
High Temperature Epoxy Foam: Optimization of Process Parameters

Abstract
:
1. Introduction
2. Materials and Methods
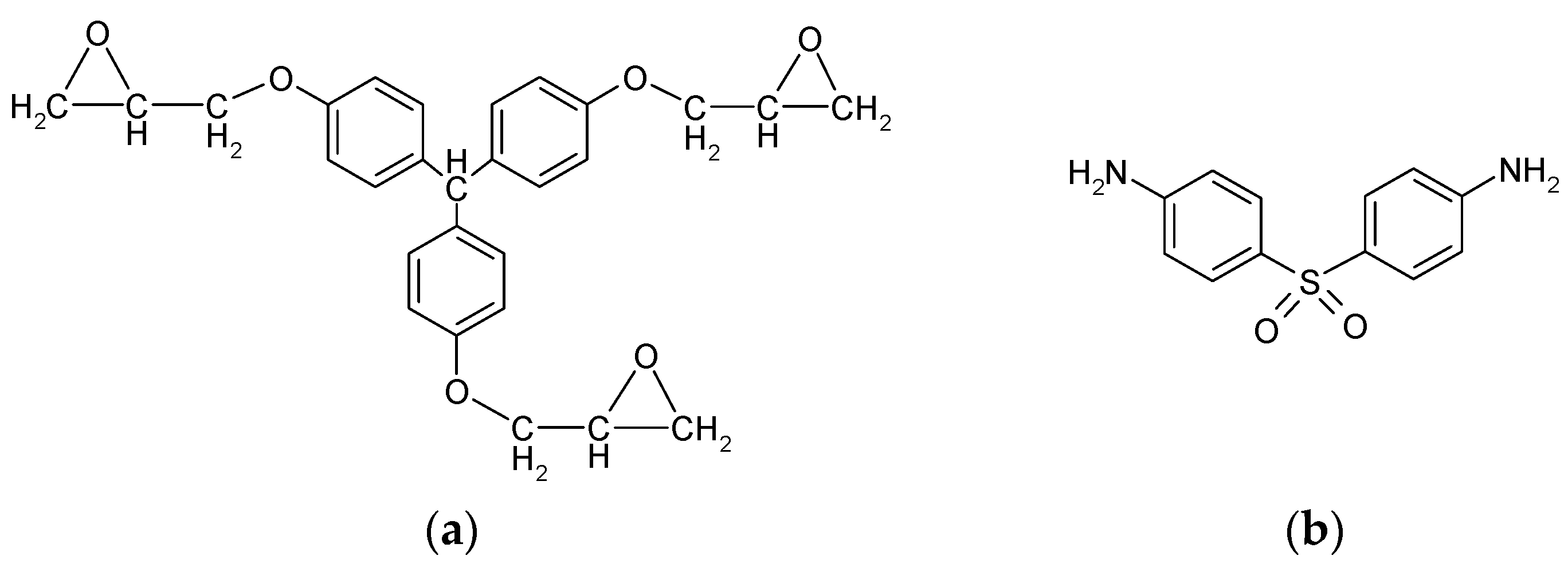
2.1. Materials
2.2. Preparation of Epoxy Resin
2.3. Preparation of Epoxy Foam
2.4. Scientific Characterization
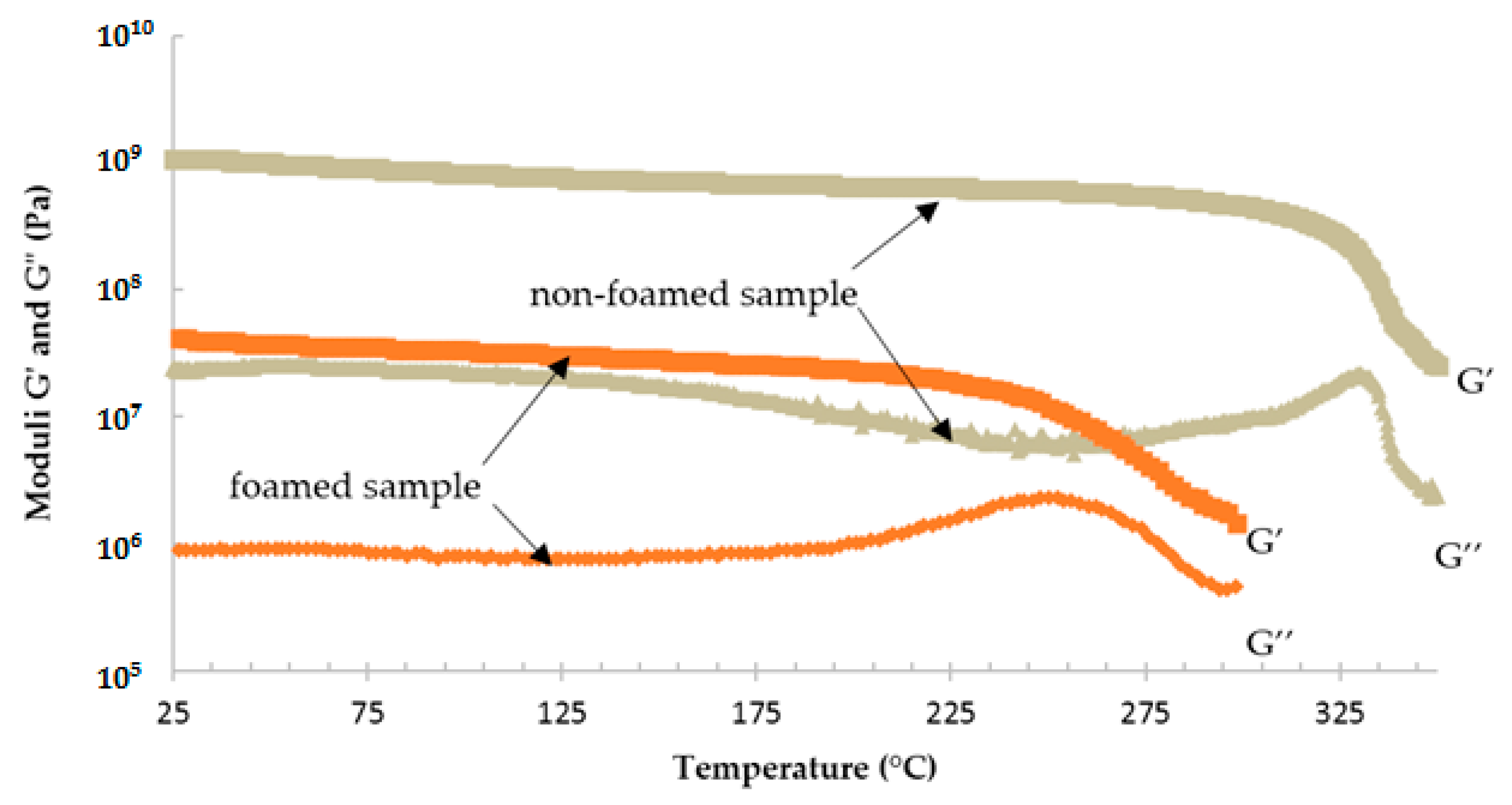
3. Results and Discussion
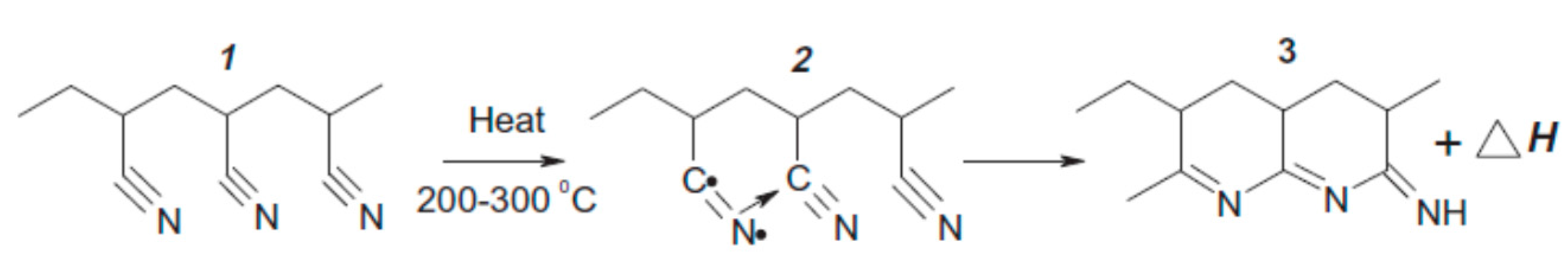
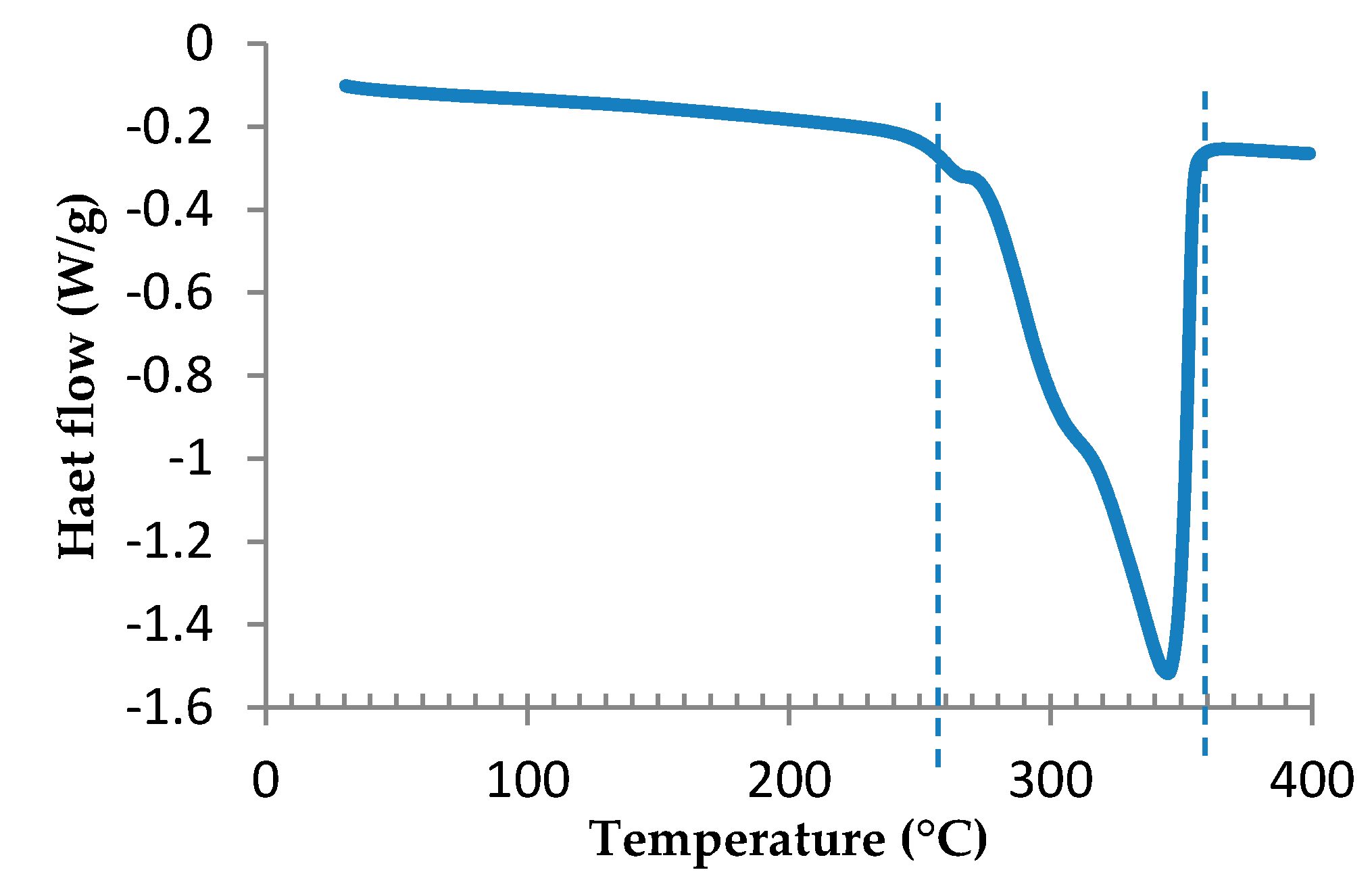
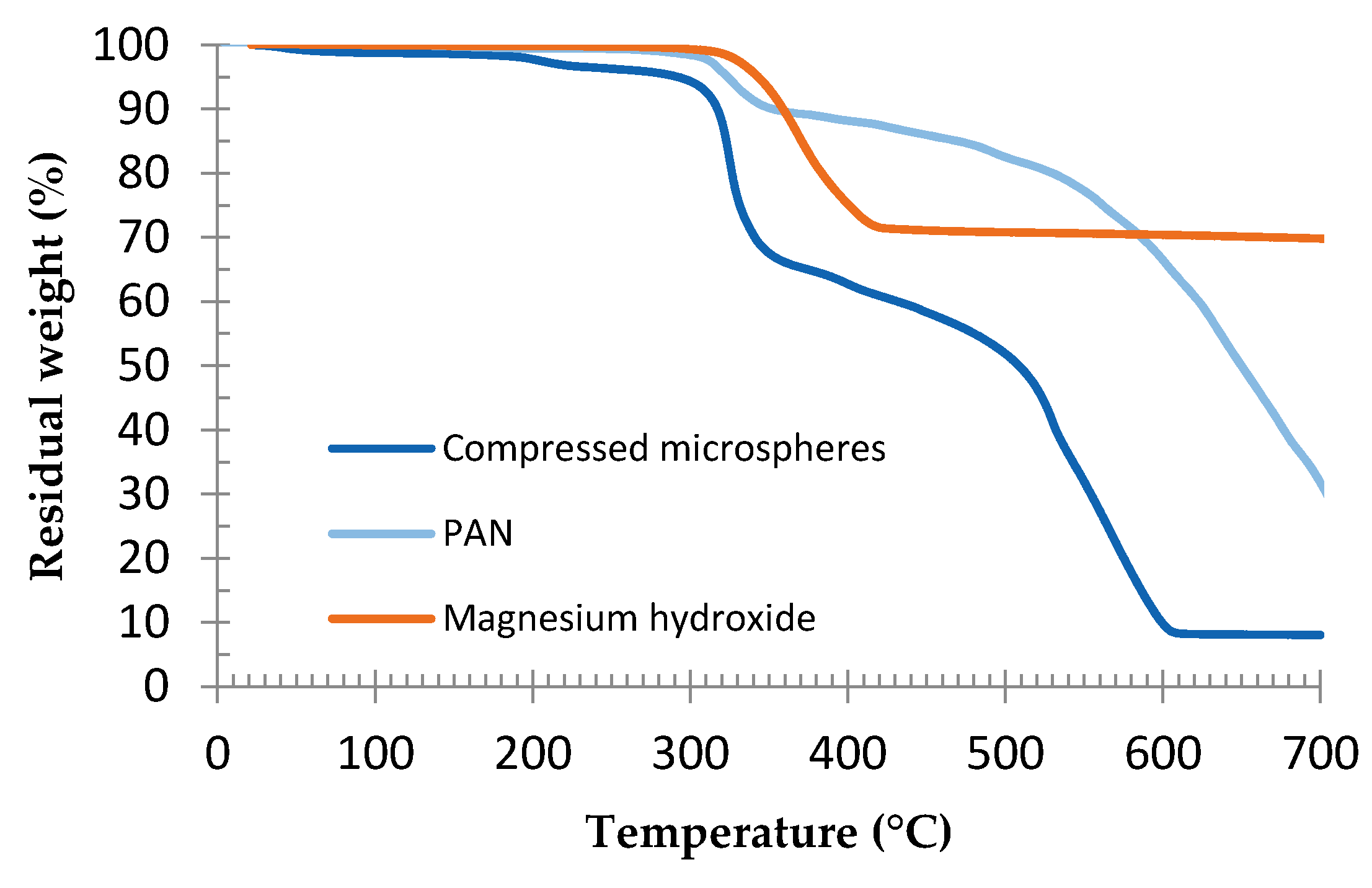
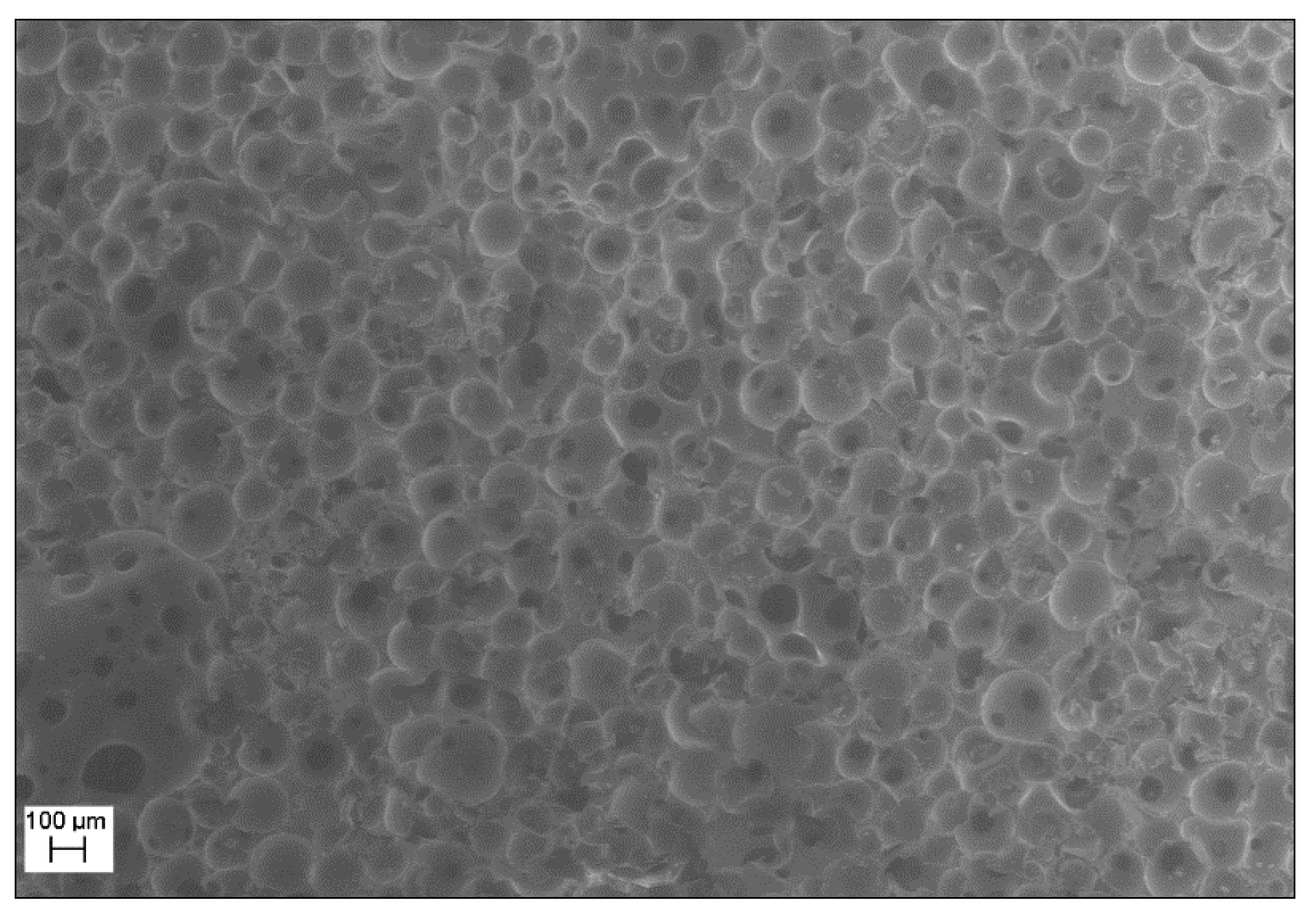
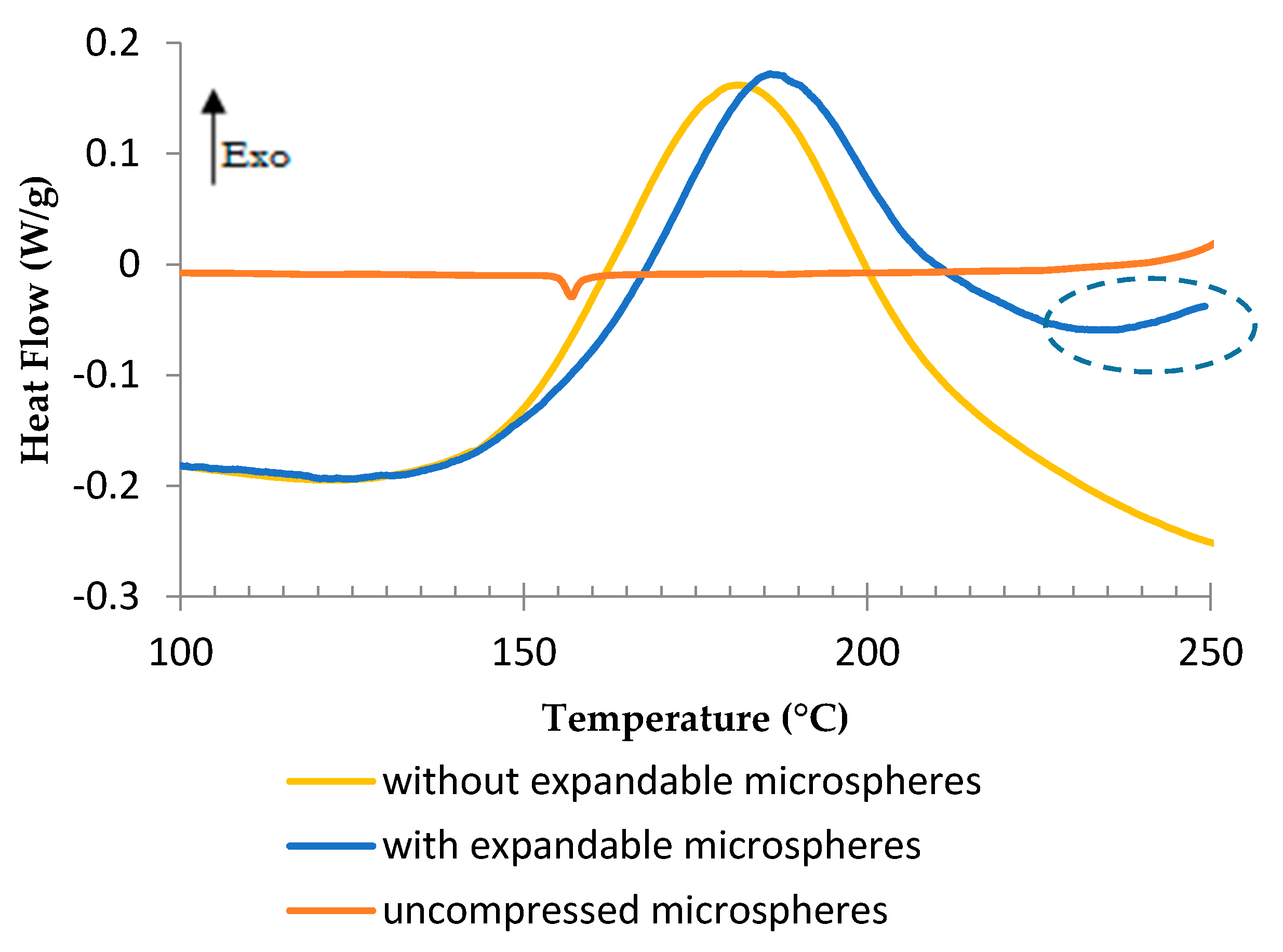
3.1. Preliminary Study of the Epoxy Formulation and Expandable Microspheres Characterization
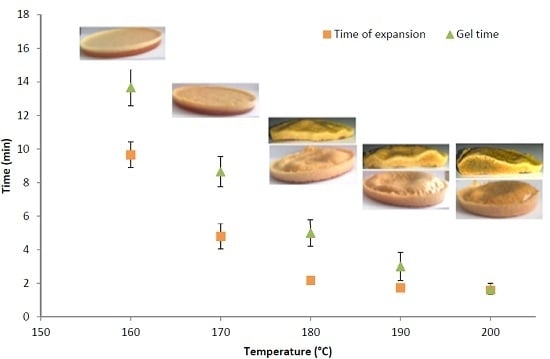
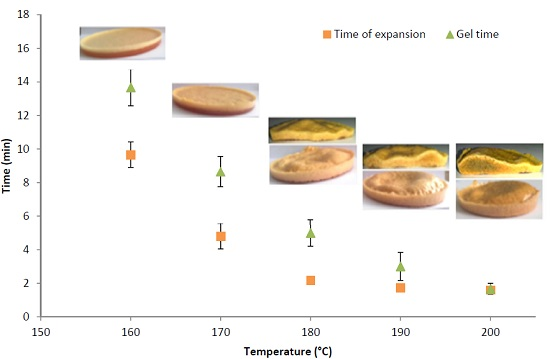
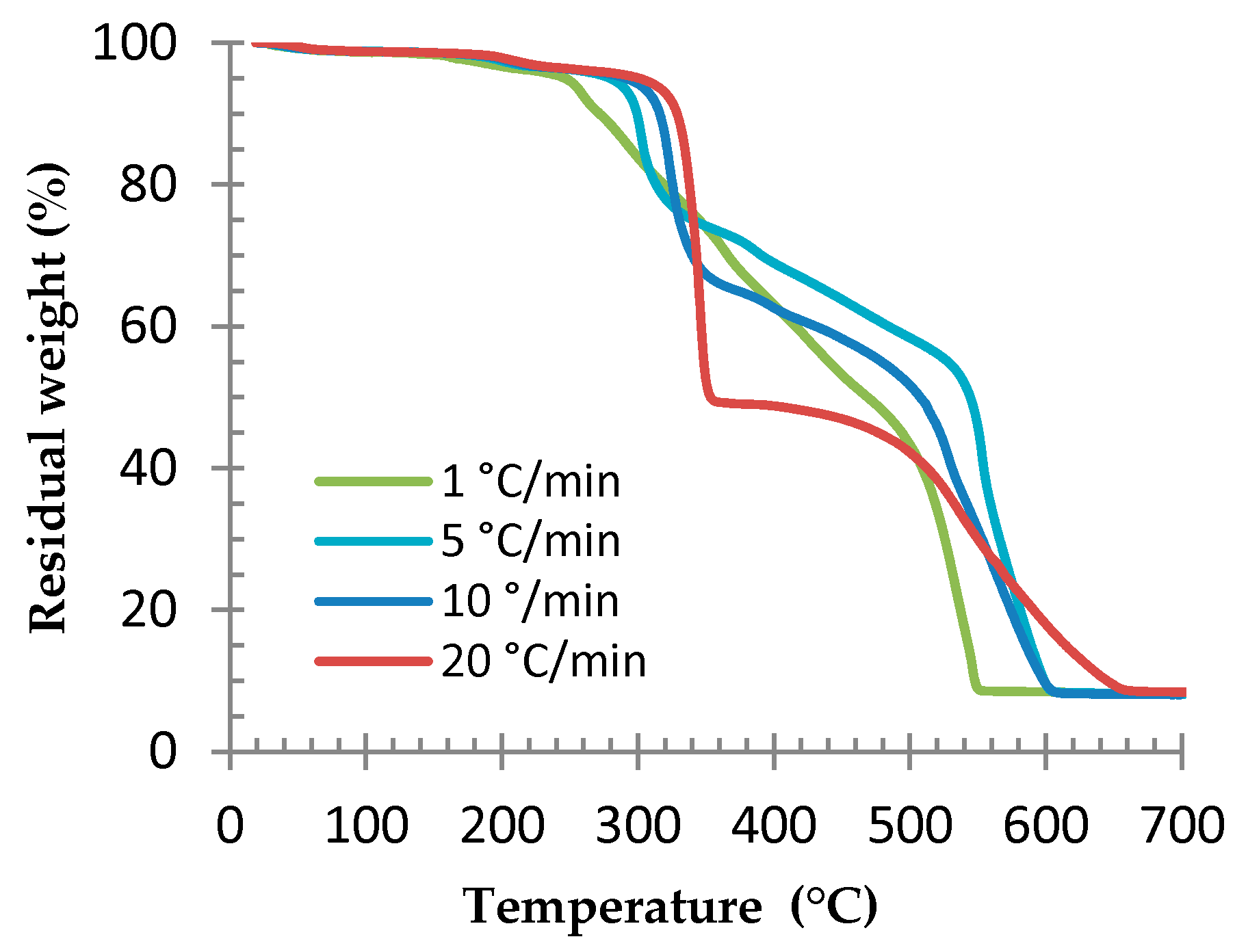
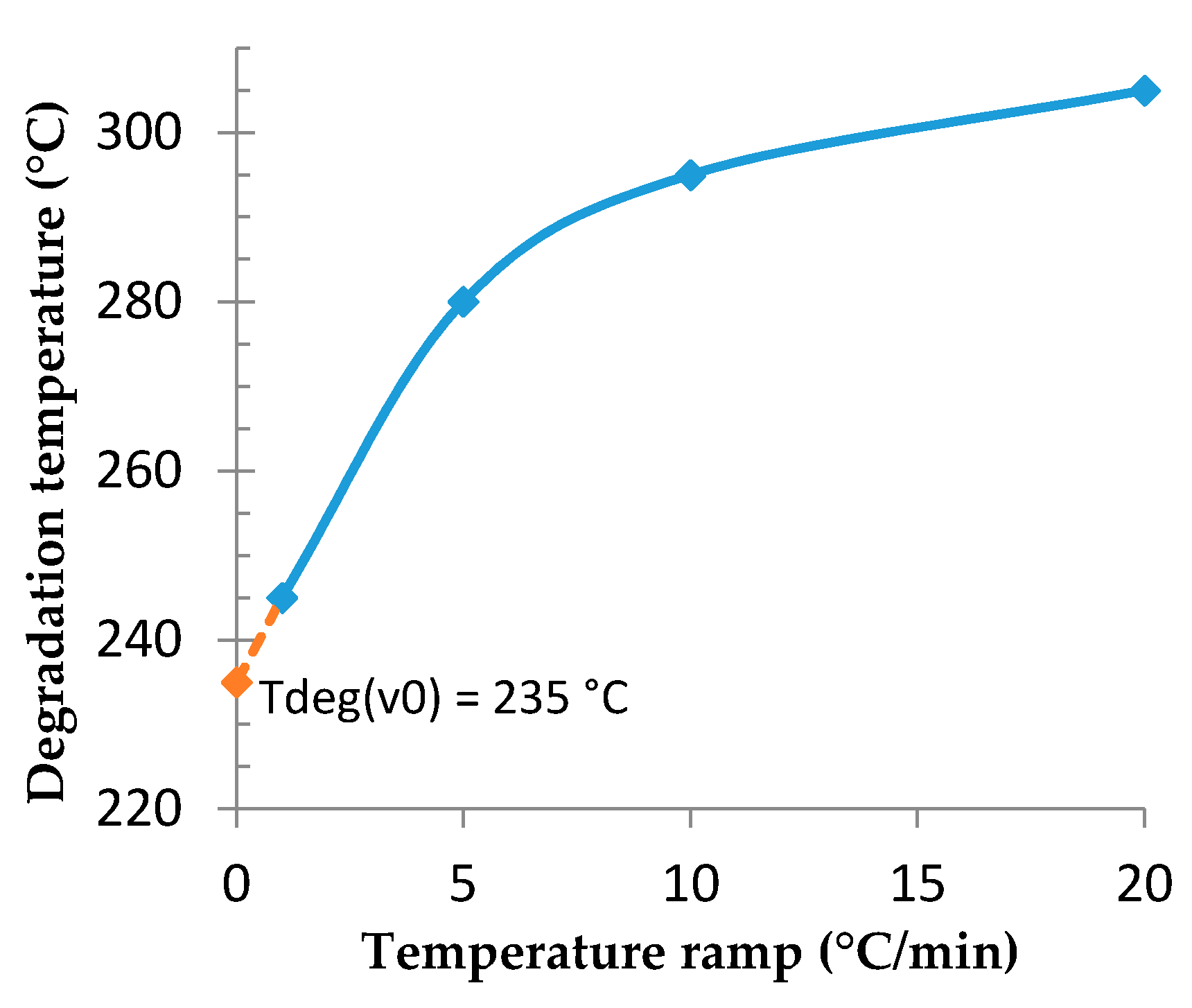
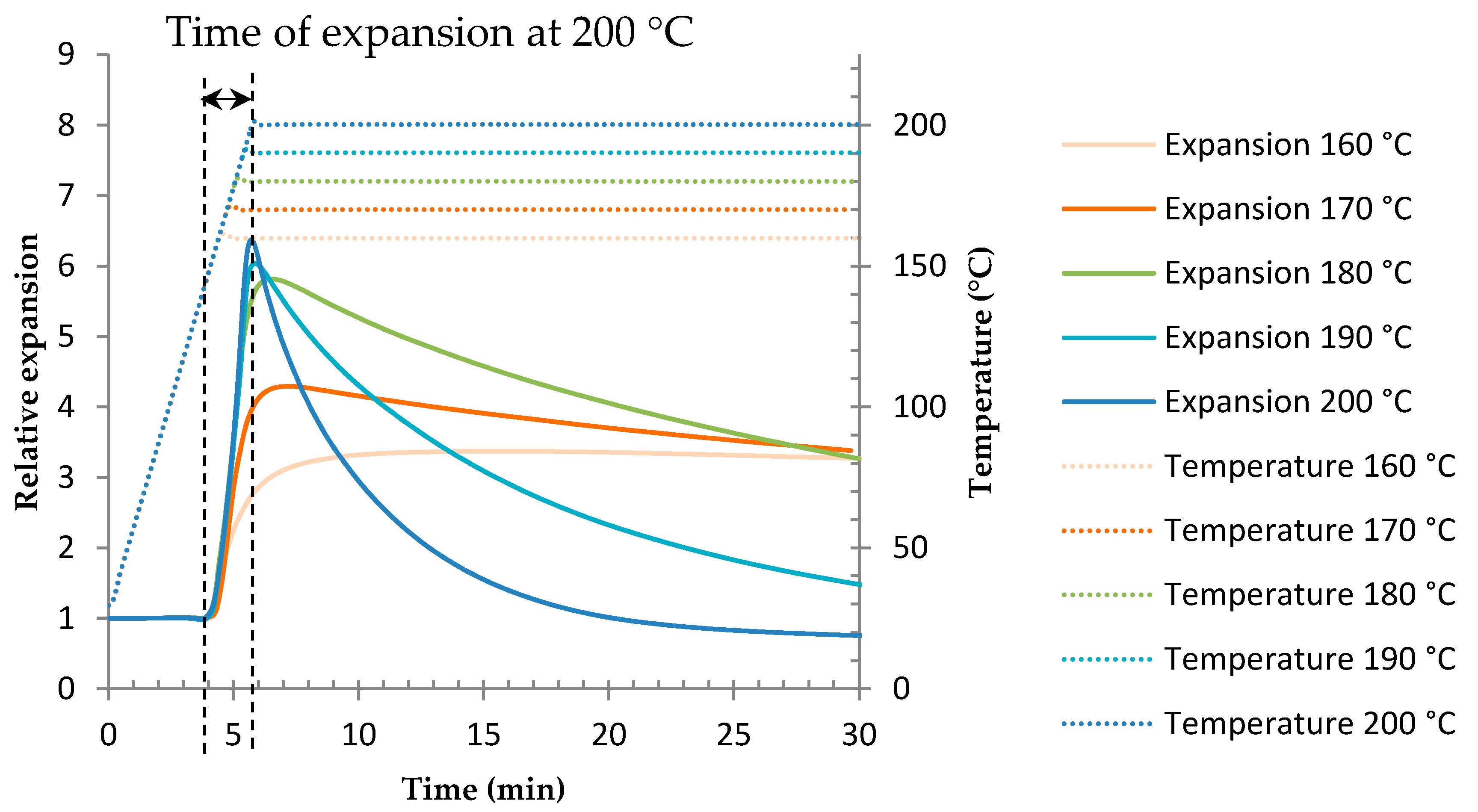
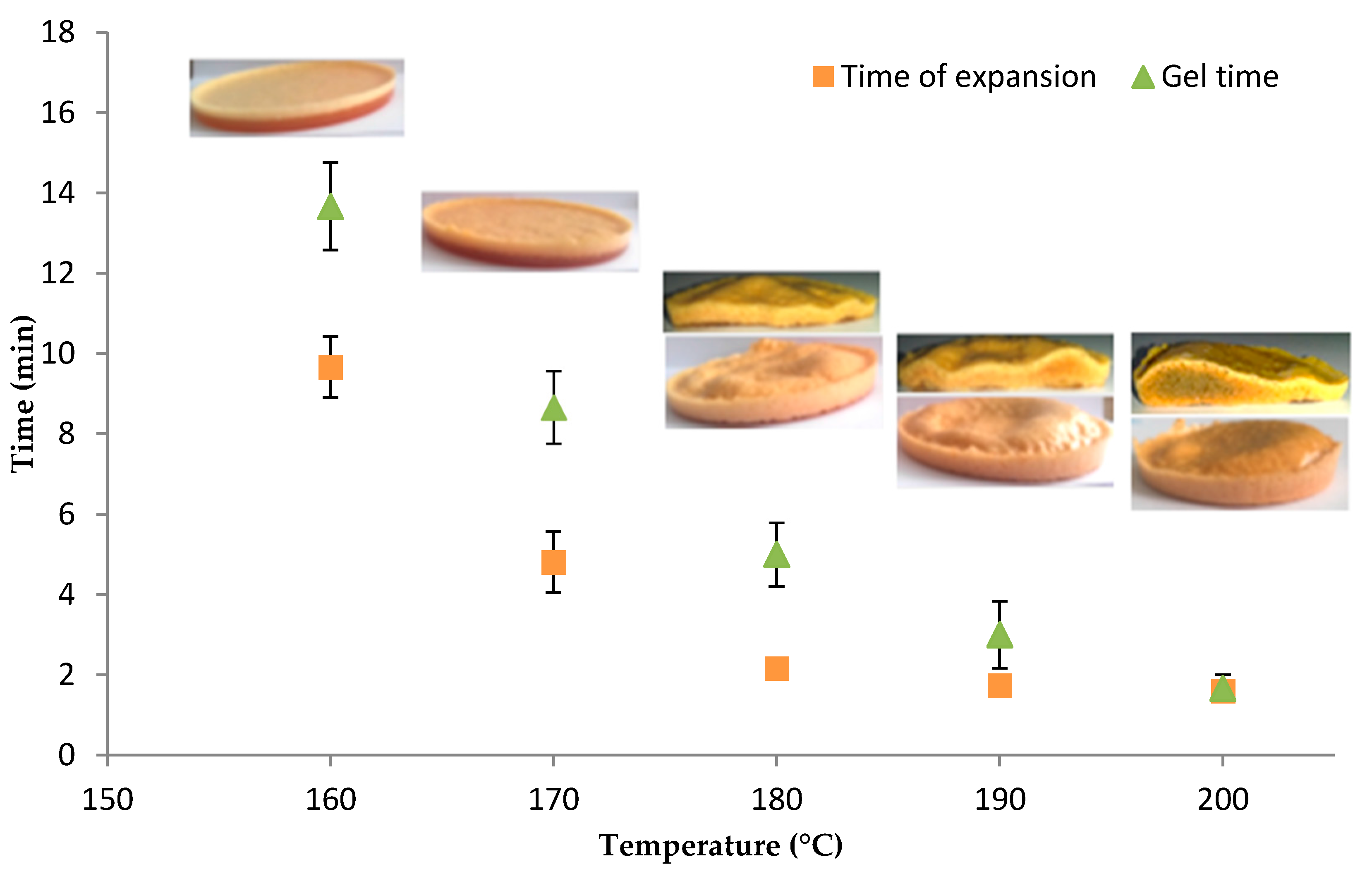
3.2. Determination of the Optimal Foaming Temperature
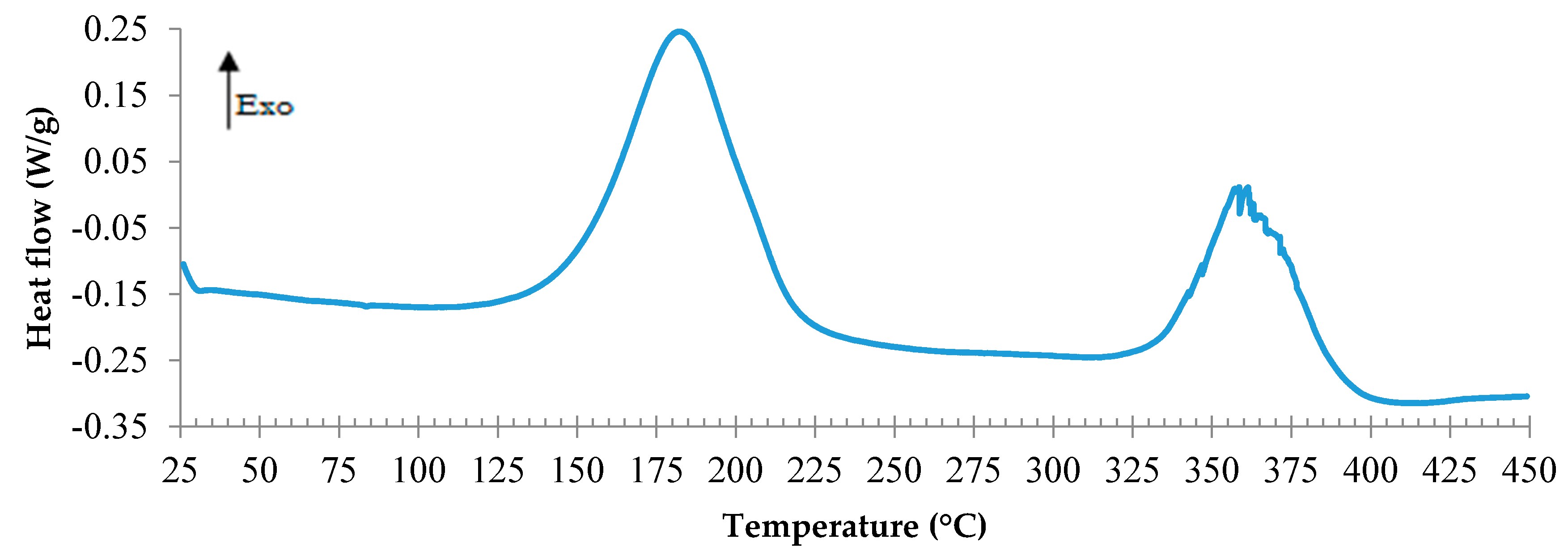
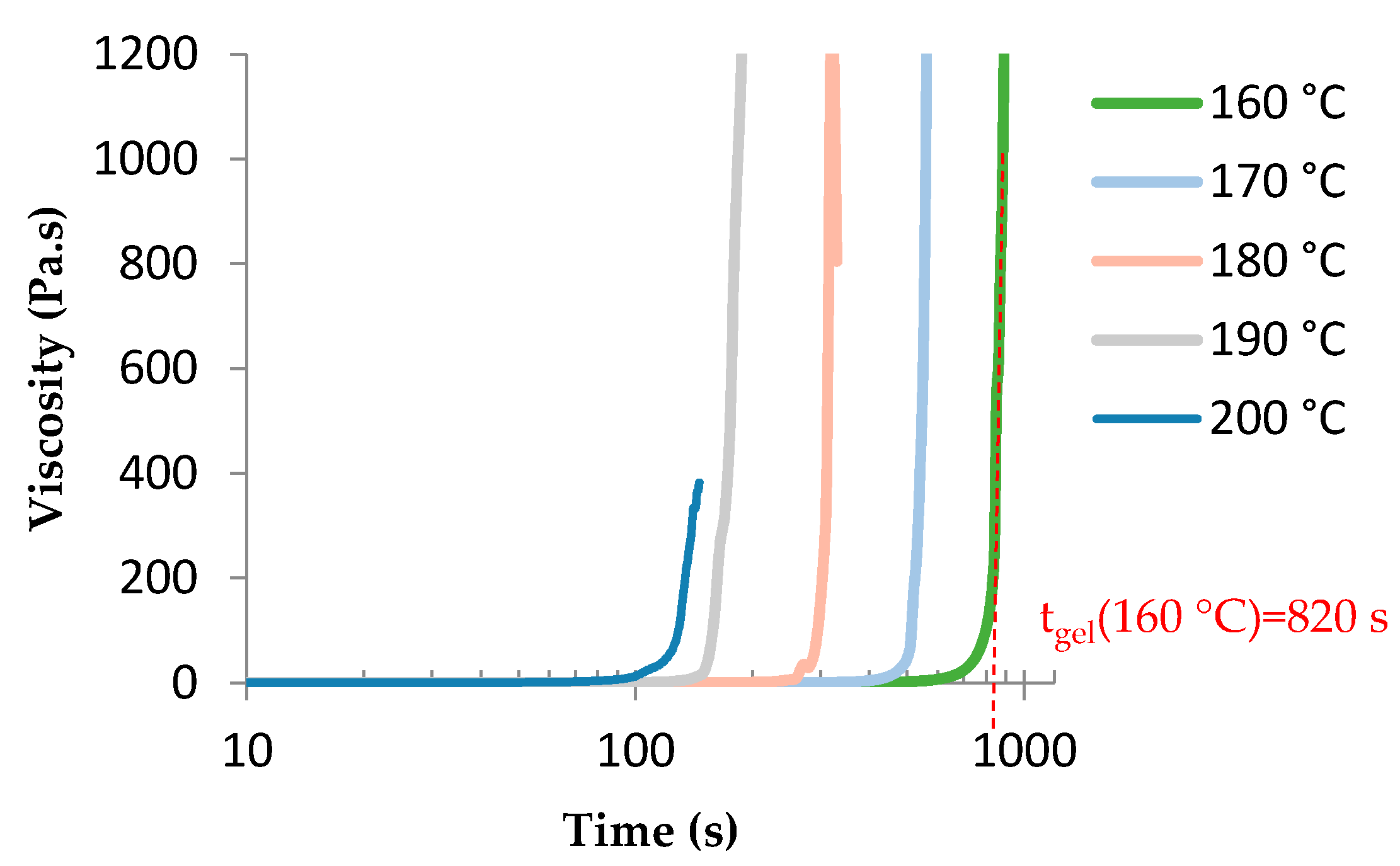
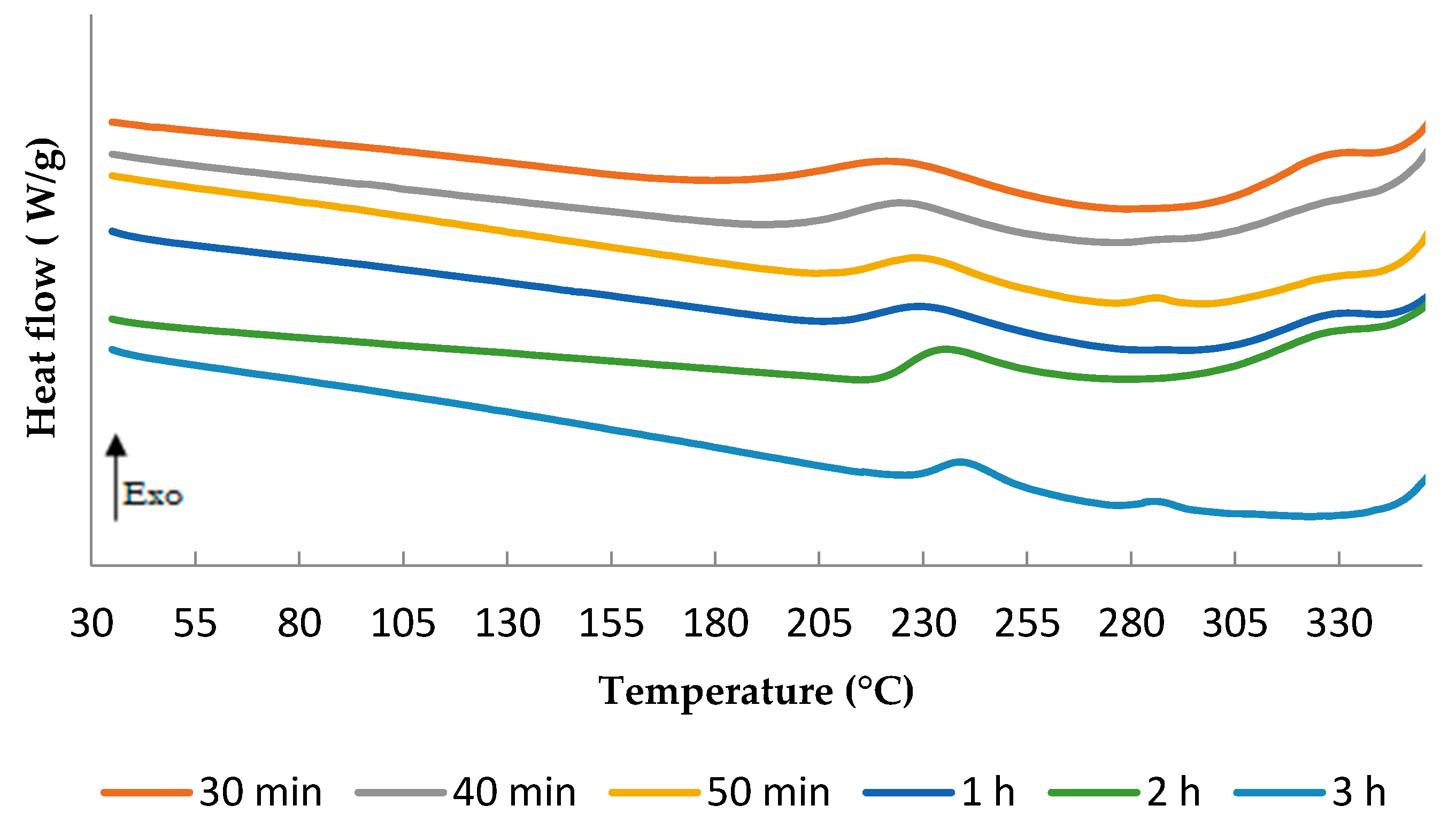
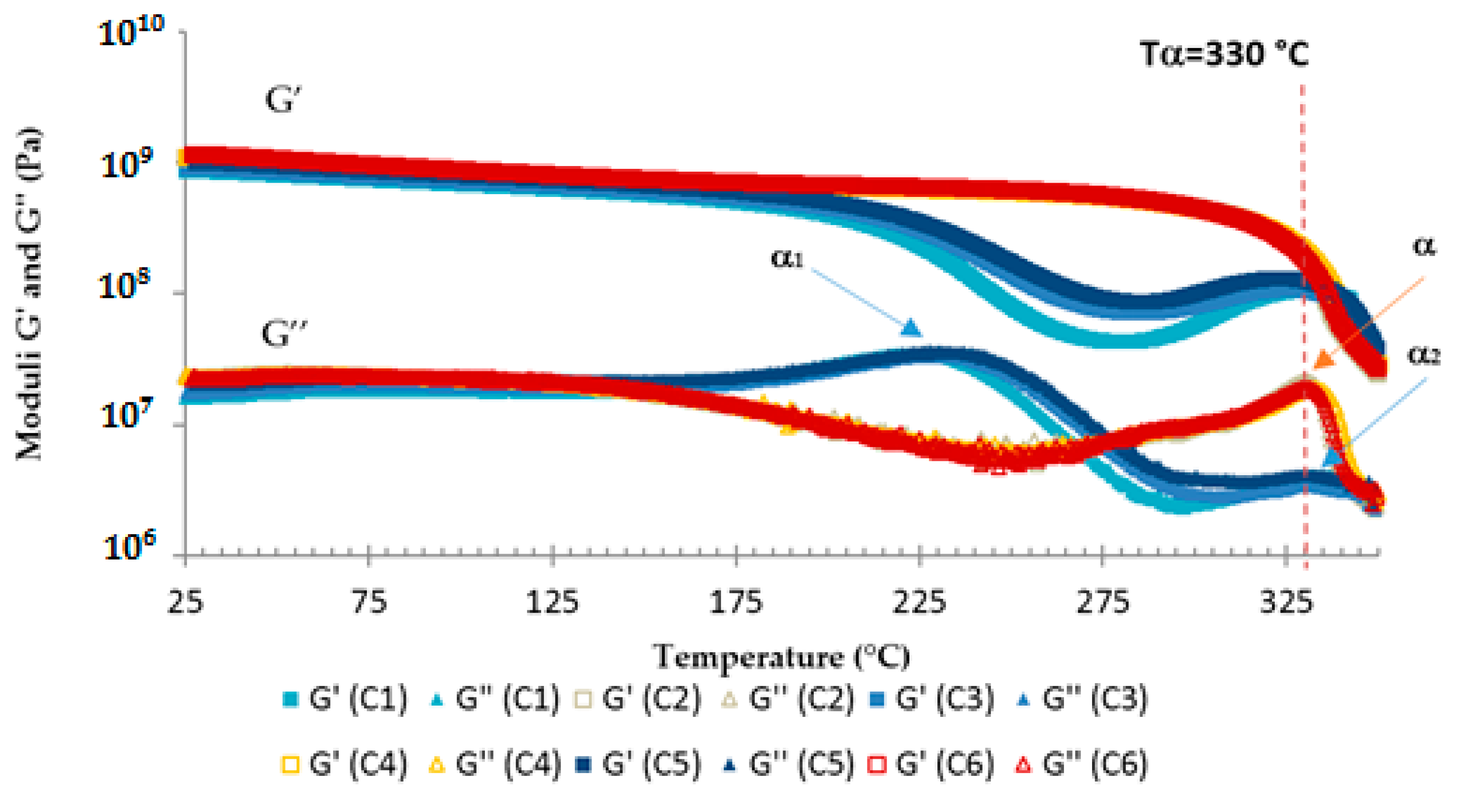
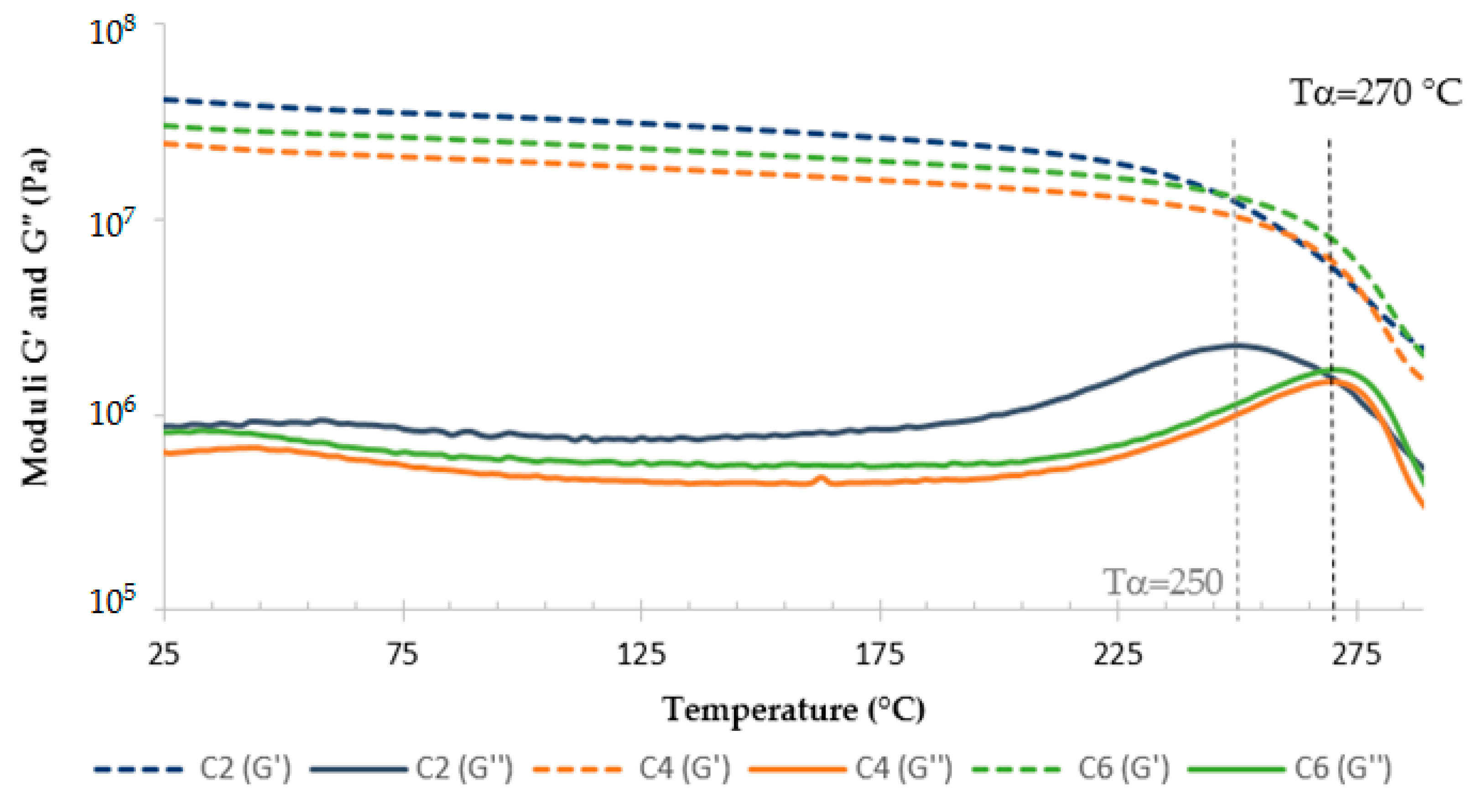
