1. Introduction
Viscoelasticity is present in numerous materials, appearing within the majority, if not all, biological tissues [
1,
2,
3]. It describes the simultaneous viscous and elastic response of a material, and is typically observed not only due to the properties of the structural components of a material (such as fibers or filaments), but also the fluid flow between these components, and the chemical (e.g., ionic) interactions of any fluid and the structural components [
3,
4,
5]. Hydrogels are viscoelastic materials that are commonly used in tissue engineering (TE), however in compression it is only their elastic properties that are typically characterized, and viscoelasticity in compression is rarely used as a deterministic criterion for scaffold design.
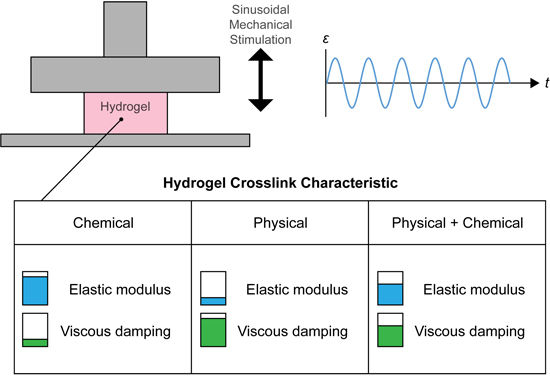
Viscoelastic materials have a more complex behavior than elastic materials, meaning that the standard compressive elastic (Young’s) modulus does not capture the nature of the material being tested—it only provides a general overview of stiffness. Viscoelastic characteristics including elastic modulus (), equilibrium modulus (), elastic storage and loss moduli ( and respectively) and loss factor/tangent () may provide a more accurate quantification of material response to loading. and describe the raw stiffness of the material and of the solid material components, respectively. and are indicative of the character of the elastic and viscous components of the tested materials under variable strain rates, whilst indicates the ratio of to (the tangent of the angle between the and vectors) or the proportion of viscous response to elastic response.
The tailoring of material viscoelasticity may be beneficial in the field of TE, where the emulation of the mechanical properties of various biological tissues may result in greater outcomes for tissue formation. Of the tissues typically targeted in TE, articular cartilage is a significantly viscoelastic material, exhibiting such behaviors in tension [
4,
6], compression [
1,
5,
7] and shear [
5,
8,
9], and was hence chosen for a preliminary analysis within this work.
Within cartilage TE, hydrogels are frequently used as scaffolds, since they provide a highly hydrated viscoelastic matrix and a desirable hypoxic environment, similar to the native tissue [
10]. However in all applications, hydrogel systems do exhibit several limitations, primarily encountered in the trade-off between mechanical properties and cytocompatibility [
11]. Whilst it is currently difficult to achieve cytocompatible hydrogels with mechanical properties of native tissues, more complex viscoelastic properties such as loss tangent may be a tailorable property.
Use of hydrogels in TE revolves primarily around cellular encapsulation, where a rapid liquid-to-solid phase transition is desirable. Light-activated free-radical polymerization (photopolymerization or photocrosslinking) provides rapid reaction rates, acceptable levels of cytotoxicity and results in uniform material properties [
11]. In this regard, ultraviolet (UV) light-activated photoinitiators (PIs) such as 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959; IC), have been frequently used with success.
The hydrogel systems used herein consist of biocompatible gelatin, gellan gum (GG) and their methacrylated derivatives. Gelatin is a solid polymer obtained by partial hydrolysis of connective tissues [
12]. It is an attractive material due to the retention of natural cell binding motifs such as Arg-Gly-Asp (RGD), and matrix metalloproteinase (MMP)-sensitive degradation sites [
13,
14]. Modification of gelatin via reaction with methacrylic anhydride yields gelatin methacrylamide (GelMA), a photocrosslinkable hydrogel which is associated with long-term cell viability and response [
13,
15,
16,
17].
Gellan gum is an FDA-approved food additive and polysaccharide polymer excreted by the bacterium
Pseudomonas elodea [
18,
19,
20]. GG forms aqueous solutions of low viscosities at high temperatures (>70 °C is required to fully hydrate the structure [
19]), and forms strong, hard and brittle gels upon cooling through a coil-helix transition, with helical aggregation and hydrogel stability dependent on the presence, concentration and valence of cations in the solution [
20,
21,
22]. GG is also a highly suitable material for three dimensional (3D) printing due to its relatively prominent pseudoplasticity and ionic tailorability [
23]. Modification of GG with methacrylic anhydride yields the photocrosslinkable gellan gum methacrylate (GGMA). In this study we present hydrogel blends consisting of the photocrosslinkable GelMA and GGMA, in various combinations with gellan gum (GG). We proposed that blending of polymers into multi-network gels would promote increased moduli and the combination of physical and chemical crosslinks would allow the tailoring of viscoelasticity. We assessed the effects of these manipulations on the above parameters to determine tailorability and compared the constructs with explanted cartilage tissue for biological relevance.
2. Materials and Methods
Gelatin (G2500), gellan gum (Gelzan™ CM G1910), methyl cellulose (M0512) and methacrylic anhydride (MAAh, 276685) were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA). 2-hydroxy-1-[4-(2-hydroxyethoxy)phenyl]-2-methyl-1-propanone (Irgacure 2959; IC) was purchased from BASF (BASF, Ludwigshafen, Germany). All quoted concentrations in percent are weight per volume (% w/v) unless otherwise stated.
2.1. Polymer Functionalization
Hydrogel groups comprised of methacrylic anhydride-functionalized gelatin (gelatin methacrylamide; GelMA) and gellan gum (gellan gum methacrylate; GGMA), plain gellan gum (GG) and methyl cellulose (MC) in various blends. Methyl cellulose was included during initial stages to aid hydrogel miscibility at 37 °C.
Functionalization of gelatin and GG was performed using previously published methods [
16]. In brief, gelatin and GG were dissolved in Milli-Q water (Merck Millipore, Billerica, MA, USA) at 10% and 1%, respectively. Gelatin was reacted with 0.6 g of MAAh per 1 g of gelatin for 1 h at 50 °C [
16]. GG was reacted with 8 g of MAAh per 1 g of GG for 6 hours at 60 °C, with the pH constantly monitored and adjusted to 8.0 with 5
M NaOH [
24]. After the prescribed time, both solutions were centrifuged to remove insoluble MAAh, and then dialyzed (14 kDa cutoff) against Milli-Q water to remove any remaining MAAh and methacrylic acid. The GG solution was dialyzed at 60 °C. After dialysis, GelMA was filtered through 0.2 µm filters (Merck Millipore) in aseptic conditions, pH adjusted to 7.4, lyophilized with filters providing the only air exchange then stored at −20 °C in sealed, sterile tubes. The GGMA was processed in the same way apart from sterile filtration, which was not undertaken to avoid macromer loss due to its variable and high (~200–500 kDa) molecular weight.
2.2. Degree of Functionalization
The degree of functionalization (DOF) of GelMA and GGMA was assessed using proton nuclear magnetic resonance (
1H NMR) spectroscopy. A Varian Direct Drive NMR spectrometer (Agilent Technologies, Palo Alt, CA, USA), operating at 400 MHz for hydrogen, was used to record
1H NMR spectra. Samples were dissolved at 1.0% (10 mg·mL
−1) in D
2O at a temperature of 50 °C. Spectra for quantitation were recorded at 50 °C, with 32 scans and a recycle delay of 30 s. For GelMA, methacrylamide shifts were normalized against the aromatic signal of phenylalanine, which occurred at a chemical shift of
7.4 ppm [
25]. DOF was defined as the proportion of modified lysine groups of gelatin, as previously described for collagen I methacrylamide [
25]. The area obtained from the integral of intensities of the protons present at the methacrylamide carbon-carbon double bond (
) occurring at δ 5.6 and 5.8 ppm was normalized to proton number (
), and the area found after integrating the peak of the aromatic groups (
) was normalized to the protons interacting with the aromatic ring (
). The prevalence of modified groups and aromatic residues were then normalized to their prevalence by number in porcine gelatin in order to allow for the quantitation of total methacrylamide to total possible lysine groups (Equation (1)) [
26]:
For GGMA, chemical shifts were normalized against the signal from the methyl group of rhamnose at
1.45 ppm [
18]. The DOF, which in this case was defined as the fraction of modified hydroxyl groups per repeating unit, was determined as previously described [
18,
24]. In brief, the average area found from integrating the proton peak at the double bond (
5.8 and 6.3 ppm;
) was normalized to the number of protons at the double bond (
) and the area found from integrating the proton peak of the methyl group of rhamnose (
) was normalized to the number of protons in the methyl group of rhamnose (
); the normalized double bond value was then divided by the normalized rhamnose value to give the number of functional groups, which was assessed as a fraction of
, the total number of reactive hydroxyl groups (10) present in a repeating monomer unit of GG, finally resulting in the percentage of DOF, as illustrated in Equation (2):
The calculation was then repeated using the signal from the methyl group on the methacrylate functional unit (occurring at ~ 2.09 ppm), with the mean of these taken for the overall DOF value for GGMA.
