargylmaleimide) Copolymer
In a typical reaction, AIBN (19.7 mg, 0.12 mmol), CMS (1.70 mL, 12.0 mmol), NPM (1.62 g, 12.0 mmol), DDMAT (218.8 mg, 0.6 mmol) and toluene (6 mL) were introduced into a test tube. The solution was degassed with argon for 30 min. The test tube was sealed and kept in a 65 °C oil bath under stirring for 2 and 4 h. The reaction mixture was diluted with tetrahydrofuran (THF) and then poured into diethyl ether (200 mL) to precipitate out P(CMS-alt-NPM) copolymer. The resulting P(CMS-alt-NPM) with a polymerization time of 2 and 4 h is referred to as P(CMS-alt-NPM)1 and P(CMS-alt-NPM)2, respectively. About 1.4 g and 2.3 g of P(CMS-alt-NPM)1 and P(CMS-alt-NPM)2 were obtained after filtering and drying under reduced pressure for 24 h.
2.4. Azidation of PFC6Br
PFC6Br (250 mg, 0.3 mmol of repeating units) and NaN3 (117.0 mg, 1.8 mmol) were suspended in dry N,N-dimethylformamide (DMF, 10 mL) and heated at 60 °C for 24 h. After that, CH2Cl2 (100 mL) was added into the mixture. The resulting precipitate was filtered, and the obtained filtrate was washed thrice with doubly-distilled water (100 mL) and dried over anhydrous MgSO4. The solution volume was further reduced to 5 mL using the rotary evaporator (Buchi Co., Essen, Germany). The obtained solution was poured into methanol (100 mL) under vigorous stirring to precipitate out the product, poly[(9,9-dihexylfluorene)-alt-(9,9-bis-(6-azidohexyl)fluorene) (PFC6N3). The precipitate was collected by centrifugation and dried under reduced pressure for 24 h.
2.5. PEGylated Fluorescent Nanoparticle Formation using One-Pot Atom Transfer Radical Polymerization and “Click Chemistry”
In a typical reaction, PEGMMA (2.38 g, 5.0 mmol), CuCl (49.5 mg, 0.5 mmol), PFC6N3 (186.5 mg, 0.25 mmol of repeating units), P(CMS-alt-NPM)1 (143.5 mg, 0.5 mmol of repeating units) and THF (30 mL) were introduced into a 50-mL round bottle flask. The flask was degassed with argon for 20 min, and PMDETA (86.5 mg, 0.5 mmol) was added quickly. After the addition was complete, the flask was sealed and stirred at 50 °C for 12 h. The solution was concentrated using the rotary evaporator and precipitated into diethyl ether (100 mL). The residual monomer, oligopolymer and the copper catalyst system were removed by dialyzing against THF for 4 days and doubly-distilled water for another 4 days. The solid NPs were obtained by lyophilization.
2.6. Cell Cytotoxicity and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium Bromide (MTT) Assay
The cytotoxicity of the PEGylated fluorescent NPs was evaluated by determining the viability of KB cells, a cell line derived from carcinoma of the nasopharynx, after incubation in media containing the PEGylated fluorescent NPs at concentrations of 0.25, 0.5, 0.75 and 1.0 mg/mL. Control experiments were carried out using the complete growth culture medium without the fluorescent NPs. Cell viability testing was carried out via the reduction of the MTT reagent. The MTT assay was performed in a 96-well plate following the standard procedure with minor modifications. These cells were seeded at a density of 1 × 104 cells per well and incubated at 37 °C for 24 h before the medium was replaced with one containing the PEGylated fluorescent NPs. The cells were incubated at 37 °C for another 24 or 48 h in the medium. The culture medium in each well was then removed, and 90 μL of the medium and 10 μL of MTT solution (5 mg/mL in PBS) were added to each well. After 4 h of incubation at 37 °C, the medium was removed, and the formazan crystals were solubilized with 100 μL of dimethyl sulfoxide (DMSO) for 15 min. The optical absorbance was then measured at 560 nm on a microplate reader (Tecan GENios, Mannedorf, Switzerland). The results were expressed as percentages relative to that obtained in the control experiment.
2.7. Cell Labeling by the Fluorescent Nanoparticles
For cell labeling experiments, KB cells were seeded on cover slips for 24 h. The medium was then aspirated and replaced with a medium containing the PEGylated fluorescent NPs at a concentration of 0.2 mg/mL. The cells were cultured for another 4 h and then washed with sterilized doubly-distilled water exhaustively to remove the free NPs completely. The cells were then fixed in 4% (v/v) formaldehyde aqueous solution for 30 min. Following fixation, the cells were observed on a Nikon A1R inverted microscope (Nikon Inc., Tokyo, Japan) equipped with an Andor iXon electron multiplying charge coupled device (EMCCD, Andor Technology, Tokyo, Japan). The cells were excited using a mercury lamp (EXFO Life Sciences, Mississauga, Canada) and a DAPi filter (λex = 340–380 nm).
2.8. Characterization
The chemical structures of the obtained monomers and polymers were characterized by 1H NMR spectroscopy on a Bruker DRX 400 MHz spectrometer (Bruker UK Ltd., Coventry, UK) using d-CDCl3 as the solvent. Gel permeation chromatography (GPC) was performed on a Waters GPC system, equipped with a Waters 1515 isocratic HPLC pump, a Waters 717 plus Autosampler injector, a Waters 2414 refractive index detector and a PL gel 10-µm Mixed-B column (S/N 10M-MB-D7-9K8, Agilent Technologies, Santa Clara, CA, USA), using THF as the eluent at a flow rate of 1.0 mL min−1. The calibration curve was generated using polystyrene molecular weight standards. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos AXIS Ultra HSA spectrometer (Kratos Analytical Ltd., Manchester, UK) equipped with a monochromatized Al Kα X-ray source (1468.71 eV photons). Field emission transmission electron microscopy (FETEM) images were obtained from a JEOL JEM-2010 FETEM (JEOL Ltd., Tokyo, Japan). The UV-visible absorption spectra in the wavelength range of 300 to 450 nm were obtained from a Hitachi U2800 spectrophotometer (Hitachi High-Tech, Tokyo, Japan). The fluorescence spectra were measured on a Shimadzu RF-5031PC spectrophotometer (Shimadzu Co., Kyoto, Japan), with an excitation wavelength of 370 nm. Fourier transform infrared (FTIR) spectroscopy analysis was carried out on a Bio-Rad FTS-135 spectrophotometer (Bio-Rad Laboratories Inc., Cambridge, MA, USA).