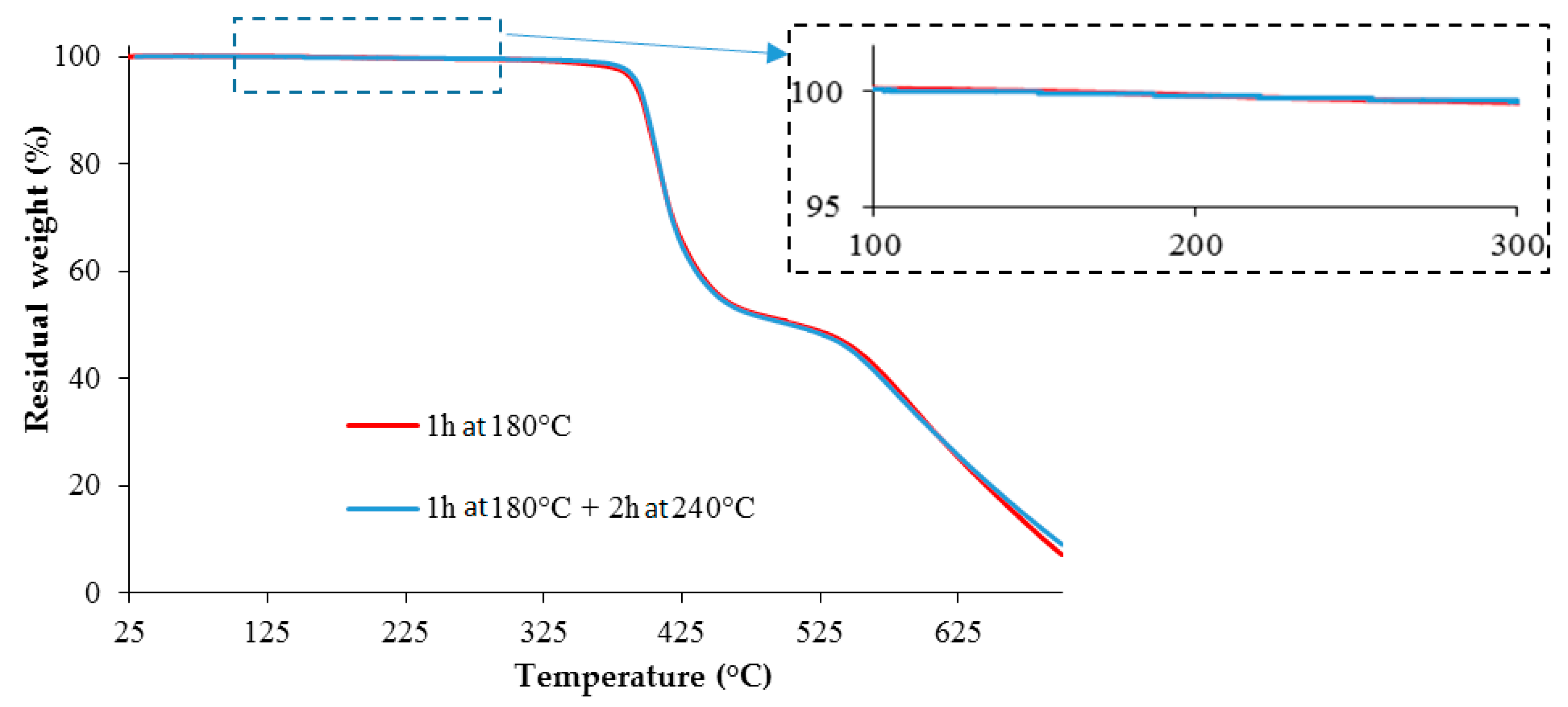
3.3. TETM/DDS Cure Cycle Optimization
- C1: 1 h at180 °C + 1 h at 240 °C
- C2: 1 h at 180 °C + 2 h at 240 °C
- C3: 2 h at 180 °C + 1 h at 240 °C
- C4: 2 h at 180 °C + 2 h at 240 °C
- C5: 3 h at 180 °C + 1 h at 240 °C
- C6: 3 h at 180 °C + 2 h at 240 °C
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TETM | Triglycidyl ether of tris(4-hydroxyphenyl)methane |
| DDS | Diaminodiphenylsulfone |
| DSC | Differential scanning calorimetry |
| TGA | Thermogravimetric analysis |
| TMA | Thermomecanical analysis |
| SEM | Scanning electronic microscopy |
References
- Mazzon, E.; Habas-Ulloa, A.; Habas, J.-P. Lightweight rigid foams from highly reactive epoxy resins derived from vegetable oil for automotive applications. Eur. Polym. J. 2015, 68, 546–557. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Jiang, T.; Zhang, C.; He, L. Cell morphology, bubbles migration, and flexural properties of non-uniform epoxy foams using chemical foaming agent. J. Appl. Polym. Sci. 2014, 131, 41175. [Google Scholar] [CrossRef]