2.3. Rheological Characterization
Hydrogel groups (detailed in
Table 1; mixed in phosphate buffered saline (PBS; Invitrogen, Carlsbad, CA, USA) or an isotonic blend of NaCl and
d-mannose (24 and 252 mM, respectively [
23]; henceforth N+M) were analyzed using a Rotational Rheometer RHEOTEST
® RN 4.1 (RHEOTEST Medingen GmbH, Ottendorf-Okrilla, Germany) with a precise thermal control unit. Testing consisted of multiple measures on each sample for analysis of gelation temperature (
) and viscosity (
across the temperature sweep. Firstly, the rheometer was set up with the 1° cone in cone and plate configuration, and the idle temperature was set to 60 °C to avoid gelation prior to testing. Hydrogel blends were made, and 500 µL of premixed, preheated hydrogel solution was pipetted onto the rheometer platform. The chamber was closed and a constant shear rate (
) of 50 S
−1 was applied whilst temperature was reduced from 60 to 20 °C at a rate of 1 °C·20 S
−1, with time (
), temperature (
) and
monitored constantly.
Table 1.
Summary of hydrogel groups used for rheological characterization. GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate; MC = methyl cellulose.
Table 1.
Summary of hydrogel groups used for rheological characterization. GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate; MC = methyl cellulose.
| Hydrogel groups assessed rheologically |
|---|
| Abbreviation | Constituents (% w/v) |
|---|
| GelMA | GG | GGMA | MC |
|---|
| GelMA | 10 | 0 | 0 | 0 |
| GelMA 0.25% GG | 10 | 0.25 | 0 | 0 |
| GelMA 0.25% GG 0.25% MC | 10 | 0.25 | 0 | 0.25 |
| GelMA 0.25% GGMA | 10 | 0 | 0.25 | 0 |
| GelMA 0.5% GGMA | 10 | 0 | 0.5 | 0 |
| GelMA 1.0% GGMA | 10 | 0 | 1 | 0 |
| GelMA 1.0% GGMA 0.25% MC | 10 | 0 | 1 | 0.25 |
| 0.5% GGMA | 0 | 0 | 0.5 | 0 |
| 1.0% GGMA | 0 | 0 | 1 | 0 |
| 2.0% GGMA | 0 | 0 | 2 | 0 |
| 0.5% GG | 0 | 0.5 | 0 | 0 |
| 1.0% GG | 0 | 1 | 0 | 0 |
2.4. Hydrogel and Explant Preparation
2.4.1. Physical Crosslinking Prior to UV
Stock solutions of GelMA, GG, GGMA and IC were made with N+M on the day of gel formation. 10% GelMA, 1.0% and 2.0% GG and GGMA gels were made at an IC concentration of 0.05% using just the individual components at the listed concentrations; all groups are included in
Table 2. Gels were pipetted into custom-made Teflon molds (50 mm × 4 mm × 2 mm strips) and covered with glass slides. Physical crosslinking consisted of UV (immediate UV), RT + UV (room temperature (RT) for 20 min prior to UV) or 4 + UV (4 °C for 10 min prior to UV), with UV crosslinking initiated by irradiation at 365 nm for a total energy of 1500 mJ·cm
−2, in a CL-1000L crosslinker (UVP, Upland, CA, USA). GG-only gels were not mixed with IC but only subjected to the temperature conditions described above. Further, GG and GGMA were not assessed with immediate UV as GG cannot crosslink through this method and thus would offer no means of comparison with the GGMA. After crosslinking, gel strips were cut to 4 mm × 4 mm × 2 mm and stored in Dulbecco’s Modified Eagle Medium (DMEM) at 37 °C overnight to allow for ionic equilibration prior to mechanical testing. Testing consisted of elastic modulus measurement, as described in the Mechanical Theory and Analyses section below (
Section 2.6.).
Table 2.
Summary of hydrogel groups used to test the effects of physical crosslinking prior to chemical crosslinking through ultraviolet (UV) radical polymerization. All gels were mixed with NaCl + mannose. GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate. UV = immediate UV crosslinking; RT + UV = 20 min physical crosslinking at room temperature (RT) prior to UV crosslinking; 4 + UV = 10 min physical crosslinking at 4 °C prior to UV crosslinking. UV-only group was not included in the GG/GGMA comparisons as GG cannot be UV crosslinked. Crosslink method groups are indicated by either “o” (included) or “×” (excluded).
Table 2.
Summary of hydrogel groups used to test the effects of physical crosslinking prior to chemical crosslinking through ultraviolet (UV) radical polymerization. All gels were mixed with NaCl + mannose. GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate. UV = immediate UV crosslinking; RT + UV = 20 min physical crosslinking at room temperature (RT) prior to UV crosslinking; 4 + UV = 10 min physical crosslinking at 4 °C prior to UV crosslinking. UV-only group was not included in the GG/GGMA comparisons as GG cannot be UV crosslinked. Crosslink method groups are indicated by either “o” (included) or “×” (excluded).
| Hydrogel groups assessed for physical crosslinking |
|---|
| Abbreviation | Constituents (% w/v) | Crosslink method |
|---|
| GelMA | GG | GGMA | UV | RT + UV | 4 + UV |
|---|
| GelMA | 10 | 0 | 0 | × | o | o |
| GelMA | 10 | 0 | 0 | o | × | o |
| GelMA | 10 | 0 | 0 | o | o | × |
| 1.0% GG | 0 | 1 | 0 | o | × | o |
| 1.0% GG | 0 | 1 | 0 | o | o | × |
| 1.0% GGMA | 0 | 0 | 1 | o | × | o |
| 1.0% GGMA | 0 | 0 | 1 | o | o | × |
| 2.0% GGMA | 0 | 0 | 2 | o | × | o |
| 2.0% GGMA | 0 | 0 | 2 | o | o | × |
2.4.2. Formation of Composite Hydrogel Blends
Stock solutions of GelMA, GG, GGMA and IC were made with N+M on the day of gel formation. Specific hydrogel precursor solutions were prepared containing 0.05% IC and formed into gel strips; the gel compositions are detailed in
Table 3. Gels were formed as above, using the RT + UV protocol. Gels were cut and equilibrated as above prior to mechanical testing.
Table 3.
Summary of hydrogel groups used for mechanical characterization. All gels were mixed with NaCl + mannose. HDC = human donor cartilage; GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate.
Table 3.
Summary of hydrogel groups used for mechanical characterization. All gels were mixed with NaCl + mannose. HDC = human donor cartilage; GelMA = gelatin methacrylamide; GG = gellan gum; GGMA = gellan gum methacrylate.
| Hydrogel groups assessed mechanically |
|---|
| Abbreviation | Constituents (% w/v) |
|---|
| GelMA | GG | GGMA |
|---|
| HDC 1 | N/A: Human donor 1 |
| HDC 2 | N/A: Human donor 2 |
| GelMA | 10 | 0 | 0 |
| GelMA 0.25% GG | 10 | 0.25 | 0 |
| GelMA 0.25% GGMA | 10 | 0 | 0.25 |
| GelMA 0.5% GG | 10 | 0.5 | 0 |
| GelMA 0.5% GGMA | 10 | 0 | 0.5 |
| GelMA 1.0% GG | 10 | 1 | 0 |
| GelMA 1.0% GGMA | 10 | 0 | 1 |
| 1.0% GG | 0 | 1 | 0 |
| 1.0% GGMA | 0 | 0 | 1 |
| 2.0% GG | 0 | 2 | 0 |
| 2.0% GGMA | 0 | 0 | 2 |
2.4.3. Explant Preparation
Full-thickness articular cartilage explants were cut from macroscopically normal regions of femoral condyles of two osteoarthritic female donors undergoing total knee replacement with patient consent and ethical approval from The Prince Charles Hospital and Queensland University of Technology and stored in PBS at 4 °C overnight prior to testing. Cross-sectional area dimensions were cut similar to the hydrogel samples (4 mm × 4 mm) whilst height was kept maximized to include the full thickness of the tissue in each case. Mean heights of human donor cartilage (HDC) samples were HDC 1: 2.72 ± 0.19 mm; HDC 2: 3.39 ± 0.37 mm;
n = 5 for both cases. Whilst typical cartilage relaxation time for equilibrium conditions is longer than the 10 min stress-relaxation described below [
1], it was decided that to maintain controlled testing between hydrogels and cartilage explants the cartilage testing would be performed in the same way as the hydrogel testing.
