
Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Solution Treatments
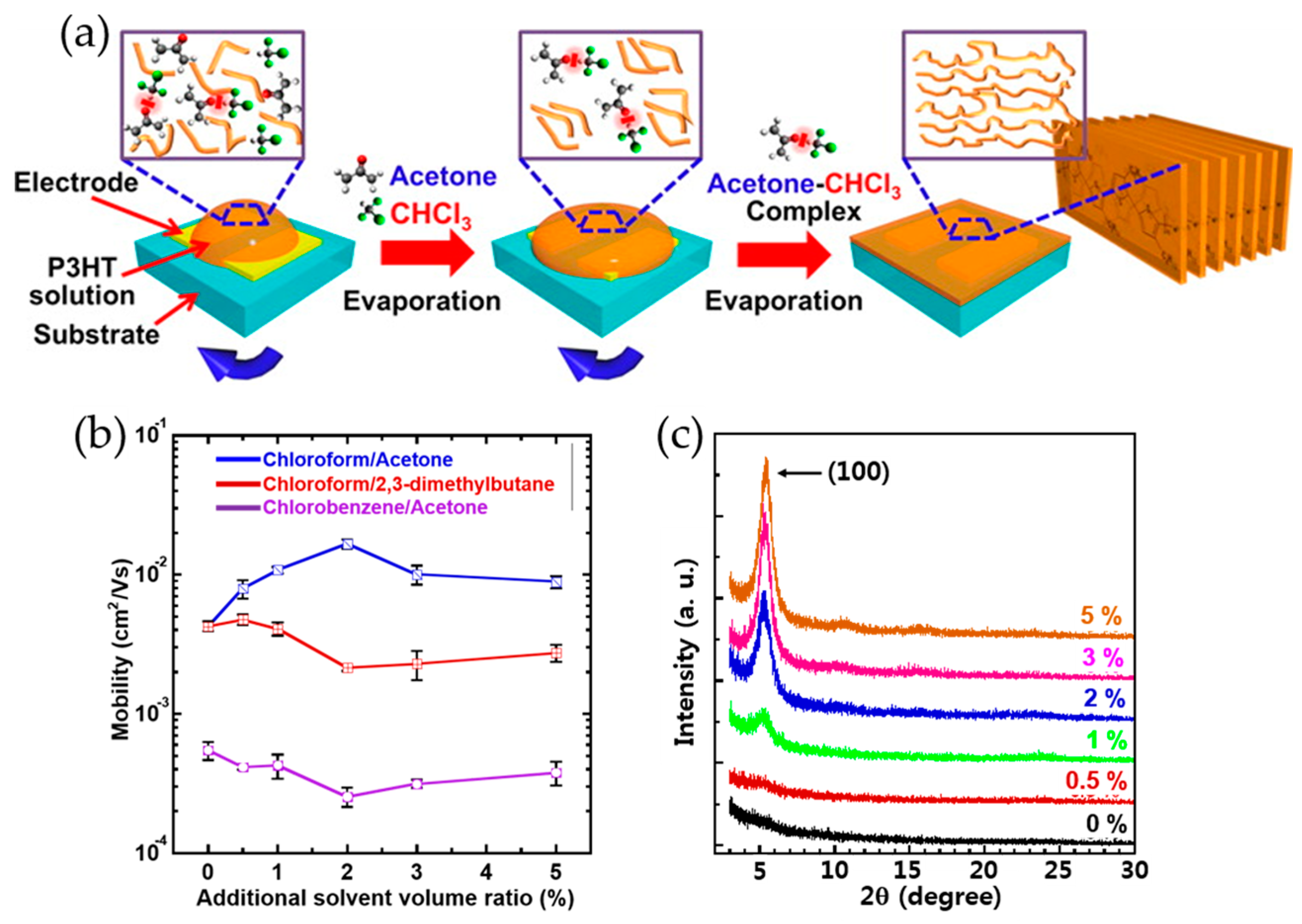
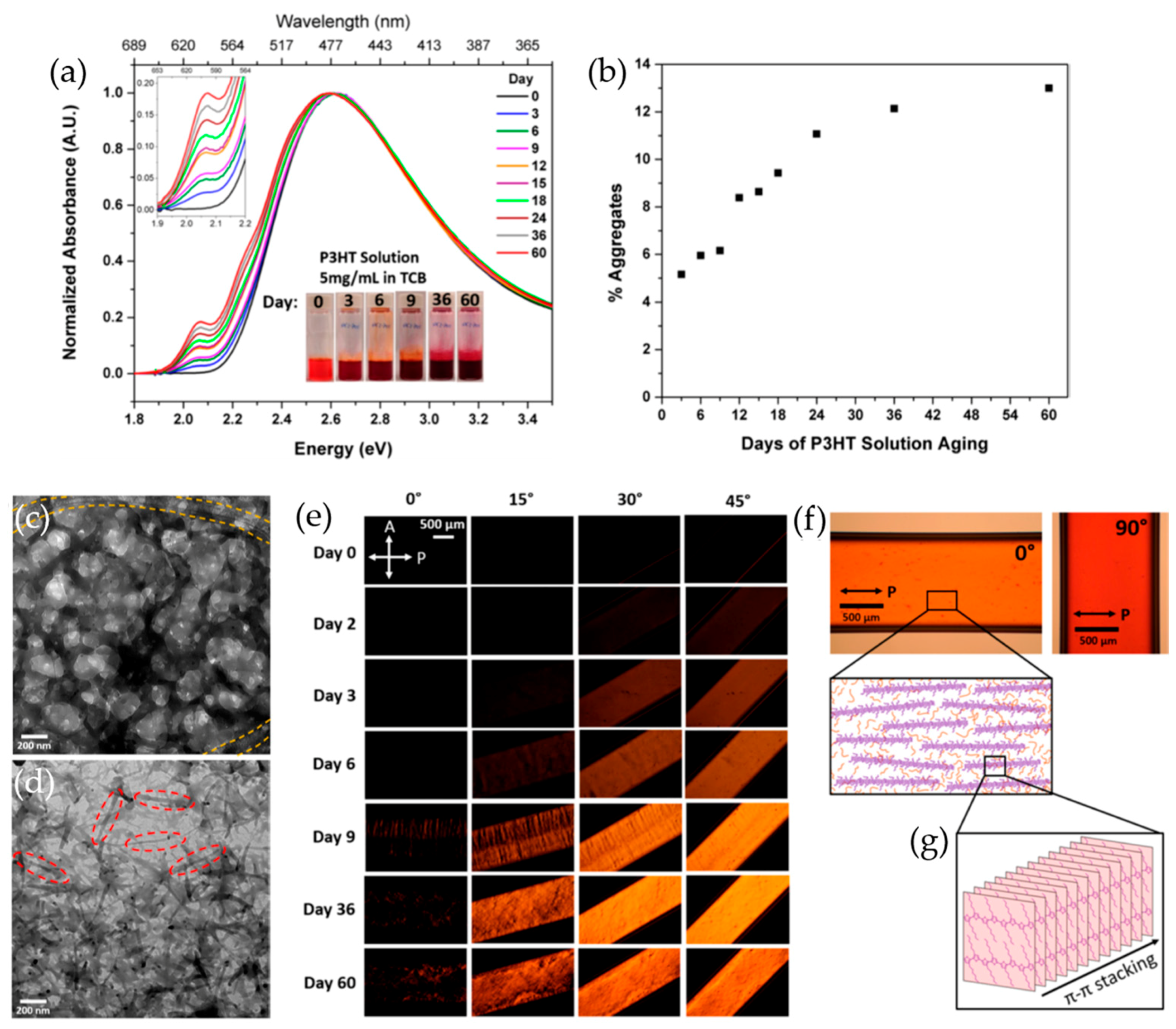
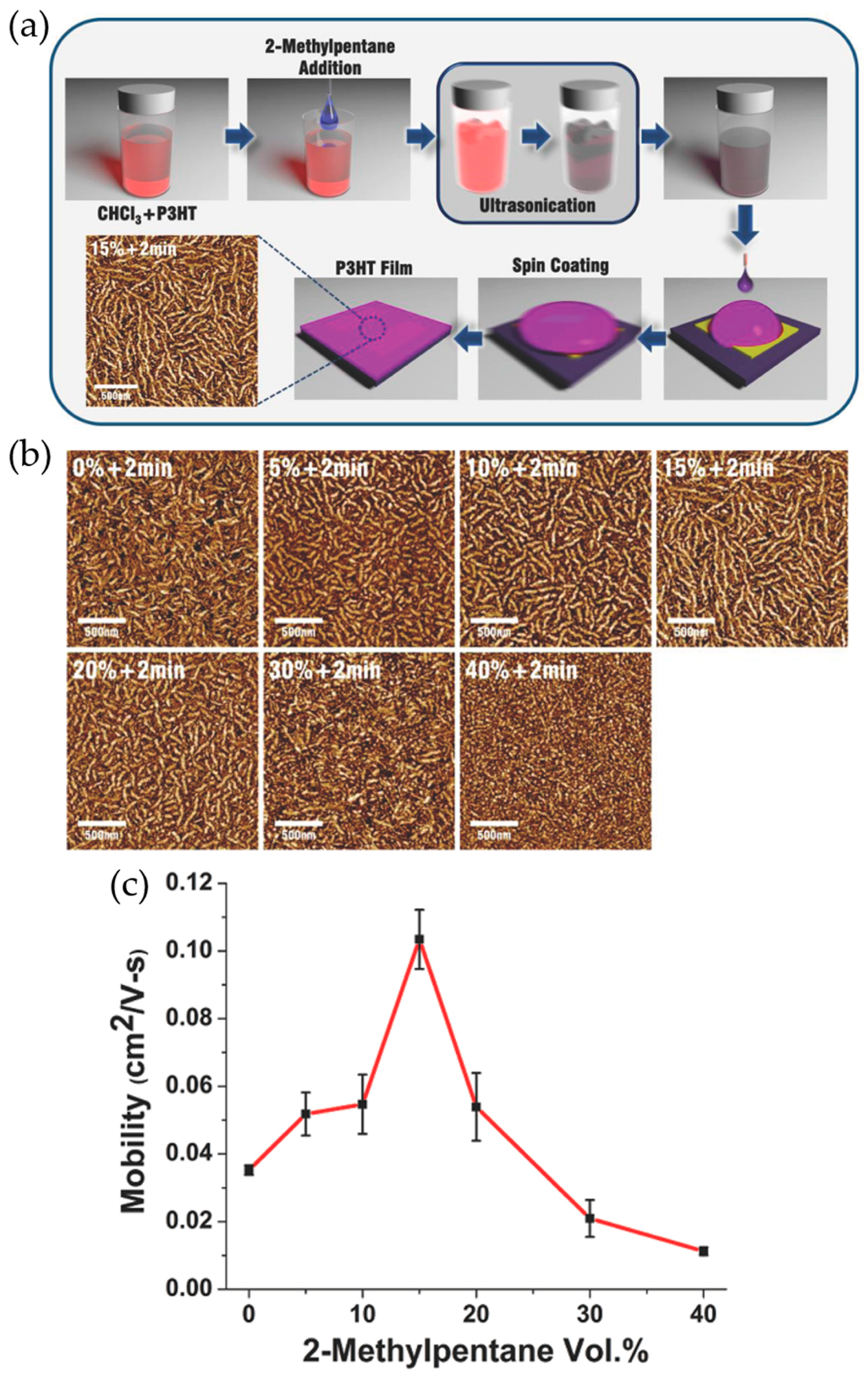
2.1. Solvent Solubility Tuning
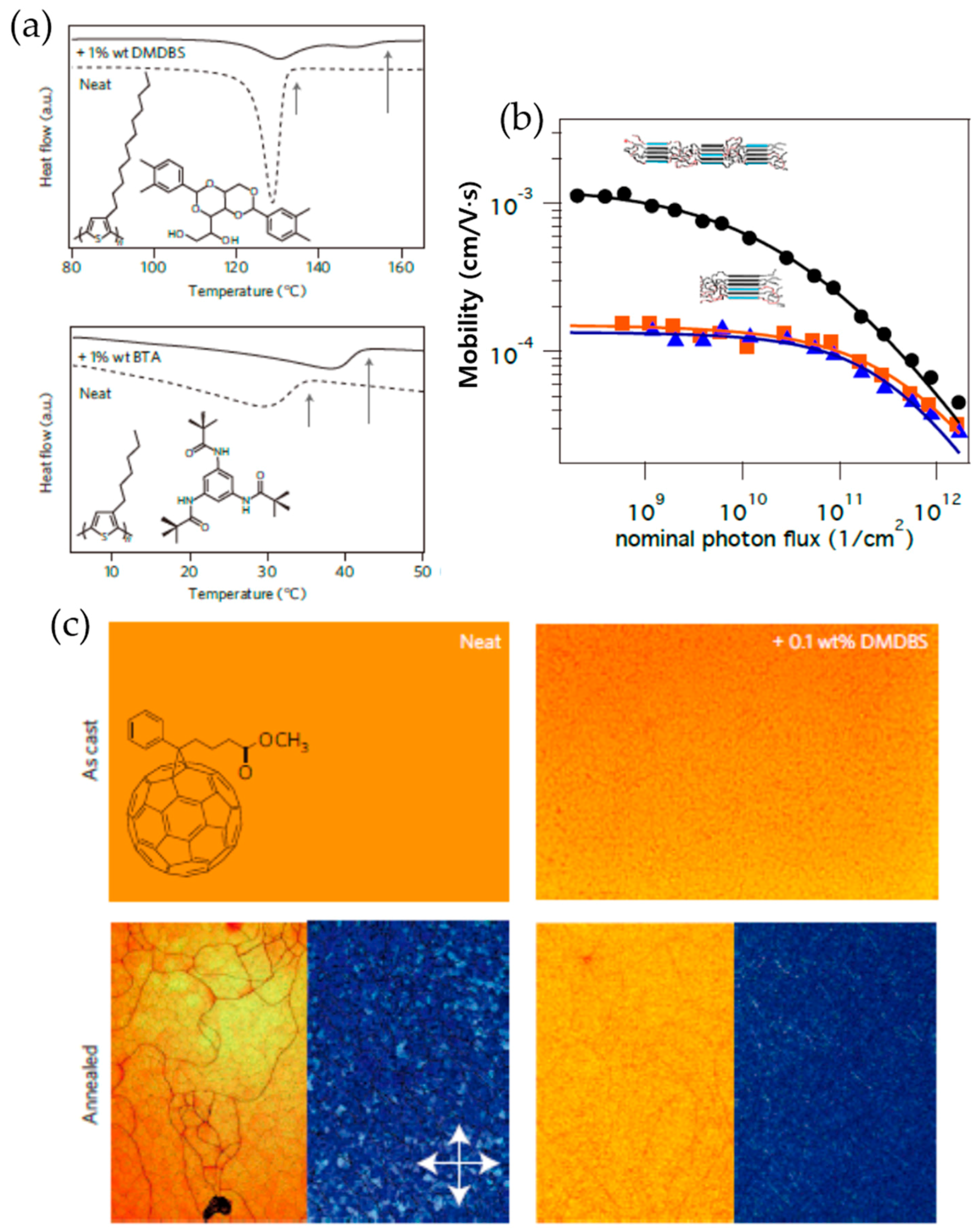
2.2. Nucleation-Inducing Agent
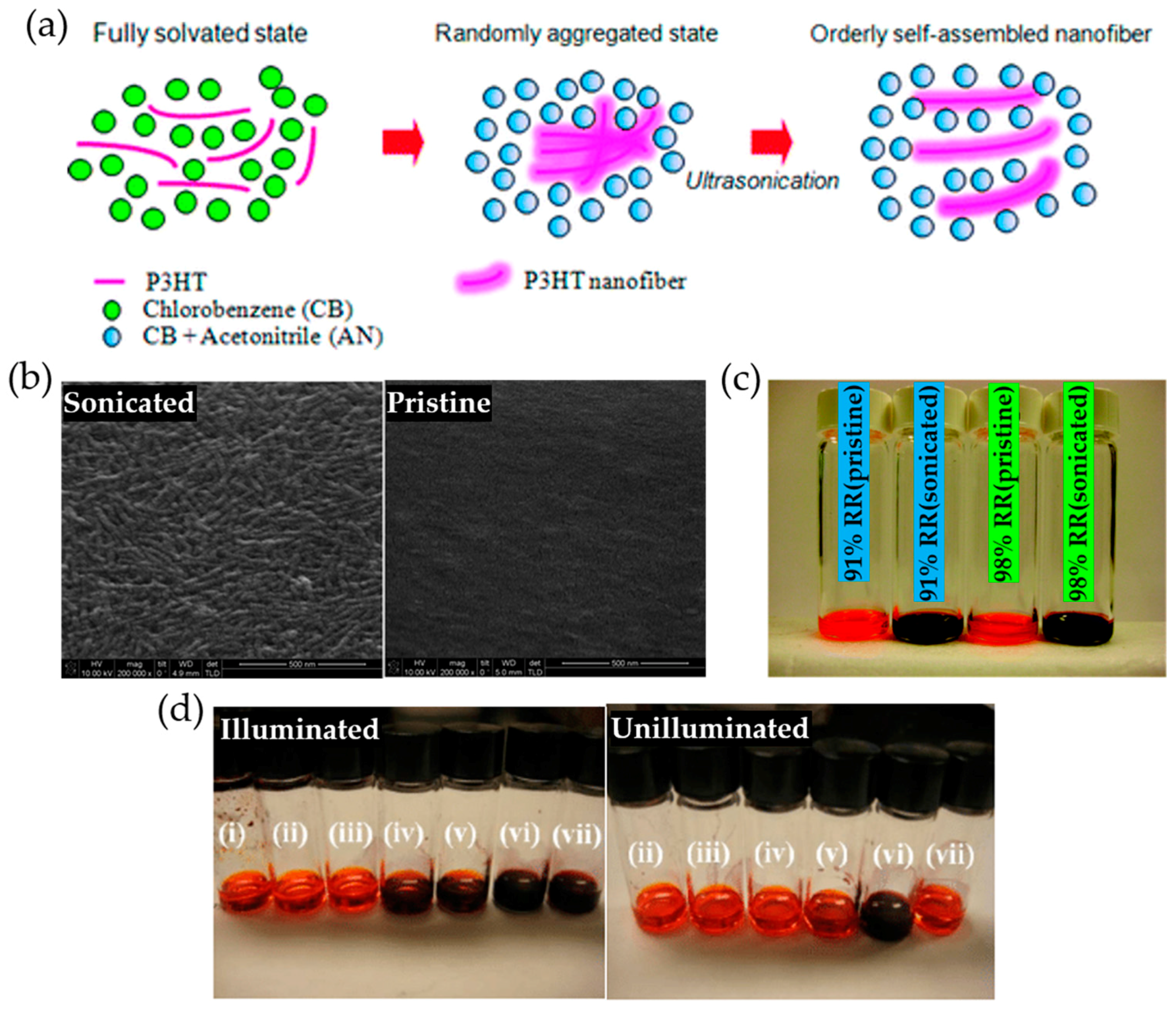
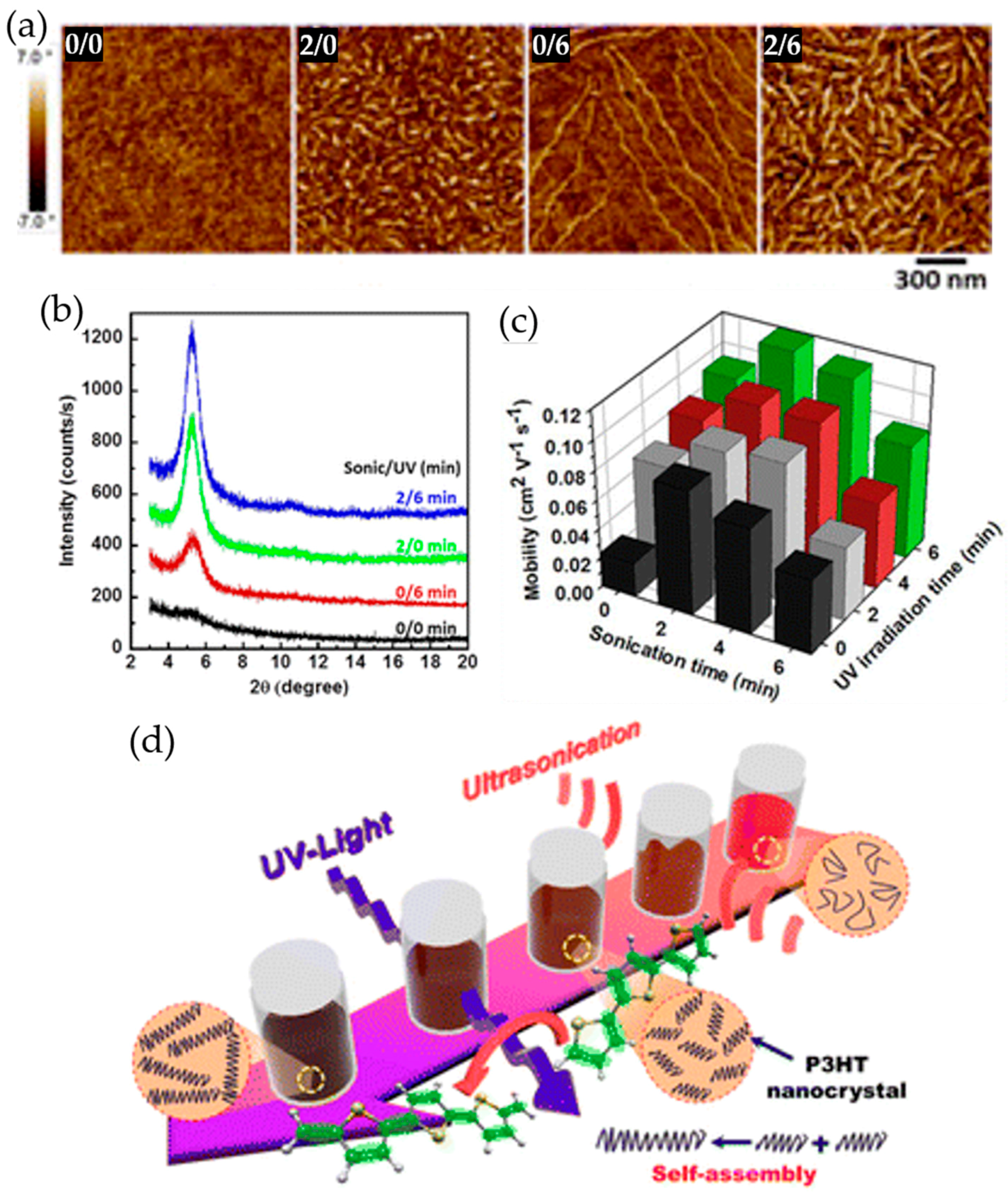
2.3. Ultrasonic Treatment
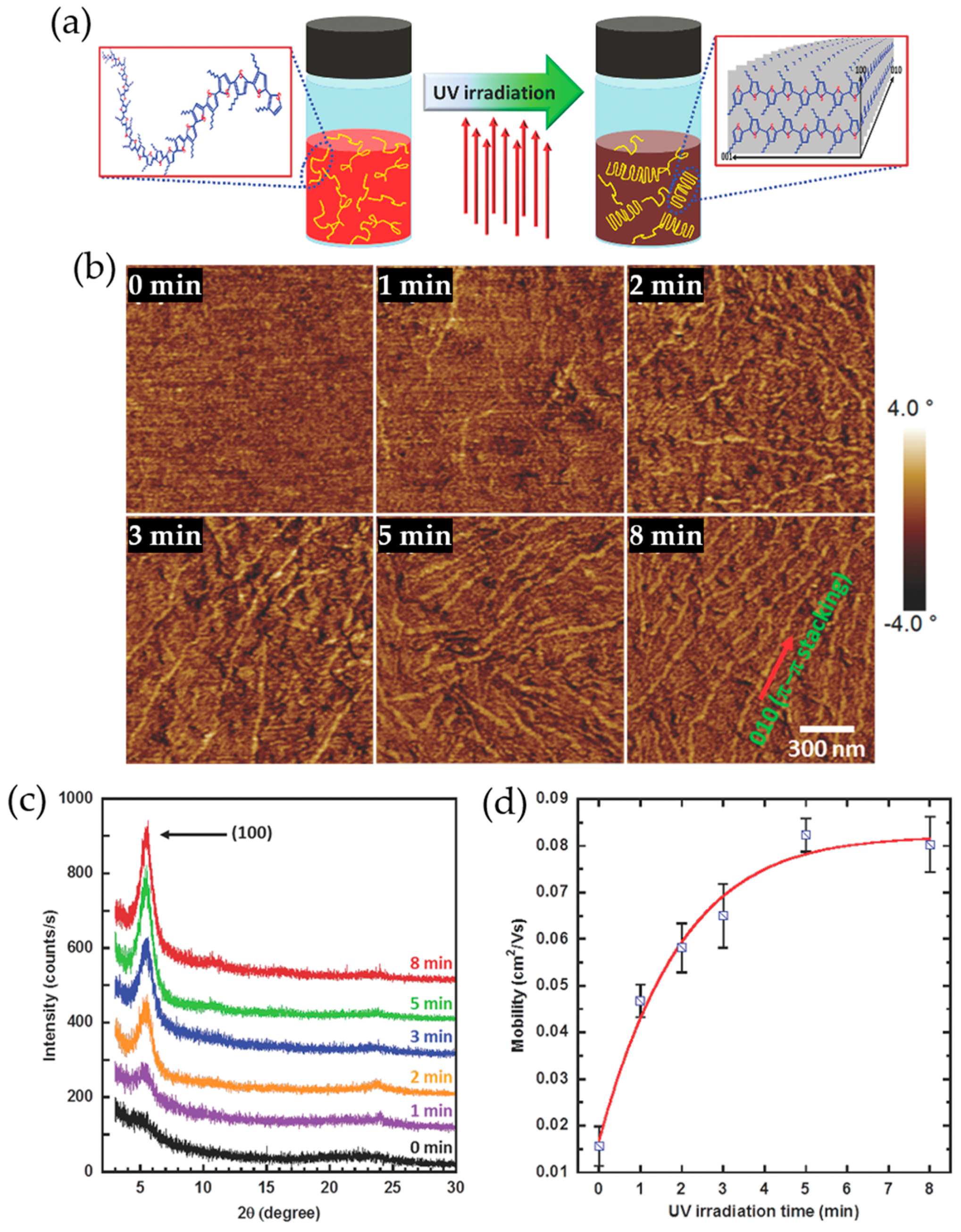
2.4. UV Irradiation
3. Film Deposition Techniques
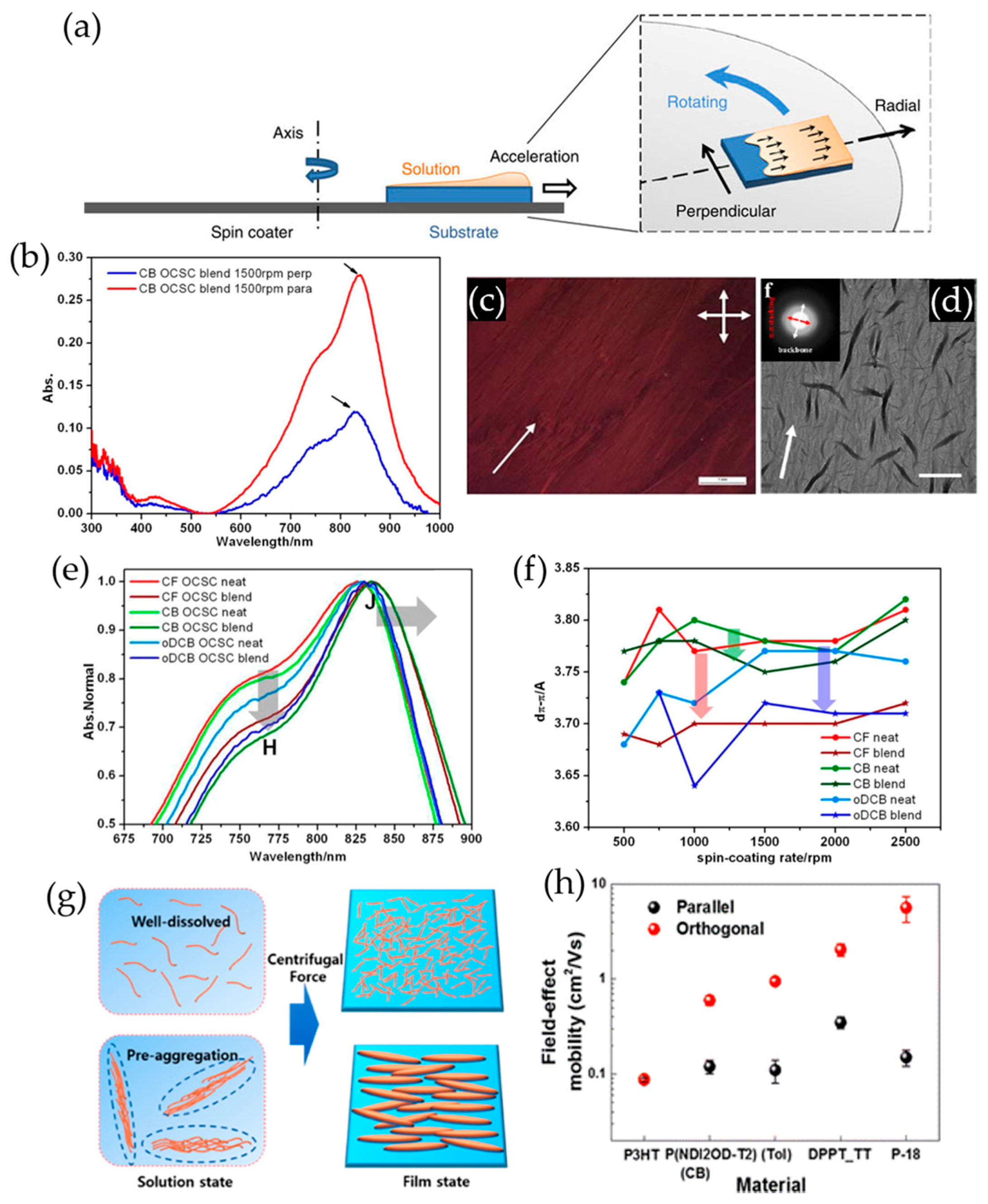
3.1. Template-Guided Solution-Shear Coating
3.2. Capillary Force-Assisted Film Deposition
3.3. Centrifugal Force-Driven Film Deposition
3.4. Mechanical Force-Assisted Film Alignment
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kim, B.G.; Jeong, E.J.; Chung, J.W.; Seo, S.; Koo, B.; Kim, J.S. Molecular Design Principle of Lyotropic Liquid-Crystalline Conjugated Polymers with Directed Alignment Capability for Plastic Electronics. Nat. Mater. 2013, 12, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, M.; Yamada, T.; Matsui, H.; Minemawari, H.; Tsutsumi, J.; Horii, Y.; Chikamatsu, M.; Azumi, R.; Kumai, R.; Hasegawa, T. Simple Push Coating of Polymer Thin-Film Transistors. Nat. Commun. 2012, 3, 1176. [Google Scholar] [CrossRef] [PubMed]
- Sirringhaus, H. Device Physics of Solution-Processed Organic Field Effect Transistors. Adv. Mater. 2005, 17, 2411–2425. [Google Scholar] [CrossRef]
- Knopfmacher, O.; Hammock, M.L.; Appleton, A.L.; Schwartz, G.; Mei, J.; Pei, J.; Bao, Z. Highly Stable Organic Polymer Field-Effect Transistor Sensor for Selective Detection in the Marine Environment. Nat. Commun. 2014, 5, 2954. [Google Scholar] [CrossRef] [PubMed]
- Noriega, R.; Rivnay, J.; Vandewal, K.; Koch, F.P.V.; Stingelin, N.; Smith, P.; Toney, M.F.; Salleo, A.A. General Relationship between Disorder, Aggregation and Charge Transport in Conjugated Polymers. Nat. Mater. 2013, 12, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef] [PubMed]
- Tessler, N.; Medvedev, V.; Kazes, M.; Kan, S.; Banin, U. Efficient Near-Infrared Polymer Nanocrystal Light-Emitting Diodes. Science 2002, 295, 1506–1508. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, A.; Dam, H.F.; Krebs, F.C.; Edman, L. Ambient Fabrication of Flexible and Large-Area Organic Light-Emitting Devices Using Slot-Die Coating. Nat. Commun. 2012, 3, 1002. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, L.; Niu, X.; Yu, Z.; Pei, Q. Elastomeric Polymer Light-Emitting Devices and Displays. Nat. Photonics 2013, 7, 817–824. [Google Scholar] [CrossRef]
- Zaumseil, J.; Friend, R.H.; Sirringhaus, H. Spatial Control of the Recombination Zone in an Ambipolar Light-Emitting Organic Transistor. Nat. Mater. 2006, 5, 69–74. [Google Scholar] [CrossRef]
- Scharber, M.C. On the Efficiency Limit of Conjugated Polymer: Fullerene-Based Bulk Heterojunction Solar Cells. Adv. Mater. 2016, 28, 1994–2001. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Sun, C.; Dong, S.; Jiang, X.F.; Wu, S.; Wu, H.; Yip, H.L.; Huang, F.; Cao, Y. n-Type Water/alcohol-Soluble Naphthalene Diimide-Based Conjugated Polymers for High-Performance Polymer Solar Cells. J. Am. Chem. Soc. 2016, 138, 2004–2013. [Google Scholar] [CrossRef] [PubMed]