3. Results and Discussion
Poly(
p-chloromethyl styrene-
alt-
N-propargylmaleimide) (P(CMS-
alt-NPM)) copolymer was prepared via reversible addition-fragmentation chain transfer (RAFT) polymerization of
p-chloromethyl styrene (CMS) and
N-propargylmaleimide (NPM) at 65 °C using 2,2’-azoisobutyronitrile (AIBN) as the initiator and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) as the chain transfer agent. The copolymerization of a strongly-electron-accepting with an electron-donating monomer produces an alternate copolymer [
37,
38].
N-substituted maleimide (electron-accepting monomer) and styrene (electron-donating monomer) comprise an extensively-studied system [
39,
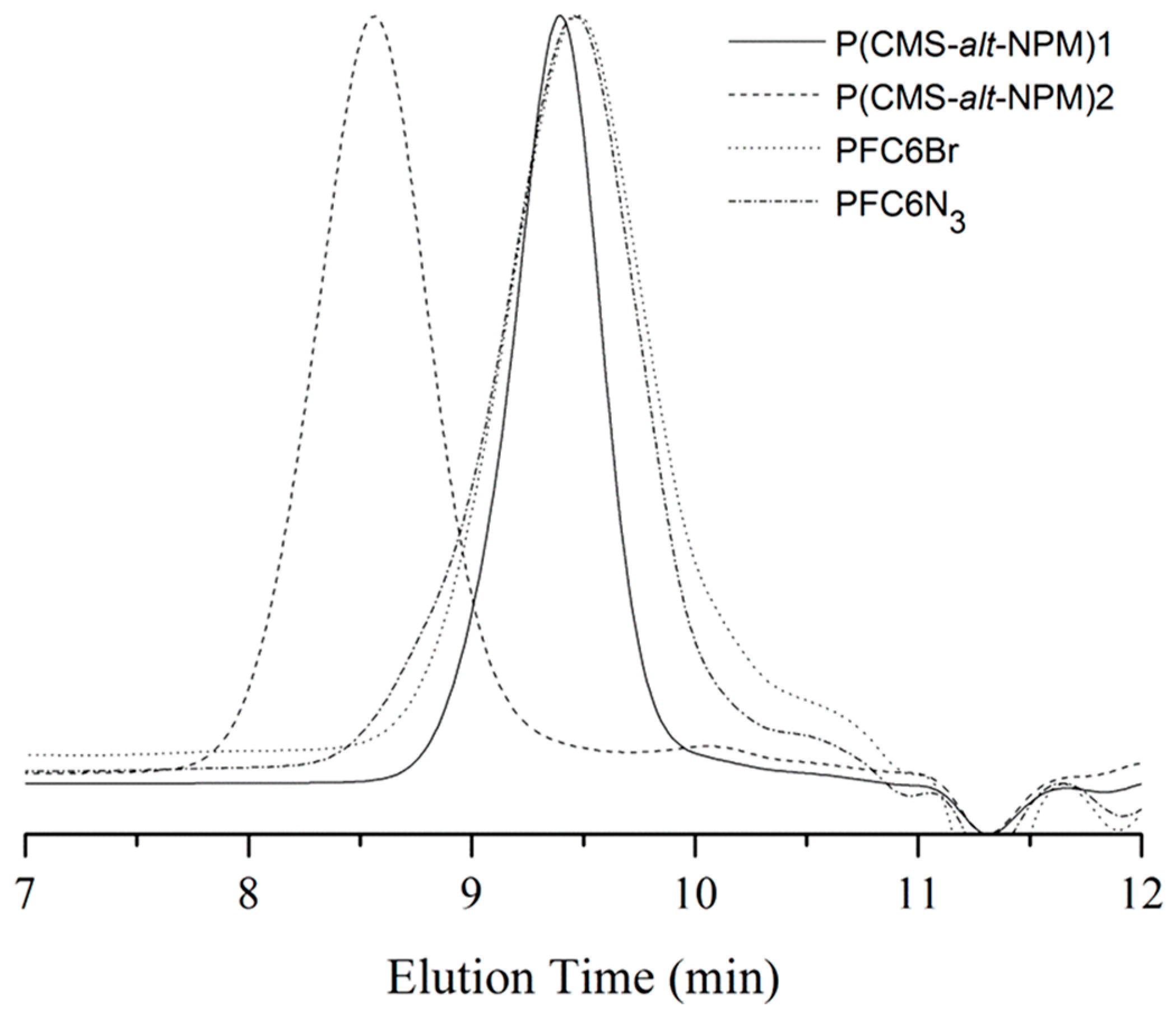
40]. Thus, the RAFT polymerization of CMS and NPM has a strong tendency toward alternative polymerization. The number-average molecular weight (
Mn) of P(CMS-
alt-NPM)1 is about 2600 g/mol, with a polydispersity index (PDI) of about 1.23 from gel permeation chromatography (GPC) measurement (
Figure 1). With the increase in polymerization time from 2 to 4 h, the
Mn and PDI of P(CMS-
alt-NPM)2 (
Figure 1) increase to 15,500 g/mol and 1.45, respectively. That may be due to the alkynyl groups in the side chain being able to induce a cross-linking reaction upon heating [
41]. Thus, the (P(CMS-
alt-NPM)1) with a short polymerization time and low polymerization degree was chosen for the subsequent characterization and functionalization.