2.5. Mechanical Characterization
Mechanical testing was performed using an Instron MicroTester (Model 5848; Instron, Norwood, MA, USA). Each sample was placed in a 37 °C PBS trough into which an aluminum plunger was submerged for compressive displacement. The testing regimen consisted of a preliminary height measurement (force () = −0.01 N as height criterion), followed by: a 0.5% strain per second (·s−1) ramp to 16 percent strain (% ) for compressive elastic modulus measurement ( over 10%–15%) after which the sample was unloaded to 0%; a 0.5%·s−1 ramp to 10% where a 10 min stress ()–relaxation was held; sinusoidal compression over a range of frequencies (for calculating compressive storage () and loss () moduli), consisting of an exponential decrease from 5.12 to 0.01 , 2% amplitude, midpoint at 10%; and a final 0.5%·s−1 ramp to 15% for a second 10 min stress-relaxation hold ( over 10%–15%). For the characterization of crosslinking reagents, testing only consisted of elastic modulus measurement. It should be noted that sinusoidal testing was performed with decreasing frequency to reduce stress-relaxation that may occur during low-frequency tests. In all cases one sample was used for all measurements, with the entire regimen repeated per sample (n = 5 for all groups).
2.6. Mechanical Theory and Analyses
The compressive elastic moduli (
) of all samples were obtained by calculating the slope of the
function over the 10%
–15%
region. For the equilibrium modulus (
), stress at the end of each relaxation point was calculated (10%ε and 15%
), with the gradient of the linear relationship between the two points deemed to be
Hydrogels and cartilage are viscoelastic and hence have a stress-lag during any dynamic strain.
and
were calculated using viscoelastic theory (Equation (3)) [
27]:
where
is the loss angle (lag or lead) between the stress and the strain, and
and
signify stress and strain sine amplitudes, respectively. In order to obtain the data required for these equations, the raw data were processed into separate frequency cycles and curve-fitted using a MATLAB script (The MathWorks, Inc., Natick, MA, USA), which gave outputs of amplitude, frequency and phase angle for each curve. These values were then adjusted to account for machine inertia and noise, and analyzed, resulting in values for
and
. The value for the loss tangent (
) was obtained by taking the tangent of the loss angle or the ratio of
to
(Equation (4)):
2.7. Cell Isolation and Expansion
The full procedure for isolating chondrocytes is described elsewhere [
28]. In brief, cartilage samples were removed from macroscopically normal regions of the femoral condyle of a patient undergoing full knee replacement surgery, with patient consent and ethical approval from the Prince Charles Hospital and Queensland University of Technology. After isolation, chondrocytes were cultured and expanded in low-glucose DMEM with 10% fetal bovine serum (Lonza, Waverly, Australia), 2 mM glutamax, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 0.1 mM non-essential amino acids (NEAAs), 0.5 µg·mL
−1 amphotericin B (Fungizone
®), 50 U·mL
−1 penicillin G sodium, 50 µg·mL
−1 streptomycin (all Invitrogen, Scoresby, VIC, Australia), 0.4 mM
l-proline and 0.1 mM ascorbic acid (Sigma-Aldrich Pty Ltd, Castle Hill, NSW, Australia).
2.8. Cell Encapsulation and Hydrogel Culture
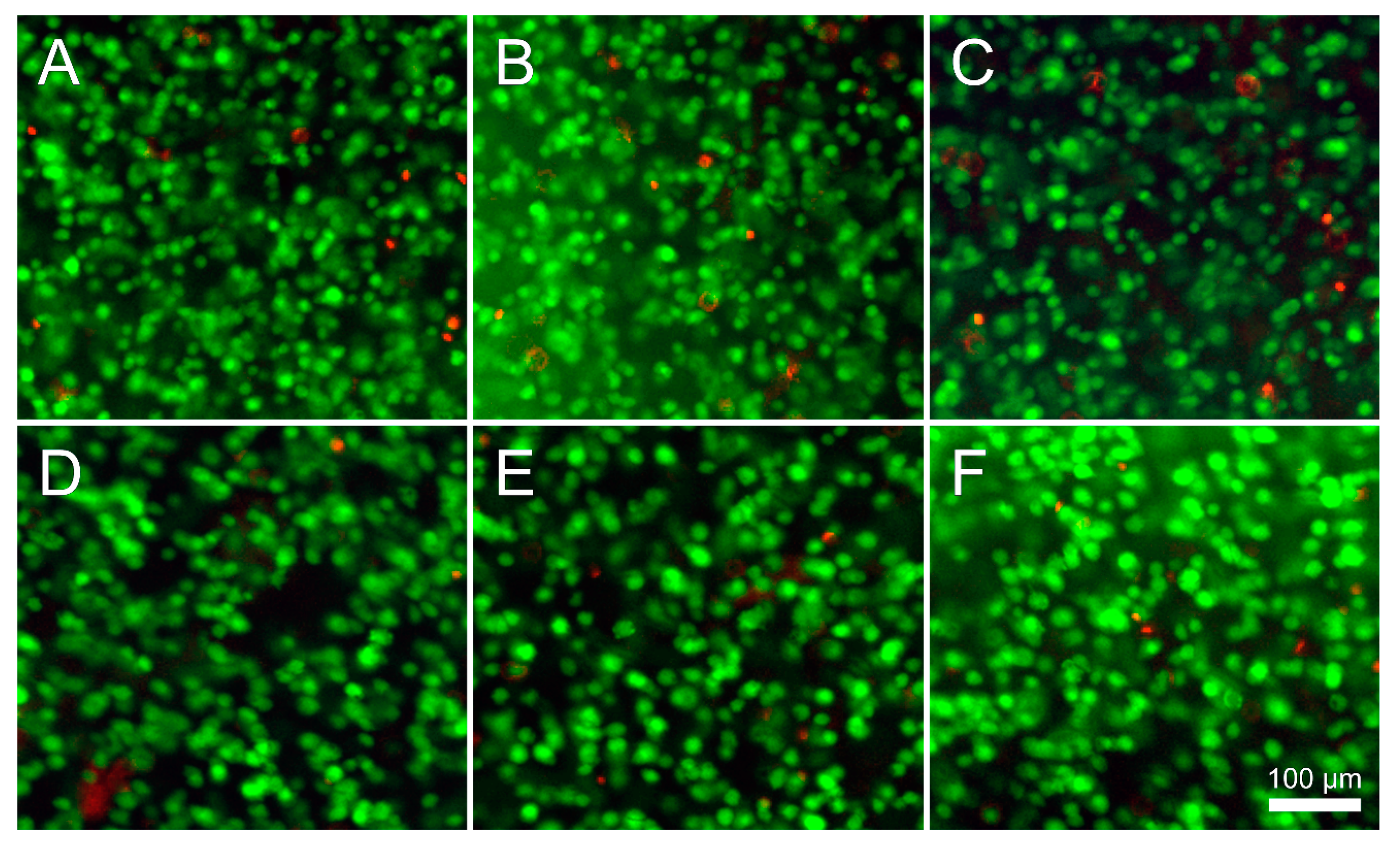
Preliminary cell culture was performed on hydrogel groups (in N+M): GelMA (10), GelMA/GG (10/0.25) and GelMA/GGMA (10/0.25) to assess cell viability in the hydrogel blends. These blends were chosen to assess the cytotoxicity of the individual components with the inclusion of the N+M solution. As previously described, all hydrogel solutions were prepared with 0.05% IC, with 2 × 105 chondrocytes·mL−1 mixed into the precursor solutions. The same crosslinking method was followed as above in the RT + UV protocol. After gels were divided into 48-well plates, they were cultured in a 5% CO2 incubator at 37 °C for 7 days in chondrogenic differentiation media (high-glucose DMEM with 2 mM glutamax, 10 mM HEPES, 0.1 mM NEAAs, 0.5 µg·mL−1 amphotericin B (Fungizone®), 50 U·mL−1 penicillin G sodium, 50 µg·mL−1 streptomycin, ITS-G (100× dilution; all Invitrogen), 1.25 mg·mL−1 bovine serum albumin (BSA), 0.4 mM l-proline, 0.1 mM ascorbic acid, 0.1 µM dexamethasone (all Sigma-Aldrich Pty Ltd, Castle Hill, NSW, Australia) and 10 ng·mL−1 transforming growth factor-β3 (TGF-β3; GroPep, Adelaide, SA, Australia). Medium was changed on day 3 of culture.