- Luo, Y.; Zhang, J.; Qi, R.; Lu, J.; Hu, X.; Jiang, P. Polylactide foams prepared by a traditional chemical compression-molding method. J. Appl. Polym. Sci. 2013, 130, 330–337. [Google Scholar] [CrossRef]
- Huang, Q.; Klotzer, R.; Seibig, B.; Paul, D. Extrusion of microcellular polysulfone using chemical blowing agents. J. Appl. Polym. Sci. 1998, 69, 1753–1760. [Google Scholar] [CrossRef]
- Rutz, B.H.; Berg, J.C. A review of the feasibility of lightening structural polymeric composites with voids without compromising mechanical properties. Adv. Colloid Interface Sci. 2010, 160, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, C.; Basso, M.C.; Pizzi, A.; Laborie, M.-P.; Celzard, A.; Fierro, V. Pine tannin-based rigid foams: Mechanical and thermal properties. Ind. Crops Prod. 2013, 43, 45–250. [Google Scholar] [CrossRef]
- Basso, M.C.; Li, X.; Fierro, V.; Pizzi, A.; Giovando, S.; Celzard, A. Green, formaldehyde-free, foams for termal insulation. Adv. Mater. Lett. 2011, 2, 378. [Google Scholar] [CrossRef]
- Basso, M.C.; Lagel, M.-C.; Pizzi, A.; Celzard, A.; Abdalla, S. First tools for tannin-furanic foams design. BioResources 2015, 10, 5233–5241. [Google Scholar] [CrossRef]