Figure 1.
Gel permeation chromatography (GPC) curves of P(CMS-alt-NPM)1, P(CMS-alt-NPM)2, poly[(9,9-dihexylfluorene)-alt-(9,9-bis(6-bromohexyl)fluorene)] (PFC6Br) and PFC6N3.
Figure 1.
Gel permeation chromatography (GPC) curves of P(CMS-alt-NPM)1, P(CMS-alt-NPM)2, poly[(9,9-dihexylfluorene)-alt-(9,9-bis(6-bromohexyl)fluorene)] (PFC6Br) and PFC6N3.
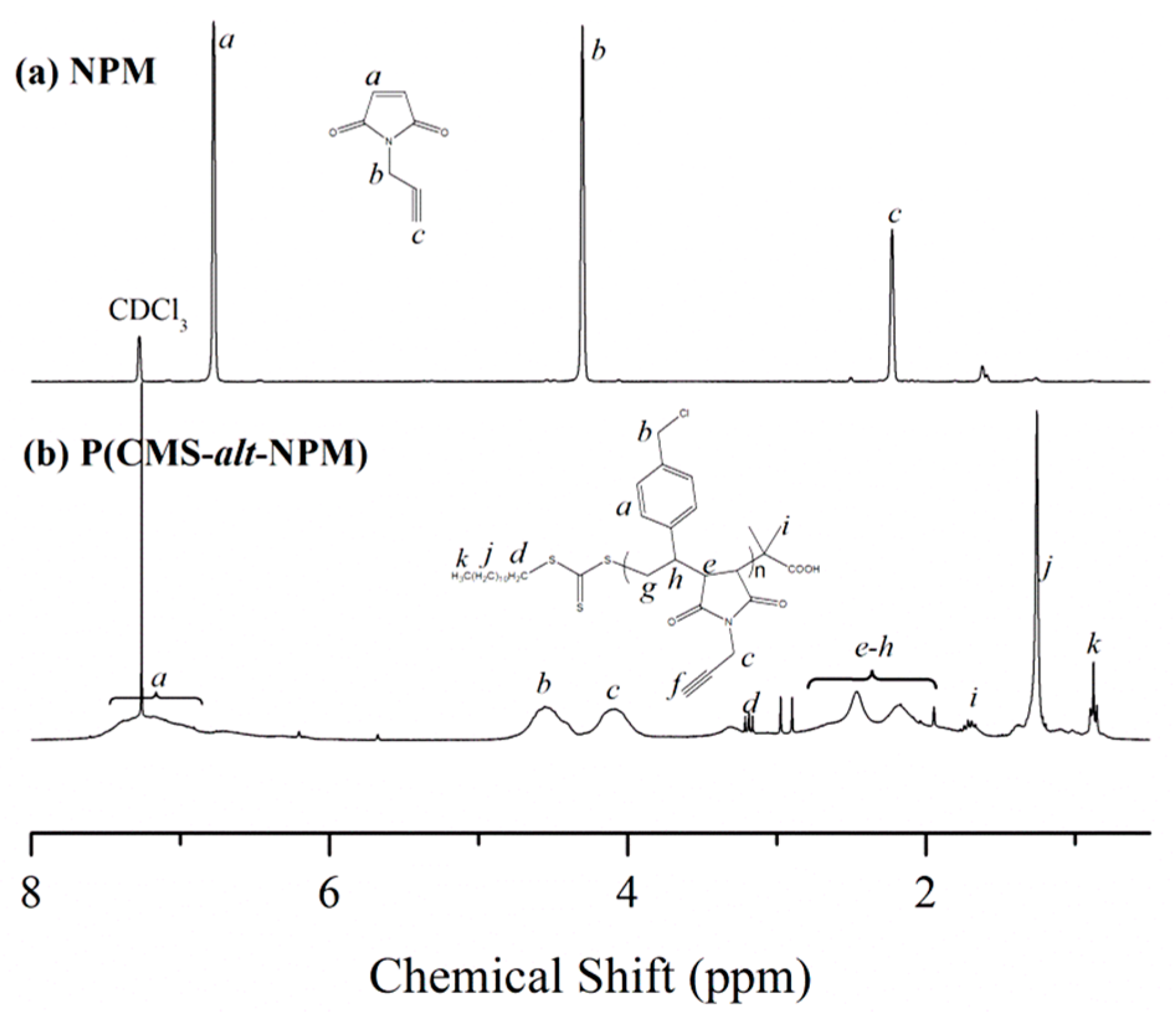
Figure 2b shows the
1H NMR spectrum of P(CMS-
alt-NPM)1 copolymer. The peaks at 4.56 and 4.09 ppm represent the methylene protons of the CMS and NPM unit adjacent to the chlorine atom and alkynyl group, respectively. The peaks in the range of 1.87 and 2.80 ppm correspond to the signals of methine and methylene protons of the main chain and alkyne signals, while the peaks from 6.01 to 7.71 ppm correspond to the aromatic protons of CMS unit. The assignment of the resonance peaks in the
1H NMR spectrum leads to the accurate evaluation of the content of each kind of monomeric unit incorporated into the copolymer chains [
37]. Thus, the copolymer ratio of CMS and NPM was calculated by measuring the integrated peak areas of the respective methylene protons of the CMS (Signal
b) and NPM (Signal
c) unit adjacent to the chlorine atom and alkynyl group, respectively. The copolymer ratio of CMS and NPM in P(CMS-
alt-NPM)1 was determined to be 1:0.95, close to that (1:1) of ideal alternative copolymers.