2.9. Cell Viability
Viability was assessed on days 1 and 7 using fluorescein diacetate (FDA) and propidium iodide (PrI; both Invitrogen, Scoresby, VIC, Australia) to stain live and dead cells, respectively. Gels were rinsed twice in fresh PBS then incubated for 5 min at 37 °C in a 5 µg·mL−1 FDA, 0.5 µg·mL−1 PrI PBS solution. Samples were imaged using fluorescence microscopy (Zeiss Axio Imager M2, Oberkochen, Germany; excitation: 488 and 568 nm, absorption 520 and 640 nm for FDA and PrI, respectively). Quantification was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.10. Statistical Analyses
Analysis of Variance (ANOVA) tests were used to assess differences, with Tukey’s post-hoc tests used to determine inter-type relationships. Tests were conducted using SPSS 21.0 with p-values <0.05 regarded as significant. Figure bars show mean ± standard deviation unless otherwise indicated. Captions state where independent sample t-tests were used instead of ANOVA.
4. Discussion
Hydrogel blends consisting of GelMA and GG or GGMA were made to establish whether viscoelastic properties were tailorable within these hydrogel systems. Further, these properties were compared to the viscoelasticity of human articular cartilage as a model biological tissue. Gels were characterized using 1H NMR and rheology prior to being subjected to a number of mechanical analyses in order to evaluate their viscoelasticity.
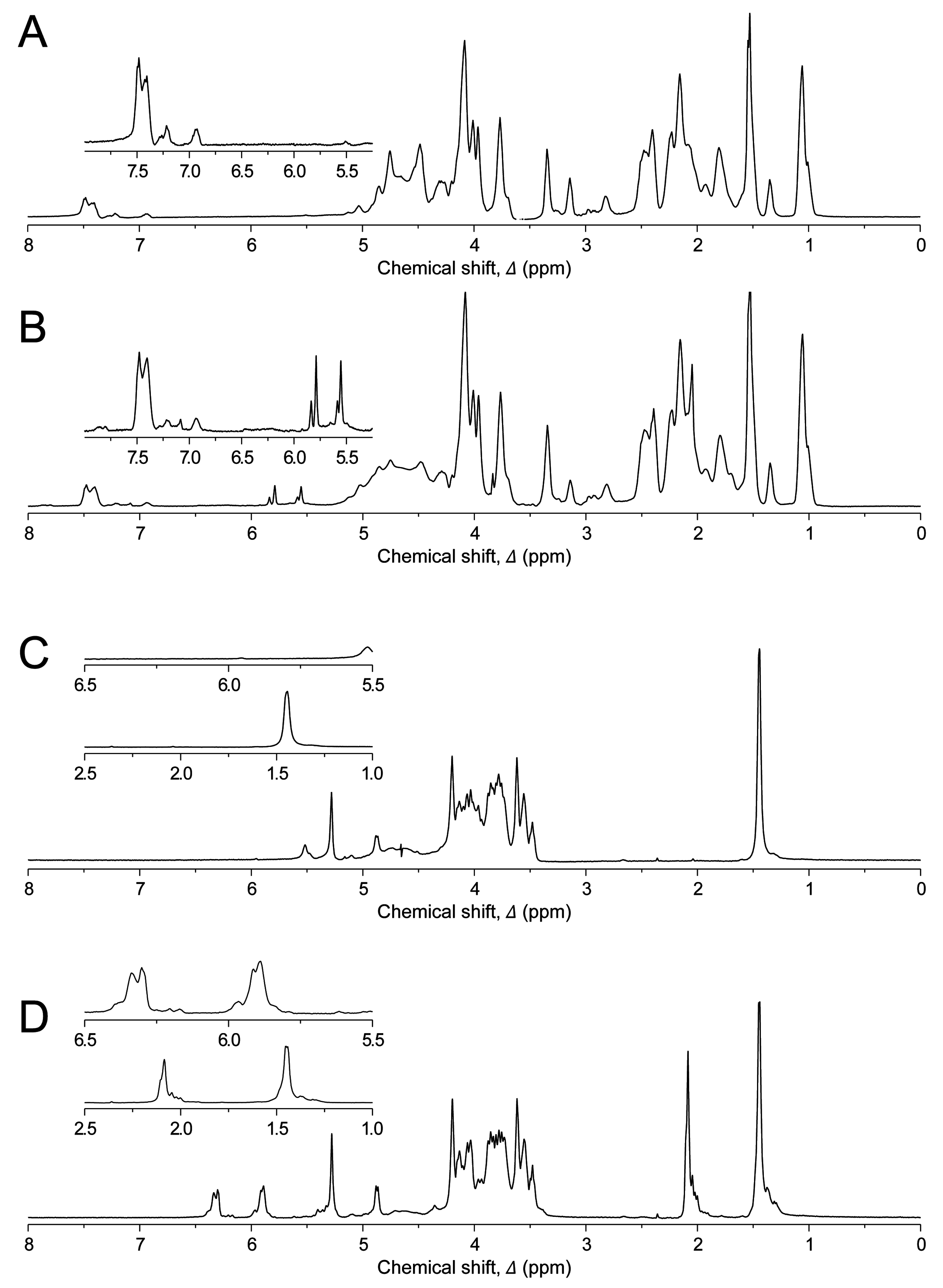
The degree of functionalization (DOF) of hydrogels was measured using
1H NMR spectroscopy (
Figure 1). GelMA was highly functionalized (76.2%) whilst GGMA was functionalized to a lesser extent (3.6%). However, lysine is not a highly prevalent amino acid in gelatin (~1%–4%) whilst there are 10 potential functional hydroxyl groups per repeating unit of GG and hence, with the higher percentage of functionalization and at concentrations used herein (10% for GelMA
vs. 1% for GGMA), the number of functional reactive groups per mL of solution would be comparable between the two gel types. The degree of crosslinking was not determined in these gels. However, based on previous experiments and modeling of GelMA hydrogels [
31], we expect that ~42% of the maximal attainable compressive modulus was reached in our experiments. Whilst UV dose could be used to increase the degree of crosslinking and hence modulus, this would likely result in greater cytotoxicity [
11,
32]. The current study focused on assessing the potential to tailor viscoelasticity in cytocompatible hydrogel systems.