- Tondi, G.; Fierro, V.; Pizzi, A.; Celzard, A. Tannin-based carbon foams. Carbon 2009, 47, 1480–1492. [Google Scholar] [CrossRef]
- Meikleham, N.E.; Pizzi, A. Acid- and alkali-catalyzed tannin-based rigid foams. J. Appl. Polym. Sci. 1994, 53, 1547–1556. [Google Scholar] [CrossRef]
- Sun, H.; Mark, J.E.; Tan, S.C.; Venkatasubramanian, N.; Houtz, M.D.; Arnold, F.E.; Lee, C.Y.-C. Microcellular foams from some high-performance composites. Polymer 2005, 46, 6623–6632. [Google Scholar] [CrossRef]
- Liu, T.; Liu, H.; Li, L.; Wang, X.; Lu, A.; Luo, S. Microstructure and properties of microcellular poly(phenylene sulfide) foams by mucell injection molding. Polym.-Plast. Technol. Eng. 2013, 52, 440–445. [Google Scholar] [CrossRef]
- Tomasko, D.L.; Li, H.; Liu, D.; Han, X.; Wingert, M.J.; Lee, L.J.; Koelling, K.W. A review of CO2 applications in the processing of polymers. Ind. Eng. Chem. Res. 2003, 42, 6431–6456. [Google Scholar] [CrossRef]
- Lacoste, C.; Basso, M.C.; Pizzi, A.; Celzard, A.; Laborie, M.-P. Natural albumin/tannin cellular foams. Ind. Crops Prod. 2015, 73, 41–48. [Google Scholar] [CrossRef]