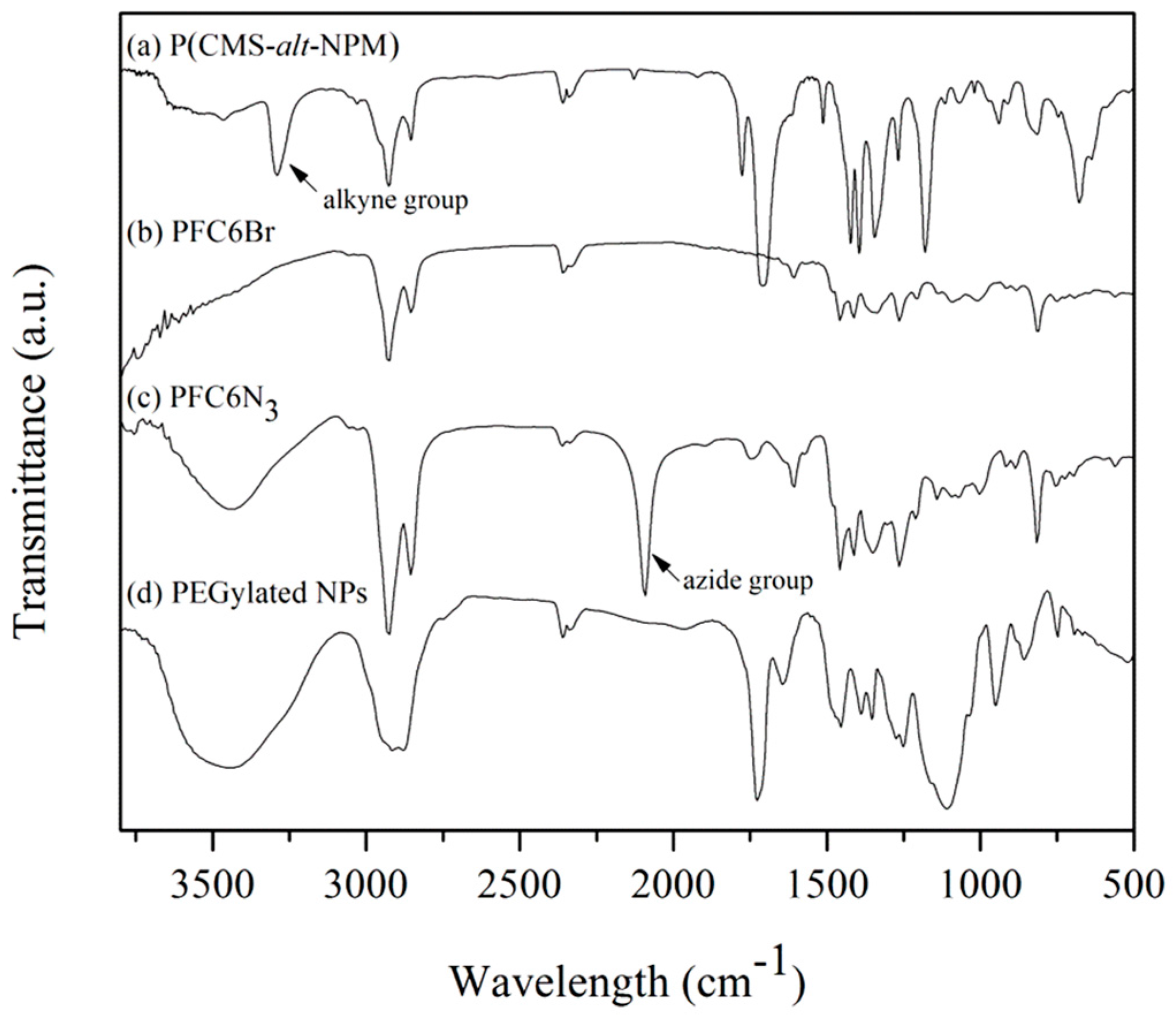
P(CMS-
alt-NPM)1 was further characterized by FTIR spectroscopy (
Figure 3a). A strong peak at about 3300 cm
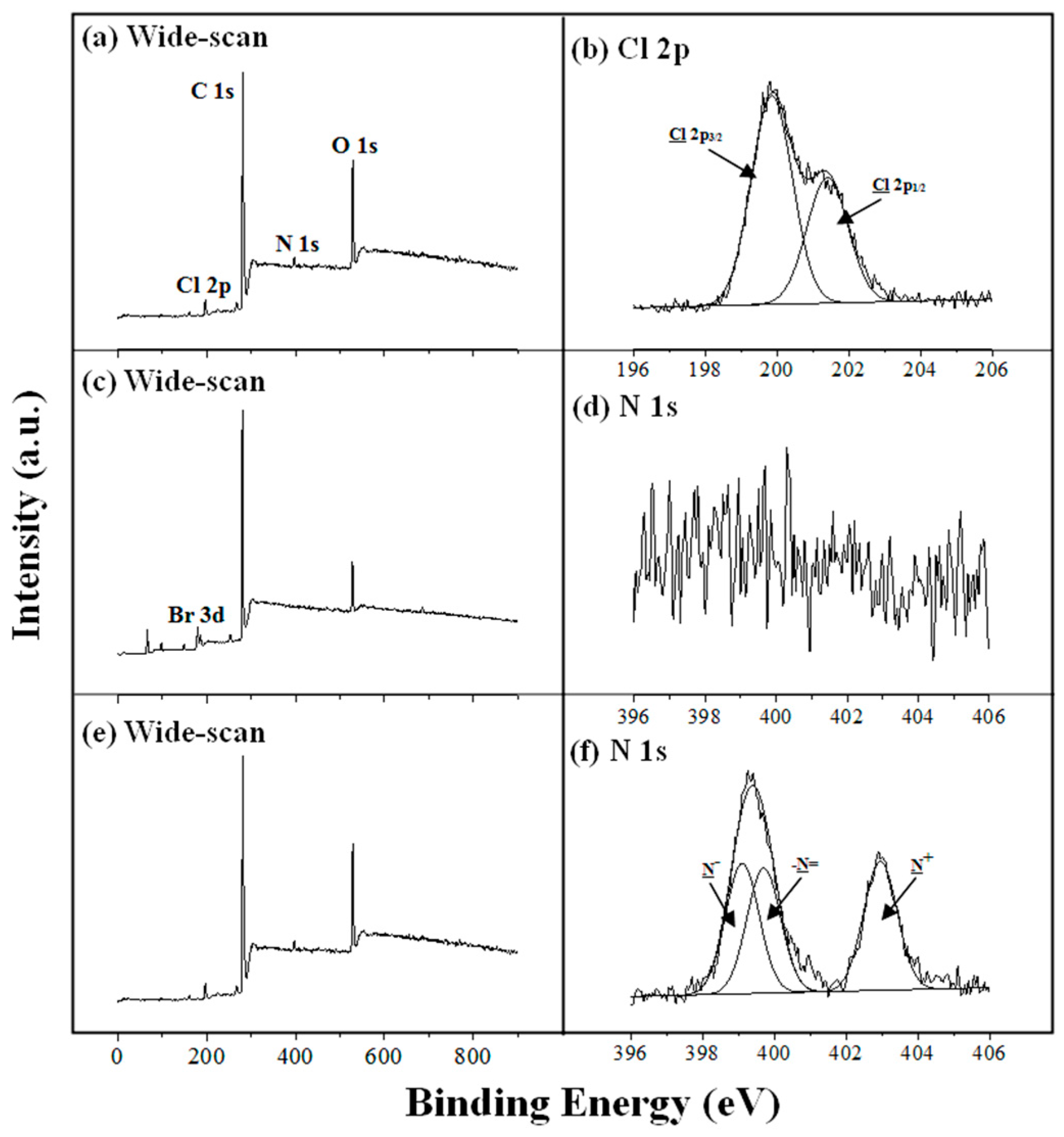
-1 attributed to alkynyl groups is discernible. The X-ray photoelectron spectroscopy (XPS) wide-scan and Cl 2p core-level spectra of P(CMS-
alt-NPM)1 are shown in
Figure 4a,b, respectively. The Cl 2p core-level spectrum of P(CMS-
alt-NPM)1 consists of the Cl 2p
3/2 and Cl 2p
1/2 peak components at the binding energies (BEs) of about 200.0 and 201.6 eV, respectively, attributable to the covalently-bonded chloride species, which can be utilized for atom transfer radical polymerization (ATRP) [
42].
Figure 2.
1H NMR spectra of (a) NPM and (b) P(CMS-alt-NPM)1 in CDCl3.
Figure 2.
1H NMR spectra of (a) NPM and (b) P(CMS-alt-NPM)1 in CDCl3.
Figure 3.
FTIR spectra of (a) P(CMS-alt-NPM)1, (b) PFC6Br, (c) PFC6N3 and (d) the PEGylated fluorescent NPs.