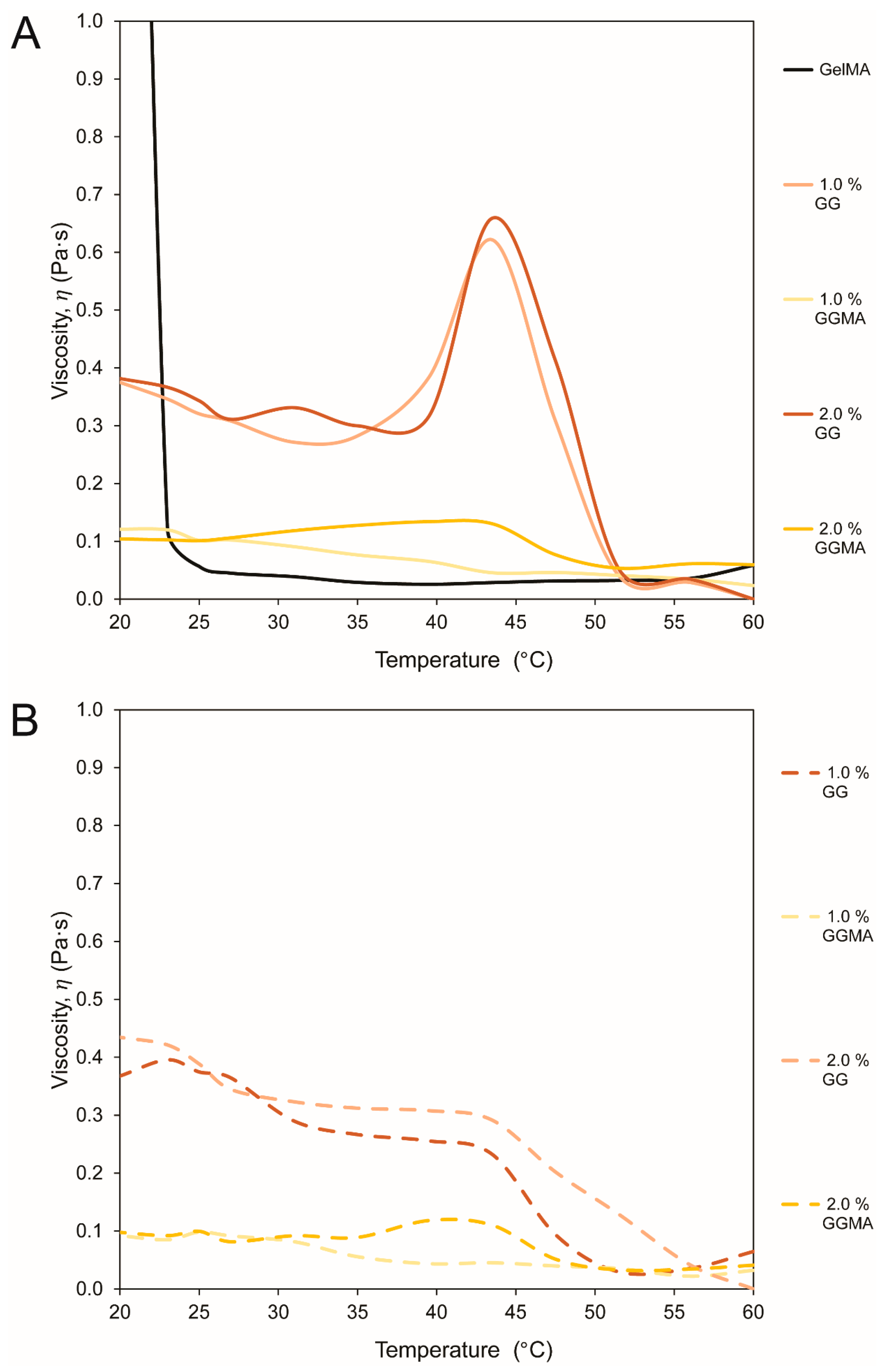
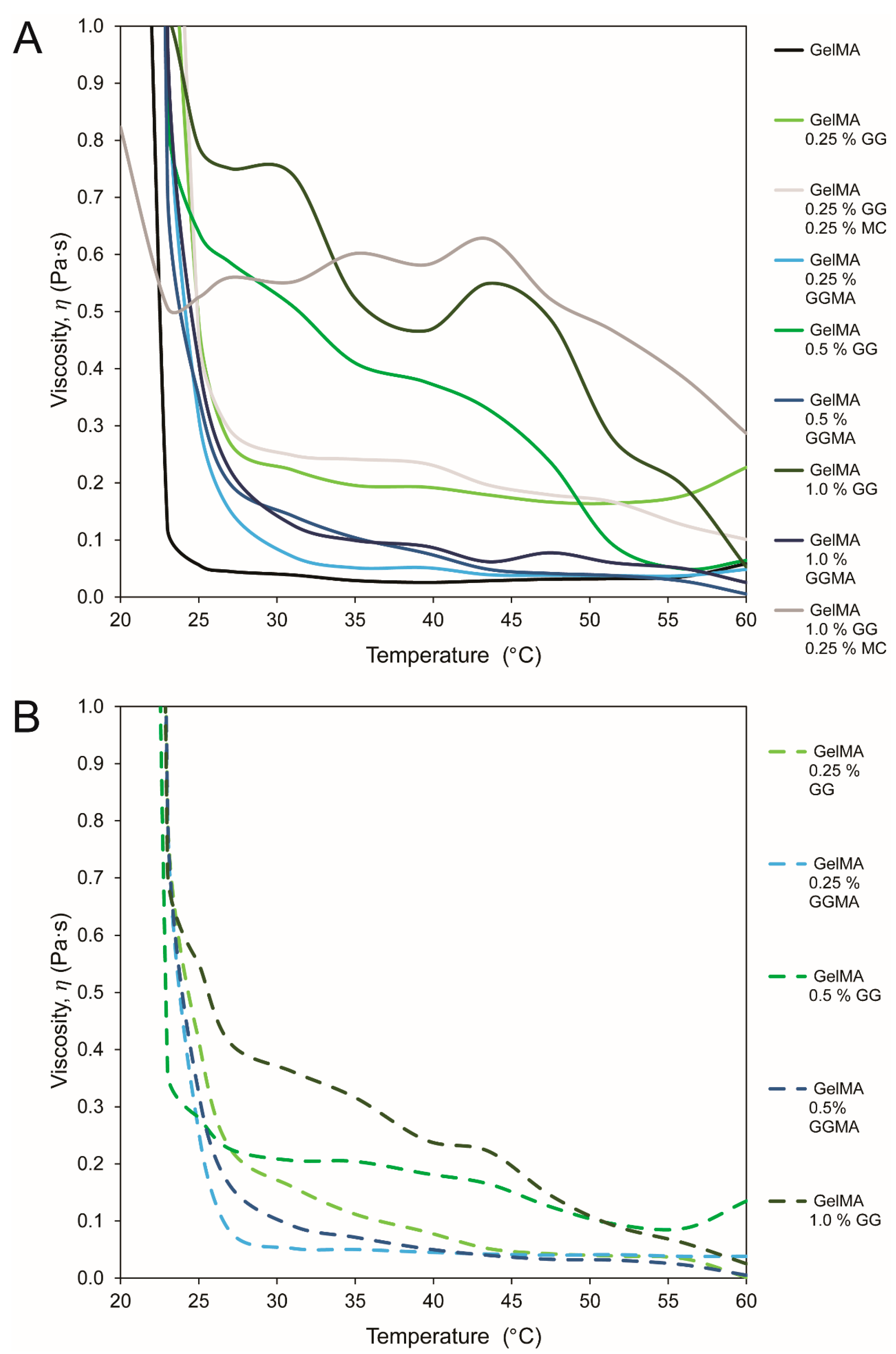
Rheological analysis (groups presented in
Table 1, data in
Figure 2 and
Figure 3) was conducted to assess reduction in gelation temperature by using PBS as a solvent
versus a 24 mM NaCl + 252 mM
d-mannose (N+M) isotonic solution, or by mixing methyl cellulose (MC) within the hydrogels. MC was included to dilute the mix during processing, with MC reported to diffuse out after crosslinking and subsequent immersion in medium [
33]. The use of MC did not achieve this outcome, in contrast to the very effective N+M, with low ion concentrations maintaining GG in a loosely crosslinked state that remained pipettable due to the pseudoplasticity of the material [
23].
GG forms gels through the initial formation of double helical junction zones which then aggregate, forming a three-dimensional network by complexation with cations and hydration of the network [
18]. Methacrylation of polysaccharides such as GG involves the modification of the hydroxyl groups, which reduces deprotonating capacity, water retention and the propensity to form coordination complexes between helices, thus reducing the gelation temperature of the substance in ion-rich solutions such as PBS when compared with standard GG [
18,
23]. Whilst this clearly reduces the ability for GGMA to form physical, ion-bound hydrogels at low gel concentrations, it also facilitates the production of hydrogel precursor solutions that are liquid at physiological temperatures, as demonstrated herein.
Whilst methacrylation of GG is one approach to achieve lower gelation temperatures, a simpler method is the use of a low-ion physiologically isotonic solvent (N+M). Furthermore, the inherent pseudoplasticity of GG, which is advantageous for 3D printing [
23], allows high fidelity deposition without flow rate irregularities as well as aiding in the stability of the printed structure. This is a disadvantage when it comes to conventional handling, as the ability to pipette the solution into molds for crosslinking is lost before the gel is completely set; hence all mechanical testing in this study was performed on gels mixed in an N+M solution.
Whilst mixing hydrogel precursors at 37 °C allows a physiological environment for cells and decreases viscosity such that processing is possible, reproducibility of mechanical properties between groups becomes a potential issue when considering varied incubation times at RT prior to the photopolymerization. This may be overcome by maintaining gels at 37 °C within molds but this is accompanied by an increased risk of cell settling due to the decreased gel viscosity at higher temperatures [
34].