- Li, X.; Pizzi, A.; Cangemi, M.; Navarrete, P.; Segovia, C.; Fierro, V.; Celzard, A. Insulation rigid and elastic foams based on albumin. Ind. Crops Prod. 2012, 37, 149–154. [Google Scholar] [CrossRef]
- Zhang, L.; Ma, J. Effect of carbon nanofiber reinforcement on mechanical properties of syntactic foam. Mater. Sci. Eng. A 2013, 574, 191–196. [Google Scholar] [CrossRef]
- Kishore, N.G.; Woldesenbet, E.; Sankaran, S. Studies on compressive failure features in syntactic foam material. J. Mater. Sci. 2001, 36, 4485–4491. [Google Scholar]
- Huang, Y.-J.; Wang, C.-H.; Huang, Y.-L.; Guo, G.; Nutt, S.R. Enhancing specific strength and stiffness of phenolic microsphere syntactic foams through carbon fiber reinforcement. Polym. Compos. 2010, 31, 256–262. [Google Scholar] [CrossRef]
- Burns, J.; Prime, R.; Barrall, E.; Oxsen, M. Chemistry of an epoxy-phenolic magnetic disk coating. In Polymers in Information Storage Technology; Springer: New York, NY, USA, 1988; pp. 237–256. [Google Scholar]
- Loera, A.G.; Cara, F.; Dumon, M.; Pascault, J.P. Porous epoxy thermosets obtained by a polymerization-induced phase separation process of a degradable thermoplastic polymer. Macromolecules 2002, 35, 6291–6297. [Google Scholar] [CrossRef]