- Peet, J.; Kim, J.Y.; Coates, N.E.; Ma, L.; Moses, D.; Heeger, A.J.; Bazan, G.C. Efficiency Enhancement in Low-Bandgap Polymer Solar Cells by Processing with Alkane Dithiols. Nat. Mater. 2007, 6, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhu, R.; Yang, Y. Polymer Solar Cells. Nat. Photonics 2012, 6, 153–161. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, K.; Coates, N.; Moses, D.; Nguyen, T.Q.; Dante, M.; Heeger, A.J. Efficient Tandem Polymer Solar Cells Fabricated by All-Solution Processing. Science 2007, 317, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, J.; Li, Z.; Mu, C.; Ma, W.; Hu, H.; Jiang, K.; Lin, H.; Ade, H.; Yan, H. Aggregation and Morphology Control Enables Multiple Cases of High-Efficiency Polymer Solar Cells. Nat. Commun. 2014, 5, 5293. [Google Scholar] [CrossRef] [PubMed]
- Mandoc, M.M.; de Boer, B.; Paasch, G.; Blom, P.W. Trap-Limited Electron Transport in Disordered Semiconducting Polymers. Phys. Rev. B 2007, 75, 193202. [Google Scholar] [CrossRef]
- Salleo, A.; Kline, R.J.; DeLongchamp, D.M.; Chabinyc, M.L. Microstructural Characterization and Charge Transport in Thin Films of Conjugated Polymers. Adv. Mater. 2010, 22, 3812–3838. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Hirate, M.; Watanabe, S.; Kuroda, S.-I. Microscopic Signature of Metallic State in Semicrystalline Conjugated Polymers Doped with Fluoroalkylsilane Molecules. Adv. Mater. 2014, 26, 2376–2383. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.F.; Sirringhaus, H.; Giles, M.; Heeney, M.; McCulloch, I. Relative Importance of Polaron Activation and Disorder on Charge Transport in High-Mobility Conjugated Polymer Field-Effect Transistors. Phys. Rev. B 2007, 76, 205204. [Google Scholar] [CrossRef]
- Rivnay, J.; Noriega, R.; Northrup, J.E.; Kline, R.J.; Toney, M.F.; Salleo, A. Structural Origin of Gap States in Semicrystalline Polymers and the Implications for Charge Transport. Phys. Rev. B 2011, 83, 121306. [Google Scholar] [CrossRef]
- Jimison, L.H.; Toney, M.F.; McCulloch, I.; Heeney, M.; Salleo, A. Charge-Transport Anisotropy Due to Grain Boundaries in Directionally Crystallized Thin Films of Regioregular Poly(3-hexylthiophene). Adv. Mater. 2009, 21, 1568–1572. [Google Scholar] [CrossRef]
- Huynh, W.U.; Dittmer, J.J.; Libby, W.C.; Whiting, G.L.; Alivisatos, A.P. Controlling the Morphology of Nanocrystal-Polymer Composites for Solar Cells. Adv. Funct. Mater. 2003, 13, 73–79. [Google Scholar] [CrossRef]
- Campoy-Quiles, M.; Ferenczi, T.; Agostinelli, T.; Etchegoin, P.G.; Kim, Y.; Anthopoulos, T.D.; Stavrinou, P.N.; Bradley, D.D.C.; Nelson, J. Morphology Evolution via Self-Organization and Lateral and Vertical Diffusion in Polymer:Fullerene Solar Cell Blends. Nat. Mater. 2008, 7, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Oosterhout, S.D.; Wienk, M.M.; van Bavel, S.S.; Thiedmann, R.; Koster, L.J.A.; Gilot, J.; Loos, J.; Schmidt, V.; Janssen, R.A. The Effect of Three-Dimensional Morphology on the Efficiency of Hybrid Polymer Solar Cells. Nat. Mater. 2009, 8, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, C.-Y.; Rose, B.D.; Jiang, Y.; Chang, M.; Chu, P.-H.; Yuan, Z.; Fuentes-Hernandez, C.; Bernard, K.; Bredas, J.-L.; et al. Molecular Engineering of Non-Halogenated Solution-Processable Bithiazole Based Electron Transport Polymeric Semiconductors. Chem. Mater. 2015, 27, 2928–2937. [Google Scholar] [CrossRef]