The formation of tertiary structures (interactions and associations between aggregates) is influenced by the rate of cooling, especially when the time taken prior to photopolymerization may vary [
35]. The hydrogels used in this study become increasingly crosslinked physically or through ionic interactions at lower temperatures without UV irradiation. Once UV exposure is initiated, the speed of radical polymerization will largely exceed that of physical crosslinking, which will also be reduced due to the ~37 °C temperatures that result from passive heat of the UV bulbs throughout the process (data not shown). The combination of various hydrogel components may also impede physical interactions and cause the aggregates of one gel type to become entrapped within the network of the other [
23]. UV irradiation immediately after precursor processing would thus form a hydrogel crosslinked predominantly through the radical reaction, rather than a combination of the thermal-setting physical crosslink followed by the chemical crosslinking.
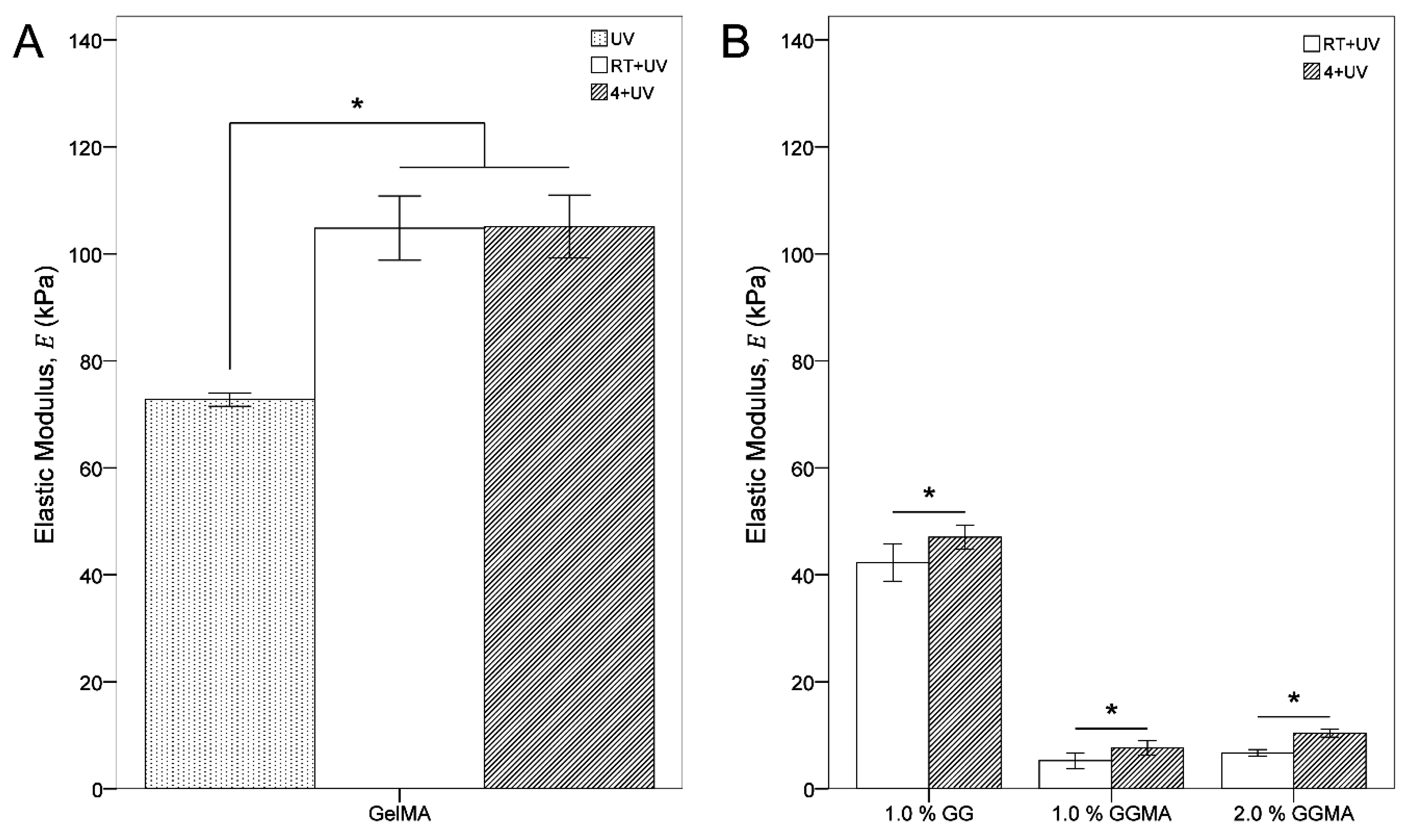
Incubation at RT or 4 °C prior to photopolymerization allows for: the maintenance of cells within the hydrogel matrix at an even distribution without cell settling; a reduction in the influence of physical gelation (which occurs at any time at RT) on mechanical properties between groups; and an increase in base mechanical properties due to the formation of more coherent tertiary structures (through these physical crosslinks). A comparison of the elastic modulus of GelMA crosslinked immediately (UV)
versus crosslinking by incubation at RT (RT + UV) and 4 °C (4 + UV) (
Table 2;
Figure 8A) illustrates the significant increase in modulus that physical crosslinking prior to chemical crosslinking may provide due to the superior structural arrangement in the hydrogel [
31]. In this system, increasing cooling rate did not appear to affect GelMA stiffness, whilst GG and GGMA were more notably (and significantly) affected (
Figure 8B). As GG/GGMA form solids through a multi-stage process reliant on ionic concentration, it may be postulated that the increased cooling rate allows for helices to become established faster, which then allows more time for ionic interactions between helices and thus a stronger hydrogel. Alternatively, helices may not have sufficient time to form discretely and instead form a disordered tangle that is stronger due to a greater degree of overlap. All of these gelation mechanisms however are very complex and extensive study beyond the scope of this work would be required to define the processes comprehensively. Nonetheless, the materials and methods used in this study were cytocompatible (viability ~90%), which is acceptable and typical of photocrosslinkable hydrogel systems using Irgacure 2959 [
15,
36,
37].