- Li, X.; Basso, M.C.; Braghiroli, F.L.; Fierro, V.; Pizzi, A.; Celzard, A. Tailoring the structure of cellular vitreous carbon foams. Carbon 2012, 50, 2026–2036. [Google Scholar] [CrossRef]
- Basso, M.C.; Giovando, S.; Pizzi, A.; Celzard, A.; Fierro, V. Tannin/furanic foams without blowing agents and formaldehyde. Ind. Crops Prod. 2013, 49, 17–22. [Google Scholar] [CrossRef]
- Basso, M.C.; Pizzi, A.; Lacoste, C.; Delmotte, L.; Al-Marzouki, F.M.; Abdalla, S.; Celzard, A. MALDI-TOF and 13C NMR analysis of tannin–furanic–polyurethane foams adapted for industrial continuous lines application. Polymers 2014, 6, 2985–3004. [Google Scholar] [CrossRef]
- Doğan, E.; Küsefoğlu, S. Synthesis and in situ foaming of biodegradable malonic acid ESO polymers. J. Appl. Polym. Sci. 2008, 110, 1129–1135. [Google Scholar] [CrossRef]
- Stefani, P.M.; Barchi, A.T.; Sabugal, J.; Vazquez, A. Characterization of epoxy foams. J. Appl. Polym. Sci. 2003, 90, 2992–2996. [Google Scholar] [CrossRef]
- Wan Hamad, W.N.F.; Teh, P.L.; Yeoh, C.K. Effect of acetic acid as catalyst on the properties of epoxy foam. Polym.-Plast. Technol. Eng. 2013, 52, 754–760. [Google Scholar] [CrossRef]
- Najib, N.N.; Ariff, Z.M.; Bakar, A.A.; Sipaut, C.S. Correlation between the acoustic and dynamic mechanical properties of natural rubber foam: Effect of foaming temperature. Mater. Des. 2011, 32, 505–511. [Google Scholar] [CrossRef]
- Morehouse, D.S. Expansible thermoplastic polymer particles containing volatile fluid foaming agent and method of foaming the same. US Patent 3615972 A, 26 October 1971. [Google Scholar]
- Jonsson, M.; Nordin, O.; Malmström, E. Increased onset temperature of expansion in thermally expandable microspheres through combination of crosslinking agents. J. Appl. Polym. Sci. 2011, 121, 369–375. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Itamura, Y.; Onimura, K.; Oishi, T. Effects of the chemical structure on the heat resistance of thermoplastic expandable microspheres. J. Appl. Polym. Sci. 2005, 96, 1306–1312. [Google Scholar] [CrossRef]
- Expancel Products—AkzoNobel Expancel—The multi-performance additive. Available online: https://www.akzonobel.com/expancel/products/ (accessed on 19 February 2016).