- Chu, P.-H.; Wang, G.; Fu, B.; Choi, D.; Park, J.O.; Srinivasarao, M.; Reichmanis, E. Synergistic Effect of Regioregular and Regiorandom Poly(3-hexylthiophene) Blends for High Performance Flexible Organic Field Effect Transistors. Adv. Electron. Mater. 2016, 2, 1500384. [Google Scholar] [CrossRef]
- Diao, Y.; Shaw, L.; Bao, Z.; Mannsfeld, S.C.B. Morphology Control Strategies for Solution-Processed Organic Semiconductor Thin Films. Energy Environ. Sci. 2014, 7, 2145–2159. [Google Scholar] [CrossRef]
- Mei, J.; Dial, Y.; Appleton, A.L.; Fang, L.; Bao, Z. Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors. J. Am. Chem. Soc. 2013, 135, 6724–6746. [Google Scholar] [CrossRef] [PubMed]
- Di, C.-A.; Lu, K.; Zhang, L.; Liu, Y.; Guo, Y.; Sun, X.; Wen, Y.; Yu, G.; Zhu, D. Solvent-Assisted Re-annealing of Polymer Films for Solution –Processable Organic Field-Effect Transistors. Adv. Mater. 2010, 22, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.-R.; Phan, H.; Luo, C.; Wang, M.; Perez, L.A.; Patel, S.N.; Ying, L.; Kramer, E.J.; Nguyen, T.-Q.; Bazan, G.C.; et al. High-Mobility Field-Effect Transistors Fabricated with Macroscopic Aligned Semiconducting Polymers. Adv. Mater. 2014, 26, 2993–2998. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, A.; Yoon, M.-H.; Marks, T.J. Gate Dielectrics for Organic Field-Effect Transistors: New Opportunities for Organic Electronics. Adv. Mater. 2005, 17, 1705–1725. [Google Scholar] [CrossRef]
- Calhoun, M.F.; Sanchez, J.; Olaya, D.; Gershenson, M.E.; Podzorov, V. Electronic Functionalization of the Surface of Organic Semiconductors with Self-Assembled Monolayers. Nat. Mater. 2008, 7, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Jager, C.M.; Schmaltz, T.; Novak, M.; Khassanov, A.; Vorobiev, A.; Hennemann, M.; Krause, A.; Dietrich, H.; Zahn, D.; Hirsch, A.; et al. Improving the Charge Transport in Self-Assembled Monolayer Field-Effect Transistors: From Theory to Devices. J. Am. Chem. Soc. 2013, 135, 4893–4900. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.D.; Lim, J.A.; Lee, H.S.; Cho, K. Interface Engineering in Organic Transistors. Mater. Today 2007, 10, 46–54. [Google Scholar] [CrossRef]
- Fu, B.; Baltazar, J.; Sankar, A.R.; Chu, P.-H.; Zhang, S.; Collard, D.M.; Reichmanis, E. Enhancing Field-Effect Mobility of Conjugated Polymers Through Rational Design of Branched Side Chains. Adv. Funct. Mater. 2014, 24, 3734–3744. [Google Scholar] [CrossRef]
- Zen, A.; Pflaum, J.; Hirschmann, S.; Zhuang, W.; Jaiser, F.; Asawapirom, U.; Rabe, J.P.; Scherf, U.; Neher, D. Effect of Molecular Weight and Annealing of Poly(3-hexylthiophene)s on the Performance of Organic Field-Effect Transistors. Adv. Funct. Mater. 2004, 14, 757–764. [Google Scholar] [CrossRef]
- Salammal, S.T.; Mikayelyan, E.; Grigorian, S.; Pietsch, U. Impact of Thermal Annealing on the Semicrystalline Nanomorphology of Spin-Coated Thin Films of Regioregular Poly(3-alkylthiophene)s as Observed by High-Resolution Transmission Electron Microscopy and Grazing Incidence X-ray Diffraction. Macromolecules 2012, 45, 5575–5585. [Google Scholar] [CrossRef]