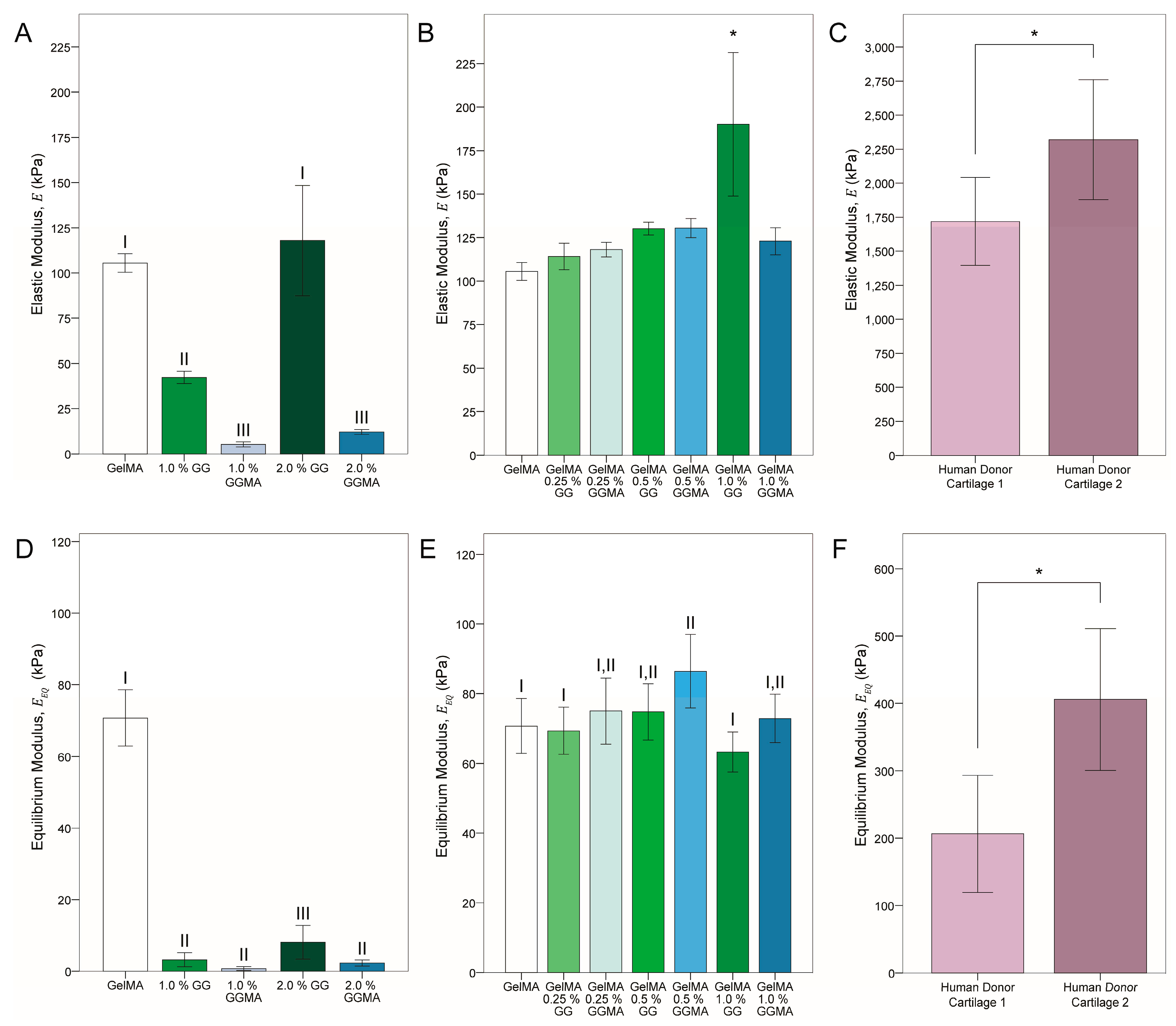
In terms of mechanical testing, GelMA hydrogels were found to be predominantly elastic. GG and GGMA was added to GelMA in various concentrations to ascertain whether viscoelasticity could be tuned using the addition of a primarily ion-bound hydrogel (GG) or a primarily covalently-bound gel (GGMA). Contrast of the two gel blends also exposed whether an interconnected covalent network (such as in GelMA/GGMA) would afford increased mechanical properties, compared with an entwined yet only physically entrapped network (such as that within GelMA/GG). Examination of the elastic moduli of the gel groups and explants (groups in
Table 3, data in
Figure 4A–C), showed that whilst inclusion of either GG or GGMA proved to increase the modulus above that of GelMA alone, the effects were predominantly less than 25% and not statistically significant, apart from the increase (~75%) in the GelMA/1.0% GG group. It may be inferred that it was only at this concentration that the GG achieved sufficient network density to enhance the overall modulus,
i.e., the GG was able to form stable crosslinks with itself, in addition to mixing through and physically entangling with the GelMA network. When the gels were made in N+M solution, the higher GG concentration produced a matrix that allowed them to recover structurally, with helical association promoted by ion reinfusion [
23].
In contrast, photocrosslinking of GGMA in a low-ion solution caused the network to become locked in place and reinfusion of ions was not able to recover modulus through network rearrangement—if recovery was possible at all, due to the modification of hydroxyl groups and hence removal of potential negative charge from deprotonation. Notably however, addition of GGMA to the GelMA did increase the modulus to a greater extent than the sum of individual moduli, with all concentrations giving a ~25% increase, similar to lower concentration GG-blended groups. Comparison with human articular cartilage donor tissues showed a dramatic 10-fold difference between hydrogels and native tissue. Variability was also found between donors, which will remain a considerable factor in the future design of tissue engineering structures, however values represented those found in literature for the elastic modulus of healthy elderly individuals (1–3 MPa) [
30].
Equilibrium modulus (
Figure 4D–F) clearly illustrated the non-covalent nature of the GG, with a drop of ~30% in GelMA and GGMA moduli but ~80% in GG modulus after relaxation for 10 min at each of two strain levels. This trend was also observed in the blended groups, with the modulus of the 1.0% GG blend being significantly lower than all other composites, apart from 0.25% GG, again suggesting cohesion of the GG network. The viscoelasticity achieved in the hydrogels was contextualized by the native tissues, which also demonstrated a dramatic decrease in modulus after relaxation.
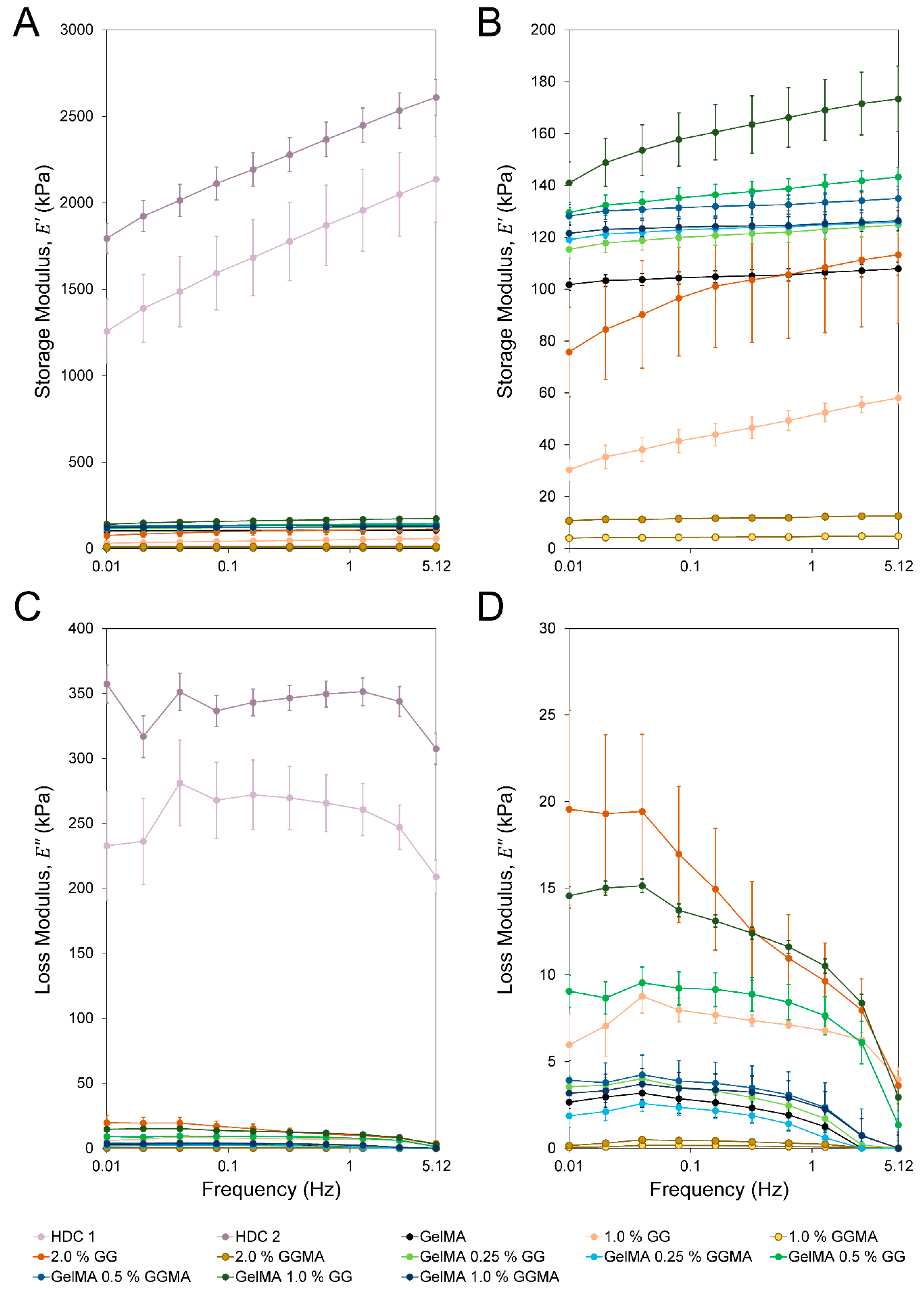
The greater physical complexity of the donor tissues was demonstrated by storage (
) and loss (
) moduli (
Figure 5A,C) where, across all frequencies, the donor explants maintained a relatively consistent loss modulus. As all hydrogels are subject to fluid flow through the matrix when compressed, some degree of hysteresis in loading must occur independent of which crosslinking modalities are used. Despite this, testing revealed that in the absence of ion-bound hydrogels such as GG, hydrogels exhibited a major frequency-dependent drop in loss modulus (e.g., GelMA, GelMA/GGMA, GGMA;
Figure 5D). Furthermore, even in hydrogel groups with relatively high loss modulus, this modulus was much more frequency-dependent when compared with the native tissues, and primarily manifested at the lowest frequencies. This may be due to the weaker bonding of the ionic hydrogels
versus the highly complex and ordered cartilage structure, in addition to the much greater water content in the hydrogels (maximum 90%
versus 75%–80% in articular cartilage) [
38]. A primarily homogenous hydrogel crosslinked through ionic interactions will exhibit a largely elastic response at high frequencies, similar to that expressed by covalently crosslinked hydrogels. The release of sufficient fluid requires a longer duration strain, which will allow the disruption of ionic associations and thus the relaxation of associations between aggregated polymer strands. The complexity of the cartilage allows it to maintain a loss modulus within ~20% of the starting value regardless of frequency, whilst a near 75% decrease is seen in 2.0% GG over 0.01–5.12 Hz.