Figure 3.
FTIR spectra of (a) P(CMS-alt-NPM)1, (b) PFC6Br, (c) PFC6N3 and (d) the PEGylated fluorescent NPs.
The bromo-bearing conjugated fluorene copolymer, PFC6Br, was synthesized via Suzuki coupling polymerization of monomers, 2,7-dibromo-9,9-bis(60-bromohexyl) fluorene and 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dihexylfluorene, with Pd(PPh
3)
4 as the catalyst. Replacement of the bromo groups in PFC6Br by treatment with sodium azide in
N,
N-dimethylformamide (DMF) led to PFC6N
3. The
Mn and PDI of PFC6Br were determined to be 2500 and 1.43, respectively, by GPC using tetrahydrofuran (THF) as the eluent (
Figure 1). After substitution of the bromo into azido groups, the
Mn and PDI of the resulting PFC6N
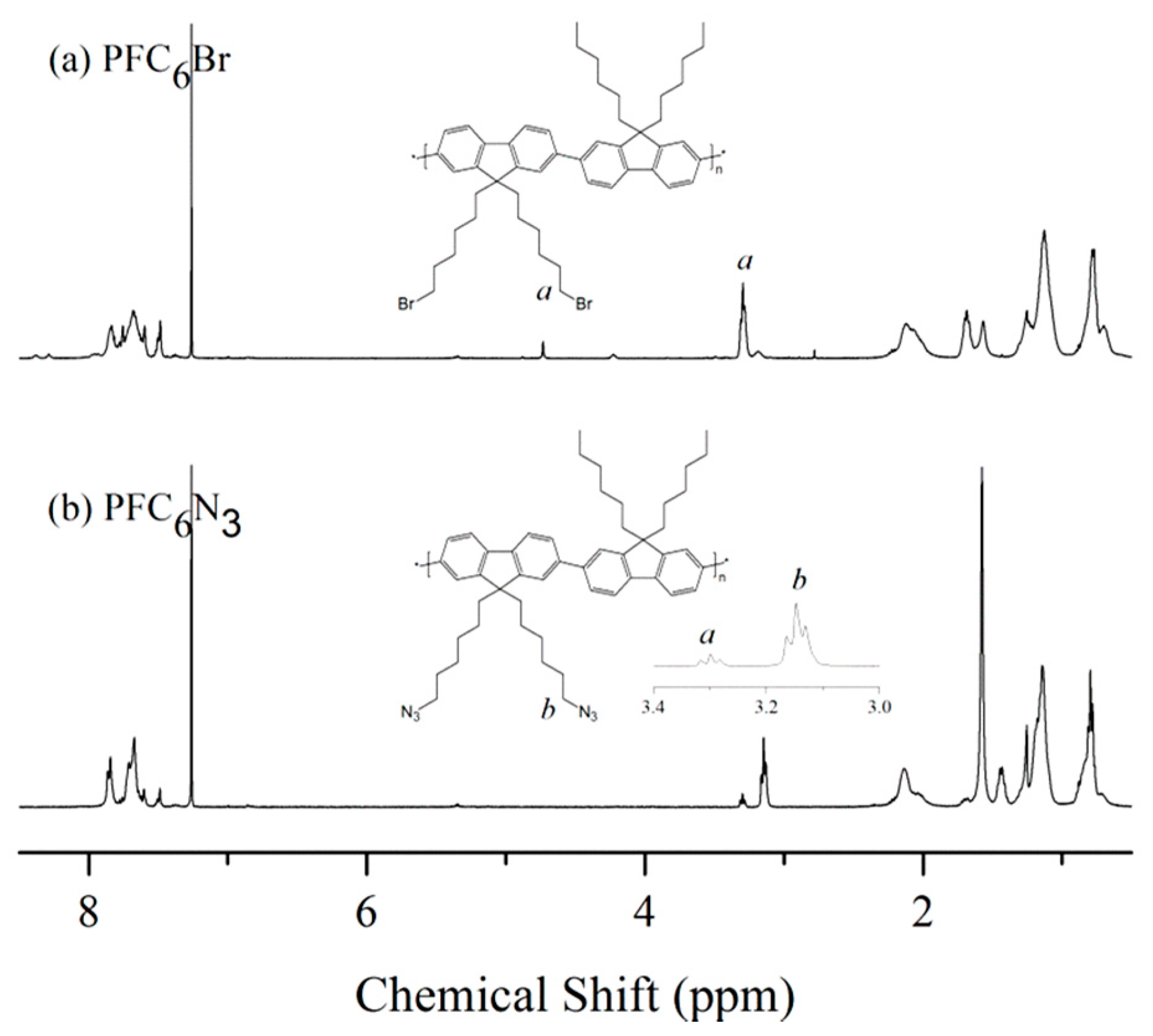
3 remain unchanged, suggesting no cross-linking or branching occurs during the azidation process.