- Sun, K.; Xiao, Z.; Lu, S.; Zajaczkowski, W.; Pisula, W.; Hanssen, E.; White, J.M.; Williamson, R.M.; Subbiah, J.; Ouyang, J.; et al. A Molecular Nematic Liquid Crystalline Material for High-Performance Organic Photovoltaics. Nat. Commun. 2015, 6, 6013. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-Y.; Yang, H.; Choi, S.-K.; Jang, S.-Y. Efficient Solvent-Assisted Post-Treatment for Molecular Rearrangement of Sprayed Polymer Field-Effect Transistors. ACS Appl. Mater. Interfaces 2012, 4, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Park, C.; Park, J.E.; Chu, K.; Choi, H.C. Vertical Crystallization of C60 Nanowires by Solvent Vapor Annealing Process. ACS Nano 2013, 7, 9122–9128. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-F.; Sun, B.; Breiby, D.W.; Nielsen, M.M.; Solling, T.I.; Giles, M.; McCulloch, I.; Sirringhaus, H. Enhanced Mobility of Poly(3-hexylthiophene) Transistors by Spin-Coating from High-Boiling-Point Solvents. Chem. Mater. 2004, 16, 4772–4776. [Google Scholar] [CrossRef]
- Aiyar, A.R.; Hong, J.-I.; Reichmanis, R. Regioregularity and Intrachain Ordering: Impact on the Nanostructure and Charge Transport in Two-Dimensional Assemblies of Poly(3-hexylthiophene). Chem. Mater. 2012, 24, 2845–2853. [Google Scholar] [CrossRef]
- DeLongchamp, D.M.; Vogel, B.M.; Jung, Y.; Gurau, M.C.; Richter, C.A.; Kirillov, O.A.; Obrzut, J.; Fischer, D.A.; Sambasivan, S.; Richter, L.J.; et al. Variations in Semiconducting Polymer Microstructure and Hole Mobility with Spin-Coating Speed. Chem. Mater. 2005, 17, 5610–5612. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.; Lee, H.H. High-Mobility Polymer Thin-Film Transistors Fabricated by Solvent-Assisted Drop-Casting. Org. Electron. 2006, 7, 256–260. [Google Scholar] [CrossRef]
- Park, M.S.; Ayiar, A.; Park, J.O.; Reichmanis, E.; Srinivasarao, M. Solvent Evaporation Induced Liquid Crystalline Phase in Poly(3-hexylthiophene). J. Am. Chem. Soc. 2011, 133, 7244–7247. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Aiyar, A.; Park, M.S.; Srinivasarao, M.; Reichmanis, E. Conducting Channel Formation in Poly(3-hexylthiophene) Field Effect Transistors: Bulk to Interface. J. Phys. Chem. C 2011, 115, 11719–11726. [Google Scholar] [CrossRef]
- Wang, S.; Kiersnowski, A.; Pisula, W.; Mullen, K. Microstructure Evolution and Device Performance in Solution-Processed Polymeric Field-Effect Transistors: The Key Role of the First Monolayer. J. Am. Chem. Soc. 2012, 134, 4015–4018. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, A.R.; Hong, J.-I.; Izumi, J.; Choi, D.; Kleinhenz, N.; Reichmanis, E. Ultrasound-Induced Ordering in Poly(3-hexylthiophene): Role of Molecular and Process Parameters on Morphology and Charge Transport. ACS Appl. Mater. Interfaces 2013, 5, 2368–2377. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, H.; Qiu, Z.; Zhang, S.-L.; Zhang, Z.-B. Small-Hysteresis Thin-Film Transistors Achieved by Facile Dip-Coating of Nanotube/Polymer Composite. Adv. Mater. 2012, 24, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Diao, Y.; Tee, B.C.-K.; Giri, G.; Xu, J.; Kim, D.H.; Becerril, H.A.; Stoltenberg, R.M.; Lee, T.H.; Xue, G.; Mannsfeld, S.C.B.; et al. Solution Coating of Large-Area Organic Semiconductor Thin Films with Aligned Single-Crystalline Domains. Nat. Mater. 2013, 12, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Khim, D.; Han, H.; Baeg, K.-J.; Kim, J.; Kwak, S.-W.; Kim, D.-Y.; Noh, Y.-Y. Simple Bar-Coating Process for Large-Area, High-Performance Organic Field-Effect Transistors and Ambipolar Complementary Integrated Circuits. Adv. Mater. 2013, 25, 4302–4308. [Google Scholar] [CrossRef] [PubMed]