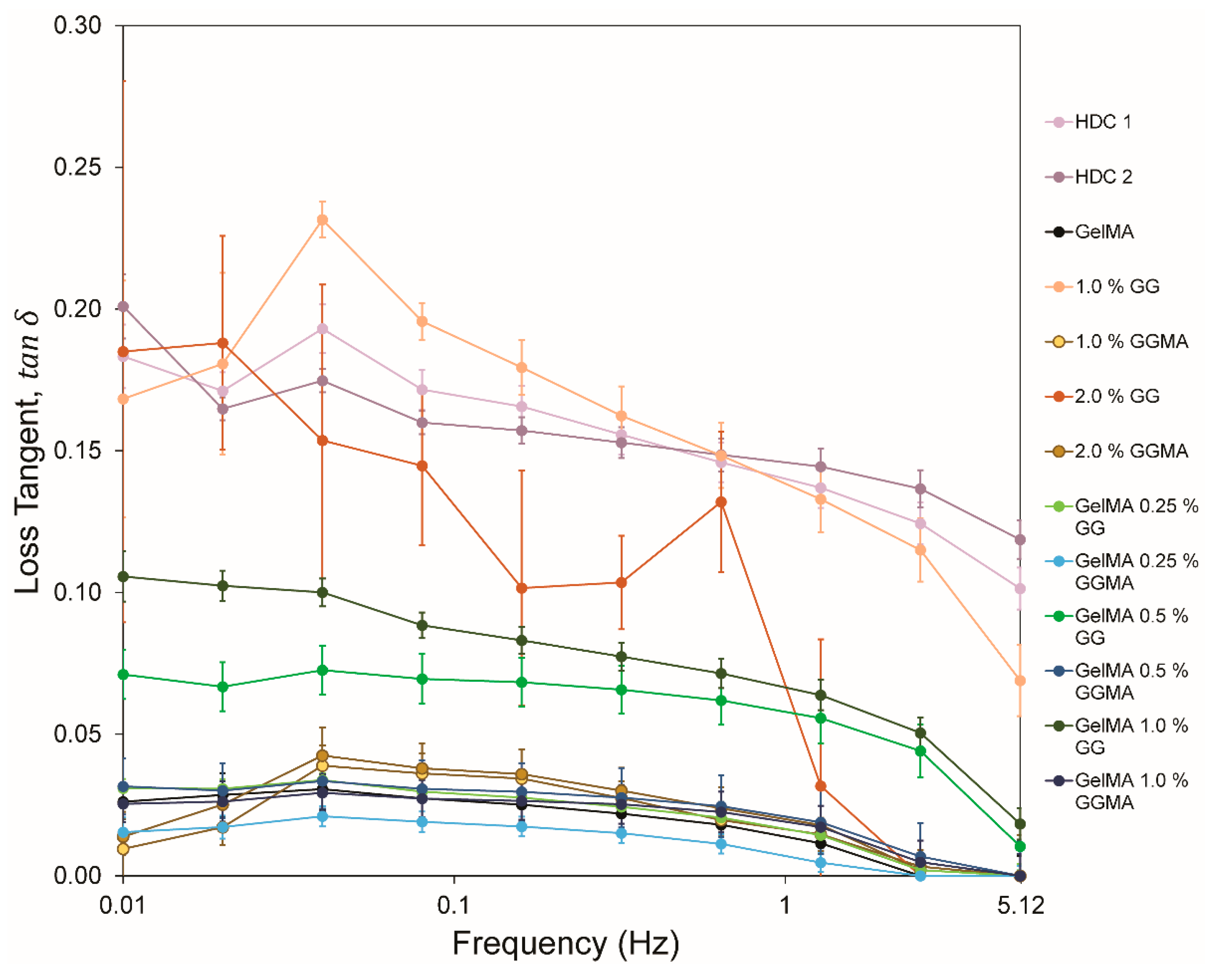
The comparison of hydrogels and cartilage is significantly simplified when the loss tangent (
) is considered (
Figure 6), as the loss tangent illustrates the ratio of loss to storage moduli. It was clear that the majority of hydrogel groups in this study did not consistently produce a large viscoelastic response, with only GelMA/0.5% GG, GelMA/1.0% GG, 1.0% GG and 2.0% GG achieving values above 0.05, or being comparable with native tissues. The consistency of loss tangent exhibited in native tissues is clear, with a decrease from ~0.2 to ~0.1 observed in the samples. Conversely the hydrogels show a much greater decrease over the frequency range, with 1.0% GG appearing closest to native values. Whilst 2.0% GG appeared somewhat variable over the range, the data suggest that stabilization of a higher concentration of GG within GelMA may reveal trends more similar to native tissue.
Analysis of double network gels has been explored in the literature primarily by the group of Gong
et al. [
39,
40,
41,
42]. However the cited systems comprise networks of a hard, brittle gel formed using photopolymerization with a soft, ductile gel at a much higher molarity infused within and then crosslinked through a second photopolymerization. This yields a loosely crosslinked ductile gel within a brittle and stiff network and allows for dramatic increases in elastic modulus, strength, strain at failure and toughness [
39]. This is unlike our system, where the hydrogel components are mixed in-solution prior to molding and crosslinking. Further, whilst these systems afford high level mechanical properties, they also tend to display the Mullins Effect, wherein hysteresis is only observable upon first-cycle loading to an initial maximum stress and subsequent cycles to the same stress level no longer show any hysteresis and hence are largely absent of viscoelasticity [
43]. Webber
et al. propose that these data are supportive of the Lake-Thomas [
44] mechanism where energy exceeds the threshold above which permanent damage is caused upon repeated loading, and that the initial hysteresis is associated with breaking of covalent links within the network. This is again in contrast with our system, where we found an increase in physical crosslinks directly increasing hysteresis, and being the largest contributor to the manifestation of viscoelasticity.
The work of the Vilgis group [
45] identifies similar trends to those observed in our work, showing that with increased covalent crosslinks there is limited hysteresis in loading and with increased physical crosslinking there is a higher stretching ratio and increased hysteresis. However, they support the theory of a Mullins effect within their system, which would indicate permanent crosslink degradation and hence a lack of viscoelastic property. This conclusion is in direct contrast to our findings, where all hysteretic data are taken from multiple cycles, with initial and terminal cycles excluded to ensure reproducibility and reduce edge effects. As these loads are reproducible throughout multiple cycles (>200 in the case of 5.12 Hz) it is clear that a Mullins Effect and Lake-Thomas microfractures [
44] are not occurring, and that the breaking and reforming of physical/ionic bonds of the GG must be a large driving factor in the establishment of viscoelasticity, as we initially indicated.
Overall, it is clear that the emulation of the biological aspect of viscoelasticity may be achievable through the use of appropriate hydrogels, ideally crosslinked through varied means to take advantage of multiple systems, such as the previously mentioned cell attachment-promoting effects of gelatin. Whilst it may be predicted that the elastic modulus of such structures will still fail to reach that of native tissues, this may in fact be desirable, as cellular stress/strain is ultimately required to stimulate matrix production and formation of cartilage
in situ. Further, it has been previously demonstrated that hydrogels with lower initial stiffness can result in constructs with higher mechanical properties after culture [
46,
47].
The work presented herein illustrated that viscoelasticity may be tailored in hydrogels depending on physical and chemical crosslinking of networks. Further study of such hydrogel systems and verification of the role of viscoelasticity in the formation of biologically relevant constructs should be explored in future.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