Figure 5a,b shows the
1H NMR spectra of PFC6Br and PFC6N
3, respectively. The peak of methylene protons at the end of side chains upfield-shifts from 3.30 to 3.15 ppm upon the conversion of bromo to azido groups. In comparison to the integrated areas of the methylene proton adjacent to the bromo and azido groups, the conversion of azidation is about 87.5%. The successful introduction of azido groups was also confirmed by the appearance of a characteristic absorbance at about 2100 cm
−1 in the FTIR spectrum of PFC6N
3 (
Figure 3c).
The XPS wide-scan and N 1s core-level spectra of PFC6N
3 are shown in
Figure 4e,f, respectively. In comparison to the XPS wide-scan spectrum of PFC6Br (
Figure 4c), the presence of the N 1s signal at about 400 eV is consistent with the replacement of the bromo moieties by azido moieties. In addition, the N 1s core-level spectra of PFC6N
3 (
Figure 4f) can be curve-fitted with three peak components with BEs at about 399.1, 400.3 and 403.8 eV, attributable to the negatively-charged nitrogen (N
−), the imine nitrogen (–N=) and the positively-charged nitrogen (N
+), respectively. In general, the azido group contains those three types of chemical environments (–N=N
+=N
−) [
43]. Thus, the N 1s core-level spectrum of PFC6N
3 indicates that azido moieties have been successfully introduced.
Figure 4.
(a) XPS wide-scan spectrum of P(CMS-alt-NPM)1, (b) Cl 2p core-level spectrum of P(CMS-alt-NPM)1, (c) XPS wide-scan spectrum of PFC6Br, (d) N 1s core-level spectrum of PFC6Br, (e) XPS wide-scan spectrum of PFC6N3, and (f) N 1s core-level spectrum of PFC6N3.
Figure 4.
(a) XPS wide-scan spectrum of P(CMS-alt-NPM)1, (b) Cl 2p core-level spectrum of P(CMS-alt-NPM)1, (c) XPS wide-scan spectrum of PFC6Br, (d) N 1s core-level spectrum of PFC6Br, (e) XPS wide-scan spectrum of PFC6N3, and (f) N 1s core-level spectrum of PFC6N3.
Figure 5.
1H NMR spectra of (a) PFC6Br and (b) PFC6N3 in CDCl3.
Figure 5.
1H NMR spectra of (a) PFC6Br and (b) PFC6N3 in CDCl3.
Intermolecular “click” cross-linking between P(CMS-
alt-NPM)1 and PFC6N
3 and the ATRP of poly(ethylene glycol) methyl ether methacrylate (PEGMMA) using P(CMS-
alt-NPM)1 as the macroinitiator, proceeded simultaneously, leading to the formation of PEGylated fluorescent NPs (
Scheme 1). After completion of the reaction and subsequent dialysis to remove unreacted starting monomers and the copper catalyst system, the PEGylated fluorescent NPs were obtained. The PEGylated fluorescent NPs were characterized by FTIR spectroscopy. The disappearance of peaks at about 3300 cm
−1 attributable to alkynyl groups and at 2100 cm
−1 attributable to azido groups and the presence of a peak at 1100 cm
−1 corresponding to –C–O–C– stretching (
Figure 3d) indicate that “click” cross-linking and ATRP of PEGMMA had been successfully carried out. The average degree of polymerization per the ATRP initiator can also be estimated by assuming that the ester absorption peak at 1724 cm
−1 and amide absorption peak at 1642 cm
−1 are originated from PEGMMA and P(CMS-
alt-NPM)1, respectively. The average number of PEGMMA repeating units per CMS is determined to be 3.7.
Figure 6a shows the transmission electron microscopy (TEM) image of PEGylated fluorescent NPs. It can be clearly seen that the PEGylated fluorescent NPs disperse well in water with no significant aggregation. A control experiment was carried out to evaluate the influence of ATRP on the formation and aqueous stability of NPs. The experiment was carried out under the same reaction condition as that used for the preparation of PEGylated fluorescent NPs, without the addition of PEGMMA. The NPs can still be formed without the participation of ATRP. However, as shown in the TEM image of the resulting NPs (
Figure 6b), they exhibit poor dispersity in solvent and have a tendency to agglomerate together. This phenomenon is probably due to the ‘click’ cross-linking between P(CMS-
alt-NPM)1 and PFC6N
3 not being able to take place only within intermolecular chains, leading to the formation of polymeric NPs, but also happening between the as-formed polymeric NPs. The presence of the ATRP of PEGMMA during the “click” cross-linking can form corona layers around the as-formed NPs, reducing the cross-linking between polymeric NPs. Thus, it is critically important to introduce surface decoration on NPs using ATRP to enhance its dispersity for further biotechnological applications.