- Giri, G.; DeLongchamp, D.M.; Reinspach, J.; Fischer, D.A.; Richter, L.J.; Xu, J.; Benight, S.; Ayzner, A.; He, M.; Fang, L.; et al. Effect of Solution Shearing Method on Packing and Disorder of Organic Semiconductor Polymers. Chem. Mater. 2015, 27, 2350–2359. [Google Scholar] [CrossRef]
- Shin, J.; Hong, T.R.; Lee, T.W.; Kim, A.; Kim, Y.H.; Cho, M.J.; Choi, D.H. Template-Guided Solution-Shearing Method for Enhanced Charge Carrier Mobility in Diketopyrrolpyrrole-Based Polymer Field-Effect Transistors. Adv. Mater. 2014, 26, 6031–6035. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Choi, D.; Egap, E. Macroscopic Alignment of One-Dimensional Conjugated Polymer Nanocrystallites for High-Mobility Organic Field-Effect Transistors. ACS Appl. Mater. Interfaces 2016, 8, 13484–13491. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Su, Z.; Egap, E. Alignment and Charge Transport of One-Dimensional Conjugated Polymer Nanowires in Insulating Polymer Blends. Macromolecules 2016, 49, 9449–9456. [Google Scholar] [CrossRef]
- Park, Y.D.; Lee, H.S.; Choi, Y.J.; Kwak, D.; Cho, J.H.; Lee, S.; Cho, K. Solubility-Induced Ordered Polythiophene Precursors for High-Performance Organic Thin-Film Transistors. Adv. Funct. Mater. 2009, 19, 1200–1206. [Google Scholar] [CrossRef]
- Chang, M.; Choi, D.; Fu, B.; Reichmanis, E. Solvent Based Hydrogen Bonding: Impact on Poly(3-hexylthiophene) Nanoscale Morphology and Charge Transport Characteristics. ACS Nano 2013, 7, 5402–5413. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhao, L.; Wang, J.; Han, W.; Yang, Y.; Qiu, F.; Lin, Z. Self-Assembly of All-Conjugated Poly(3-alkylthiophene) Diblock Copolymer Nanostructures from Mixed Selective Solvents. ACS Nano 2010, 4, 3241–3247. [Google Scholar] [CrossRef] [PubMed]
- Kleinhenz, N.; Rosu, C.; Chatterjee, S.; Chang, M.; Nayani, K.; Xue, Z.; Kim, E.; Middlebrooks, J.; Russo, P.S.; Park, J.O.; et al. Liquid Crystalline Poly(3-hexylthiophene) Solutions Revisited: Role of Time-Dependent Self-Assembly. Chem. Mater. 2015, 27, 2687–2694. [Google Scholar] [CrossRef]
- Liu, J.; Zou, J.; Zhai, L. Bottom-up Assembly of Poly(3-hexylthiophene) on Carbon Nanotubes: 2D Building Blocks for Nanoscale Circuits. Macromol. Rapid Commun. 2009, 30, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Treat, N.D.; Malik, J.A.N.; Reid, O.; Yu, L.; Shuttle, C.G.; Rumbles, G.; Hawker, C.J.; Chabinyc, M.L.; Smith, P.; Stingelin, N. Microstructure Formation in Molecular and Polymer Semiconductors Assisted by Nucleation Agents. Nat. Mater. 2013, 12, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Rosu, C.; Kleinhenz, N.; Choi, D.; Tassone, C.J.; Zhang, X.; Park, J.O.; Srinivasarao, M.; Russo, P.S.; Reichmanis, E. Protein-Assisted Assembly of π-Conjugated Polymers. Chem. Mater. 2016, 28, 573–582. [Google Scholar] [CrossRef]
- Zhao, K.; Xue, L.; Liu, J.; Gao, X.; Wu, S.; Han, Y.; Geng, Y. A New Method to Improve Poly(3-hexylthiophene) (P3HT) Crystalline Behavior: Decreasing Chains Entanglement to Promote Order-Disorder Transformation in Solution. Langmuir 2010, 26, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Kim, M.-S.; Kim, J. Ultrasonic-Assisted Nanodimensional Self-Assembly of Poly-3-hexylthiophene for Organic Photovoltaic Cells. ACS Nano 2010, 4, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Aiyar, A.R.; Hong, J.-I.; Nambiar, R.; Collard, D.M.; Reichmanis, E. Tunable Crystallinity in Regioregular Poly(3-Hexylthiophene) Thin Films and Its Impact on Field Effect Mobility. Adv. Funct. Mater. 2011, 21, 2652–2659. [Google Scholar] [CrossRef]