- Tomalino, M.; Bianchini, G. Heat-expandable microspheres for car protection production. Prog. Org. Coat. 1997, 32, 17–24. [Google Scholar] [CrossRef]
- Rumeau, N.; Buisson, A.; Trouillot, P. Novel composite materials and methods for manufacturing same. U.S. Patent 20140220841 A1, 25 August 2011. [Google Scholar]
- Samira, E.G. Influence de la structure chimique et des propriétés physiques d’un système époxyde sur la formation et la morphologie de structures poreuses. In Proceeding of the Matériaux 2014, Montpellier, France, 24–28 November 2014.
- Pascault, J.-P.; Sautereau, H.; Verdu, J.; Williams, R.J.J. Thermosetting Polymers; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Nakka, J.S.; Jansen, K.M.B.; Ernst, L.J. Tailoring the viscoelasticity of epoxy thermosets. J. Appl. Polym. Sci. 2013, 128, 3794–3806. [Google Scholar] [CrossRef]
- Halary, J.L. Structure–property relationships in epoxy-amine networks of well-controlled architecture. High. Perform. Polym. 2000, 12, 141–153. [Google Scholar] [CrossRef]
- Nakka, J.S.; Jansen, K.M.B.; Ernst, L.J. Effect of chain flexibility in the network structure on the viscoelasticity of epoxy thermosets. J. Polym. Res. 2011, 18, 1879–1888. [Google Scholar] [CrossRef]
- Pearce, E.M. Properties of polymers, their estimation and correlation with chemical structure, 2nd ed.; Van Krevelen, D.W., Nijenhuis, K., Eds.; Elsevier: New York, NY, USA, 1976. [Google Scholar]
- Shieh, J.-Y.; Ho, T.-H.; Wang, C.-S. Synthesis and modification of a trifunctional epoxy resin with amino-terminated poly(dimethylsiloxane)s for semiconductor encapsulation. Angew. Makromol. Chem. 1997, 245, 125–137. [Google Scholar] [CrossRef]
- Aronhime, M.T.; Gillham, J.K. Time-temperature-transformation (TTT) cure diagram of thermosetting polymeric systems. Adv. Polym. Sci. 1986, 78, 83–113. [Google Scholar]
- Wang, L.; Yang, X.; Zhang, J.; Zhang, C.; He, L. The compressive properties of expandable microspheres/epoxy foams. Compos. Part. B Eng. 2014, 56, 724–732. [Google Scholar] [CrossRef]
- Rose, N.; Le Bras, M.; Delobel, R.; Costes, B.; Henry, Y. Thermal oxidative degradation of an epoxy resin. Polym. Degrad. Stab. 1993, 42, 307–316. [Google Scholar] [CrossRef]
- Jonsson, M.; Nordin, O.; Kron, A.L.; Malmström, E. Thermally expandable microspheres with excellent expansion characteristics at high temperature. J. Appl. Polym. Sci. 2010. [Google Scholar] [CrossRef]
- Katircioğlu, T.Y.; Güven, O. Glass-transition behavior of poly(methacrylonitrile). J. Appl. Polym. Sci. 2001, 82, 1936–1943. [Google Scholar] [CrossRef]
- Mark, J.E. Polymer Data Handbook; Oxford University: Oxford, UK, 1999. [Google Scholar]
- Korobeinyk, A.V.; Whitby, R.L.D.; Mikhalovsky, S.V. High temperature oxidative resistance of polyacrylonitrile-methylmethacrylate copolymer powder converting to a carbonized monolith. Eur. Polym. J. 2012, 48, 97–104. [Google Scholar] [CrossRef]
- Bashir, Z.; Packer, E.J.; Herbert, I.R.; Price, D.M. Base-induced gelation of polymethacrylonitrile. Polymer 1992, 33, 373–378. [Google Scholar] [CrossRef]
- Park, C.B.; Behravesh, A.H.; Venter, R.D. Low density microcellular foam processing in extrusion using CO2. Polym. Eng. Sci. 1998, 38, 1812–1823. [Google Scholar] [CrossRef]
- Klempner, D.; Sendijareviʹc, V.; Aseeva, R.M. Handbook of Polymeric Foams and Foam Technology; Hanser Verlag: Munich, Germany, 2004. [Google Scholar]
- Matuana, L.M.; Park, C.B.; Balatinecz, J.J. Processing and cell morphology relationships for microcellular foamed PVC/wood-fiber composites. Polym. Eng. Sci. 1997, 37, 1137–1147. [Google Scholar] [CrossRef]
- Pötschke, P.; Krause, B.; Stange, J.; Munstedt, H. Elongational viscosity and foaming behavior of PP modified by electron irradiation or nanotube addition. Macromol. Symp. 2007, 254, 400–408. [Google Scholar] [CrossRef]
- Bresson, G.; Jumel, J.; Shanahan, M.E.R.; Serin, P. Statistical aspects of the mechanical behaviour a paste adhesive. Int. J. Adhes. Adhes. 2013, 40, 70–79. [Google Scholar] [CrossRef]
- Nassiet, V.; Habas, J.P.; Hassoune-Rhabbour, B.; Baziard, Y.; Petit, J.A. Correlation between viscoelastic behavior and cooling stresses in a cured epoxy resin system. J. Appl. Polym. Sci. 2006, 99, 679–690. [Google Scholar] [CrossRef]
- Grassie, N.; Scott, G. Polymer degradation and stabilisation. Polymer 1988, 29, 1725. [Google Scholar]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mixing temperature | Gel time (min) | ||||||
|---|---|---|---|---|---|---|---|
| 130 °C | 140 °C | 150 °C | 160 °C | 170 °C | 180 °C | 190 °C | |
| 100 °C | 65.6 ± 4 | 46.3 ± 3.5 | 33.8 ± 2.2 | 18.3 ± 1 | 10.3 ± 1 | 6.1 ± 0.9 | 3.3 ± 0.8 |
| 180 °C | 58.6 ± 4 | 41 ± 3.3 | 24.3 ± 2 | 13.6 ± 1 | 8.6 ± 1 | 5.0 ± 0.8 | 3.0 ± 0.8 |
| Temperature | 160 °C | 170 °C | 180 °C | 190 °C | 200 °C |
|---|---|---|---|---|---|
| Density (kg·m−3) | 909 ± 14 | 797 ± 22 | 676 ± 27 | 495 ± 38 | 462 ± 42 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Gazzani, S.; Nassiet, V.; Habas, J.-P.; Freydier, C.; Hilleshein, A. High Temperature Epoxy Foam: Optimization of Process Parameters. Polymers 2016, 8, 215. https://doi.org/10.3390/polym8060215
El Gazzani S, Nassiet V, Habas J-P, Freydier C, Hilleshein A. High Temperature Epoxy Foam: Optimization of Process Parameters. Polymers. 2016; 8(6):215. https://doi.org/10.3390/polym8060215
Chicago/Turabian StyleEl Gazzani, Samira, Valérie Nassiet, Jean-Pierre Habas, Christian Freydier, and Aline Hilleshein. 2016. "High Temperature Epoxy Foam: Optimization of Process Parameters" Polymers 8, no. 6: 215. https://doi.org/10.3390/polym8060215
APA StyleEl Gazzani, S., Nassiet, V., Habas, J. -P., Freydier, C., & Hilleshein, A. (2016). High Temperature Epoxy Foam: Optimization of Process Parameters. Polymers, 8(6), 215. https://doi.org/10.3390/polym8060215