Figure 6.
TEM images of (a) the PEGylated fluorescent NPs and (b) the resulting NPs without the conducting of ATRP.
Figure 6.
TEM images of (a) the PEGylated fluorescent NPs and (b) the resulting NPs without the conducting of ATRP.
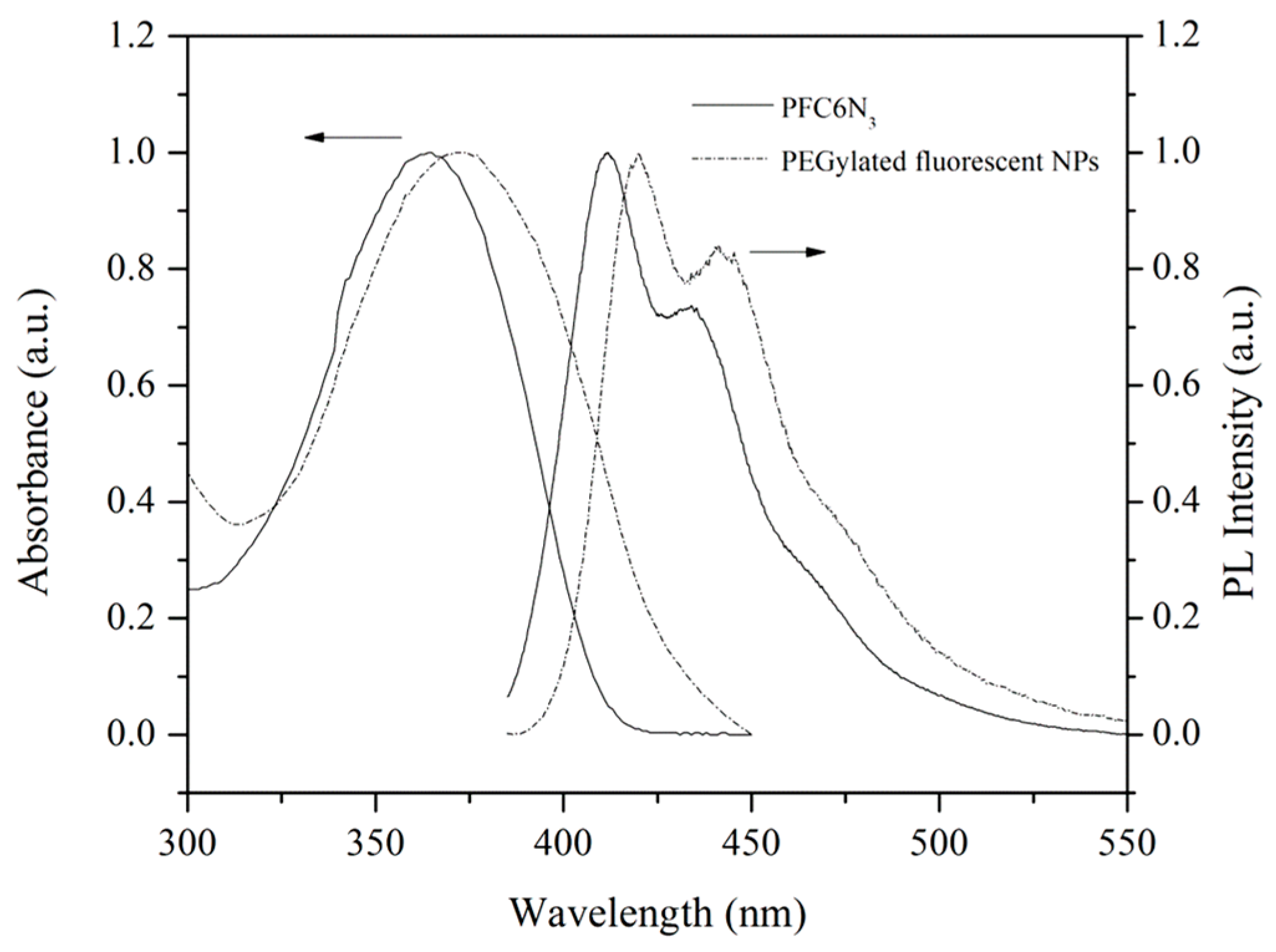
The optical properties of PEGylated fluorescent NPs, as well as their precursor, PFC6N
3, were investigated by UV-visible absorption and fluorescence spectroscopy (
Figure 7). The UV-visible absorption spectrum of PEGylated fluorescent NPs in aqueous solution shows an absorption maximum at 372 nm. The photoluminescence (PL) spectrum of PEGylated fluorescent NPs in aqueous solution exhibits an emission peak at 420 nm with a vibronic shoulder at 441 nm, when excited at a wavelength of λ
ex = 370 nm. In comparison with PFC6N
3 in THF, the peaks are red-shifted by ~ 8 and 9 nm for the respective UV–visible absorption and fluorescence spectrum. The fluorescence quantum yield (Φ
f) of PFC6N
3 in THF is about 72%, as measured in 0.10 mol·L
−1 H
2SO
4 (Φ
f = 0.546) using quinine sulfate as the standard. Unlike those of PFC6N
3 in THF, the Φ
f of PEGylated fluorescent NPs decrease to about 17%, when measured under the same conditions. These phenomena may be due to the aggregation of fluorene chromophores, and the collapse of polymer chains into nanoscale spherical particles reduces the effective conjugation length of the polymer chains [
36]. The UV–visible absorption and fluorescence spectroscopic results are thus consistent with the successful “click” cross-linking of PFC6N
3 into the NPs. The incorporated PFC6N
3 not only highlights the possibility of modifying NPs with various azido-containing functional molecules or polymers, but also allows an easy localization of PEGylated fluorescent NPs within the cells using the fluorescence techniques.