- Choi, D.; Chang, M.; Reichmanis, E. Controlled Assembly of Poly(3-hexylthiophene): Managing the Disorder to Order Transition on the Nano- through Meso-Scales. Adv. Funct. Mater. 2015, 25, 920–927. [Google Scholar] [CrossRef]
- Chang, M.; Lee, J.; Kleinhenz, N.; Fu, B.; Reichmanis, E. Photoinduced Anisotropic Supramolecular Assembly and Enhanced Charge Transport of Poly(3-hexylthiophene) Thin Films. Adv. Funct. Mater. 2014, 24, 4457–4465. [Google Scholar] [CrossRef]
- Chang, M.; Choi, D.; Wang, G.; Kleinhenz, N.; Persson, N.; Park, B.; Reichmanis, E. Photoinduced Anisotropic Assembly of Conjugated Polymers in Insulating Polymer Blends. ACS Appl. Mater. Interfaces 2015, 7, 14095–14103. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Lee, J.; Chu, P.-H.; Choi, D.; Park, B.; Reichmanis, E. Anisotropic Assembly of Conjugated Polymer Nanocrystallites for Enhanced Charge Transport. ACS Appl. Mater. Interfaces 2014, 6, 21541–21549. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Lee, W.H.; Cho, K. Recent Advances in Organic Transistor Printing Processes. ACS Appl. Mater. Interfaces 2013, 5, 2302–2315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jie, J.; Deng, W.; Shang, Q.; Wang, J.; Wang, H.; Chen, X.; Zhang, X. Alignment and Patterning of Ordered Small-Molecule Organic Semiconductor Micro-/Nanocrystals for Device Applications. Adv. Mater. 2016, 28, 2475–2503. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M. Structure and Morphology Control in Thin Films of Regioregular Poly(3-hexylthiophene). J. Polym. Sci. B Polym. Phys. 2011, 49, 1218–1233. [Google Scholar] [CrossRef]
- Lu, G.; Blakesley, J.; Himmelberger, S.; Pingel, P.; Frisch, J.; Lieberwirth, I.; Salzmann, I.; Oehzelt, M.; Pietro, R.D.; Salleo, A.; et al. Moderate Doping Leads to High Performance of Semiconductor/Insulator Polymer Blend Transistors. Nat. Commun. 2013, 4, 1588. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Park, N.; Lee, J.; Min, H.; Choi, H.H.; Lee, H.S.; Cho, K. Surface-Order Mediated Assembly of π−Conjugated Molecules on Self-Assembled Monolayers with Controlled Grain Structures. Chem. Mater. 2015, 27, 4669–4676. [Google Scholar] [CrossRef]
- Cho, S.; Lee, K.; Yuen, J.; Wang, G.M.; Moses, D.; Heeger, A.J.; Surin, M. Thermal annealing-induced enhancement of the field-effect mobility of regioregular poly(3-hexylthiophene) films. J. Appl. Phys. 2006, 100, 114503. [Google Scholar] [CrossRef]
- Fu, Y.; Lin, C.; Tsai, F.Y. High field-effect mobility from poly(3-hexylthiophene) thin-film transistors by solvent-vapor-induced reflow. Org. Electron. 2009, 10, 883–888. [Google Scholar] [CrossRef]
- Kong, J.; Hwang, I.-W.; Lee, K. Top-Down Approach for Nanophase Reconstruction in Bulk Heterojunction Solar Cells. Adv. Mater. 2014, 26, 6275–6283. [Google Scholar] [CrossRef] [PubMed]
- Samitsu, S.; Shimomura, T.; Heike, S.; Hashizume, T.; Ito, K. Effective Production of Poly(3-alkylthiophene) Nanofibers by means of Whisker Method using Anisole Solvent: Structural, Optical, and Electrical Properties. Macromolecules 2008, 41, 8000–8010. [Google Scholar] [CrossRef]
- Lee, Y.; Oh, J.Y.; Son, S.Y.; Park, T.; Jeong, U. Effects of Regioregularity and Molecular Weight on the Growth of Polythiophene Nanofibrils and Mixes of Short and Long Nanofibrils To Enhance the Hole Transport. ACS Appl. Mater. Interfaces 2015, 7, 27694–27702. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Shin, M.; Lee, T.I.; Jang, W.S.; Min, Y.; Myung, J.M.; Baik, H.K.; Jeong, U. Self-Seeded Growth of Poly(3-hexylthiophene) (P3HT) Nanofibrils by a Cycle of Cooling and Heating in Solutions. Macromolecules 2012, 45, 7504–7513. [Google Scholar] [CrossRef]
- Vargas, R.; Garza, J.; Dixon, D.A.; Hay, B.P. How Strong Is the CR–H⋅⋅⋅O=C Hydrogen Bond? J. Am. Chem. Soc. 2000, 122, 4750–4755. [Google Scholar] [CrossRef]
- Sato, K.; Li, J.-G.; Kamiya, H.; Ishigaki, T. Ultrasonic Dispersion of TiO2 Nanoparticles in Aqueous Suspension. J. Am. Ceram. Soc. 2008, 91, 2481–2487. [Google Scholar] [CrossRef]
- Rai, C.L.; Struenkmann, G.; Mueller, J.; Rao, P.G. Influence of Ultrasonic Disintegration on Sludge Growth Reduction and Its Estimation by Respirometry. Environ. Sci. Technol. 2004, 38, 5779–5785. [Google Scholar] [CrossRef] [PubMed]
- Beckett, M.A.; Hua, I. Impact of Ultrasonic Frequency on Aqueous Sonoluminescence and Sonochemistry. J. Phys. Chem. A 2001, 105, 3796–3802. [Google Scholar] [CrossRef]
- Schmitt, M.; Faber, E.; Botz, R.; Stoffers, P. Extraction of Methane from Seawater Using Ultrasonic Vacuum Degassing. Anal. Chem. 1991, 63, 529–532. [Google Scholar] [CrossRef]
- Li, J.; Sanderson, R.D. Jacobs, E.P. Ultrasonic Cleaning of Nylon Microfiltration Membranes Fouled by Kraft Paper Mill Effluent. J. Membr. Sci. 2002, 205, 247–257. [Google Scholar] [CrossRef]
- Luque de Castro, M.D.; Priego-Capote, F. Ultrsound-Assisted Crystallization (Sonocrystallization). Ultrason. Sonochem. 2007, 14, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Murthy, Z.V.P. Ultrasound Assisted Crystallization for the Recovery of Lactose in an Anti-Solvent Acetone. Cryst. Res. Technol. 2009, 44, 889–896. [Google Scholar] [CrossRef]
- Dhumal, R.S.; Biradar, S.V.; Paradka, A.R.; York, P. Ultrasound Assisted Engineering of Lactose Crystals. Pharm. Res. 2008, 25, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.G.; Kim, H.; Lee, J.W. Characteristics of Polystyrene/Polyethylene/Clay Nanocomposites Prepared by Ultrasound-Assisted Mixing Process. Polym. Eng. Sci. 2004, 44, 1198–1204. [Google Scholar] [CrossRef]
- Nish, A.; Hwang, J.-Y.; Doig, J.; Nicholas, R.J. Highly Selective Dispersion of Single-Walled Carbon Nanotubes Using Aromatic Polymers. Nat. Nanotechnol. 2007, 2, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Morgan, A.B.; Harris, J.D. Rheological Characterization of Polystyrene-Clay Nanocomposites to Compare the Degree of Exfoliation and Dispersion. Polymer 2005, 46, 8641–8660. [Google Scholar] [CrossRef]
- Higashi, T.; Yamasaki, N.; Utsumi, H.; Yoshida, H.; Fujii, A.; Ozaki, M. Anisotropic Properties of Aligned π-Conjugated Polymer Films Fabricated by Capillary Action and Their Post-Annealing Effects. Appl. Phys. Express 2011, 4, 091602. [Google Scholar] [CrossRef]