Figure 7.
UV–visible absorption and photoluminescence (PL) spectra of PFC6N3 in THF and the PEGylated fluorescent NPs in water. The excitation wavelength is 370 nm.
Figure 7.
UV–visible absorption and photoluminescence (PL) spectra of PFC6N3 in THF and the PEGylated fluorescent NPs in water. The excitation wavelength is 370 nm.
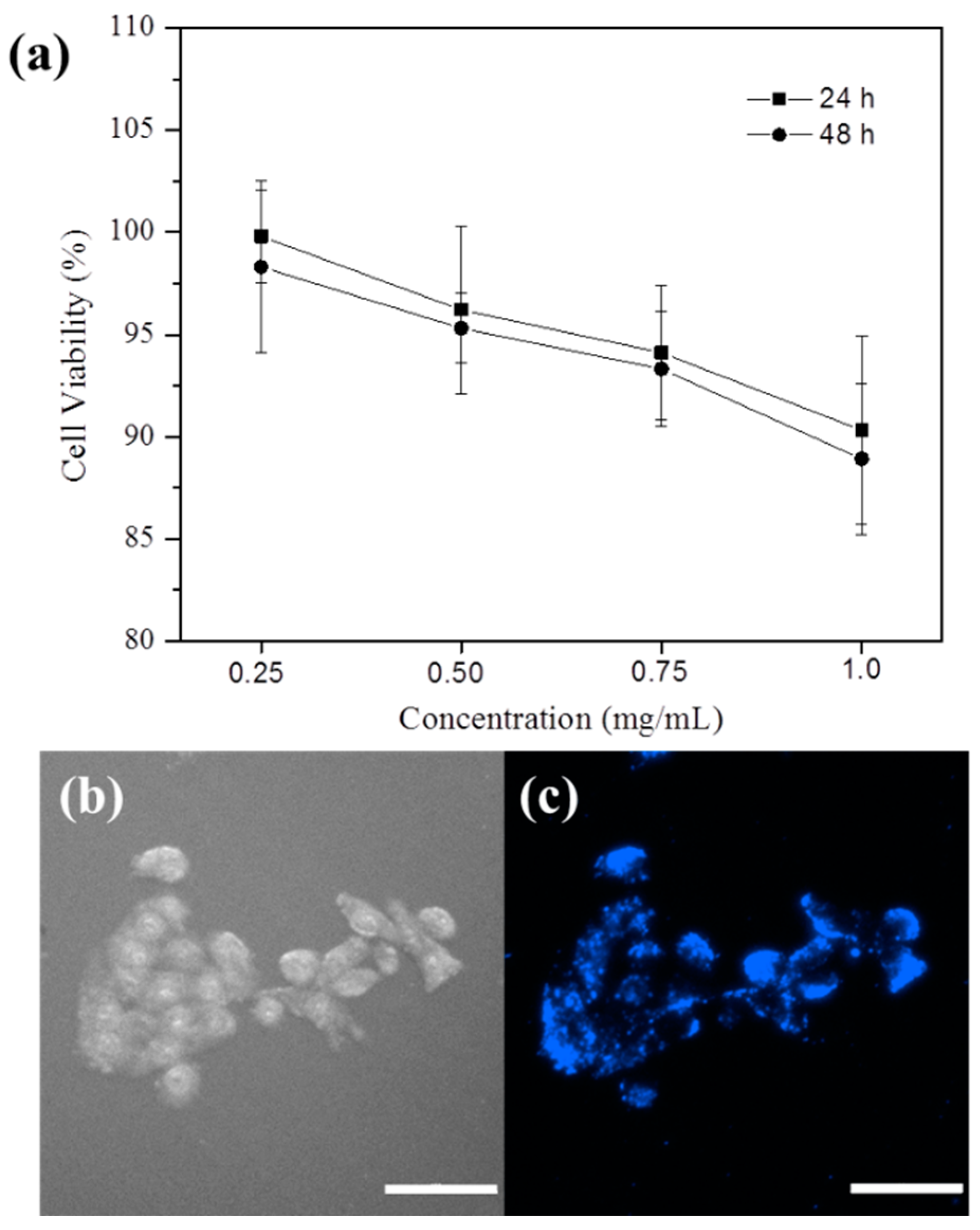
The KB nasopharynx cancer cell line was chosen for the investigation of the cytotoxicity and cellular uptake of the PEGylated fluorescent NPs. The cytotoxicity of PEGylated fluorescent NPs was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability assay.
Figure 8a summarizes the
in vitro KB cell viability after being cultured for 24 or 48 h. The cell viabilities are close to 100% at 0.25 mg/mL and above 85% at 1.0 mg/mL within the tested period, suggesting the low cytotoxicity level of the PEGylated fluorescent NPs. The cellular uptake of the PEGylated fluorescent NPs after 4 h of co-culture was imaged with fluorescence microscopy. Compared to the bright-field image (
Figure 8b), significant blue fluorescence can be observed from the image of the labelled KB cells (
Figure 8c). The weakly-fluorescent nuclei of the KB cells confirm that the PEGylated fluorescent NPs were mainly internalized and accumulated within the cytoplasm.
Figure 8.
(a) Viability of KB cells after incubation with the PEGylated fluorescent NPs of different concentrations for 24 and 48 h; (b) bright-field and (c) fluorescent images of KB cells after incubation with 0.2 mg/mL of the PEGylated fluorescent NPs for 4 h. The scale bar is 100 μm.
Figure 8.
(a) Viability of KB cells after incubation with the PEGylated fluorescent NPs of different concentrations for 24 and 48 h; (b) bright-field and (c) fluorescent images of KB cells after incubation with 0.2 mg/mL of the PEGylated fluorescent NPs for 4 h. The scale bar is 100 μm.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