- Luo, C.; Kyaw, A.K.K.; Perez, L.A.; Patel, S.; Wang, M.; Grimm, B.; Bazan, G.C.; Kramer, E.J.; Heeger, A.J. General Strategy for Self-Assembly of Highly Oriented Nanocrystalline Semiconducting Polymers with High Mobility. Nano Lett. 2014, 14, 2764–2771. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Giri, G.; Ayzner, A.L.; Zoombelt, A.P.; Mannsfeld, S.C.B.; Chen, J.; Nordlund, D.; Toney, M.F.; Huang, J.; Bao, Z. Ultra-High Mobility Transparent Organic Thin Film Transistors Grown by an Off-Centre Spin-Coating Method. Nat. Commun. 2014, 5, 3005. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, L.; Xing, R.; Liu, J.; Han, Y. Simultaneous Control over Both Molecular Order and Long-Range Alignment in Films of the Donor-Acceptor Copolymer. Langmuir 2015, 31, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-K.; Jang, S.-Y.; Pace, G.; Caironi, M.; Park, W.-T.; Khim, D.; Kim, J.; Kim, D.-Y.; Noh, Y.-Y. High-Performance Organic Field-Effect Transistors with Directionally Aligned Conjugated Polymer Film Deposited from Pre-Aggregated Solution. Chem. Mater. 2015, 27, 8345–8353. [Google Scholar] [CrossRef]
- Huang, J.; Carpenter, J.H.; Li, C.-Z.; Yu, J.-S.; Ade, H.; Jen, A.K.-Y. Highly Efficient Organic Solar Cells with Improved Vertical Donor-Acceptor Compositional Gradient via an Inverted Off-Center Spinning Method. Adv. Mater. 2016, 28, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.J.; Li, C.Y.; Xue, G.; Mann, I.K.; Zhang, D.; Wang, S.-Y.; Harris, F.W.; Cheng, S.Z.D.; Hong, S.-C.; Zhuang, X.; et al. Rubbing-Induced Molecular Reorientation on an Alignment Surface of an Aromatic Polyimide Containing Cyanobiphenyl Side Chains. J. Am. Chem. Soc. 2001, 123, 5768–5776. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kumar, S.; Lee, S.-D. Alignment of Liquid Crystals on Polyimide Films Exposed to Ultraviolet Light. Phys. Rev. E 1998, 57, 5644. [Google Scholar] [CrossRef]
- Jayathilake, H.D.; Zhu, M.H.; Rosenblatt, C.; Bordenyuk, A.N.; Weeraman, C.; Benderskii, A.V. Rubbing-Induced Anisotropy of Long Alkyl Side Chains at Polyimide Surfaces. J. Chem. Phys. 2006, 125, 064706. [Google Scholar] [CrossRef] [PubMed]
- Heil, H.; Finnberg, T.; von Malm, N.; Schmechel, R.; von Seggern, H. The Influence of Mechanical Rubbing on the Field-Effect Mobility in Polyhexylthiophene. J. Appl. Phys. 2003, 93, 1636. [Google Scholar] [CrossRef]
- Biniek, L.; Pouget, S.; Djurado, D.; Gonthier, E.; Tremel, K.; Kayunkid, N.; Zaborova, E.; Crespo-Monteiro, N.; Boyron, O.; Leclerc, N.; et al. High-Temperature Rubbing: A Versatile Method to Align π-Conjugated Polymers without Alignment Substrate. Macromolecules 2014, 47, 3871–3879. [Google Scholar] [CrossRef]
- Soeda, J.; Matsui, H.; Okamoto, T.; Osaka, I.; Takimiya, K.; Takeya, J. Highly Oriented Polymer Semiconductor Films Compressed at the Surface of Ionic Liquids for High-Performance Polymeric Organic Field-Effect Transistors. Adv. Mater. 2014, 26, 6430–6435. [Google Scholar] [CrossRef] [PubMed]












© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, M.; Lim, G.T.; Park, B.; Reichmanis, E. Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films. Polymers 2017, 9, 212. https://doi.org/10.3390/polym9060212
Chang M, Lim GT, Park B, Reichmanis E. Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films. Polymers. 2017; 9(6):212. https://doi.org/10.3390/polym9060212
Chicago/Turabian StyleChang, Mincheol, Gyun Taek Lim, Byoungnam Park, and Elsa Reichmanis. 2017. "Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films" Polymers 9, no. 6: 212. https://doi.org/10.3390/polym9060212
APA StyleChang, M., Lim, G. T., Park, B., & Reichmanis, E. (2017). Control of Molecular Ordering, Alignment, and Charge Transport in Solution-Processed Conjugated Polymer Thin Films. Polymers, 9(6), 212. https://doi.org/10.3390/polym9060212