RNA-Seq Analysis of Prickled and Prickle-Free Epidermis Provides Insight into the Genetics of Prickle Development in Red Raspberry (Rubus ideaus L.)

Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Isolation and Quality Control
2.2. RNA-Seq Assay and Illumina Sequencing
2.3. Quantitative Expression Analysis Methods
2.4. Differential Gene Expression
2.5. Functional Annotation Using Blast2GO/OmicsBox and GO Enrichment Analysis
2.6. Expression Analysis through Quantitative Reverse-Transcription PCR (qRT-PCR) Analysis
3. Results
3.1. Differentially Expressed Genes (DEGs)
3.2. GO Analysis and GO Enrichment Analysis
3.3. Putative Transcription Factors (TF) Differentially Expressed in Prickle-Free Plants
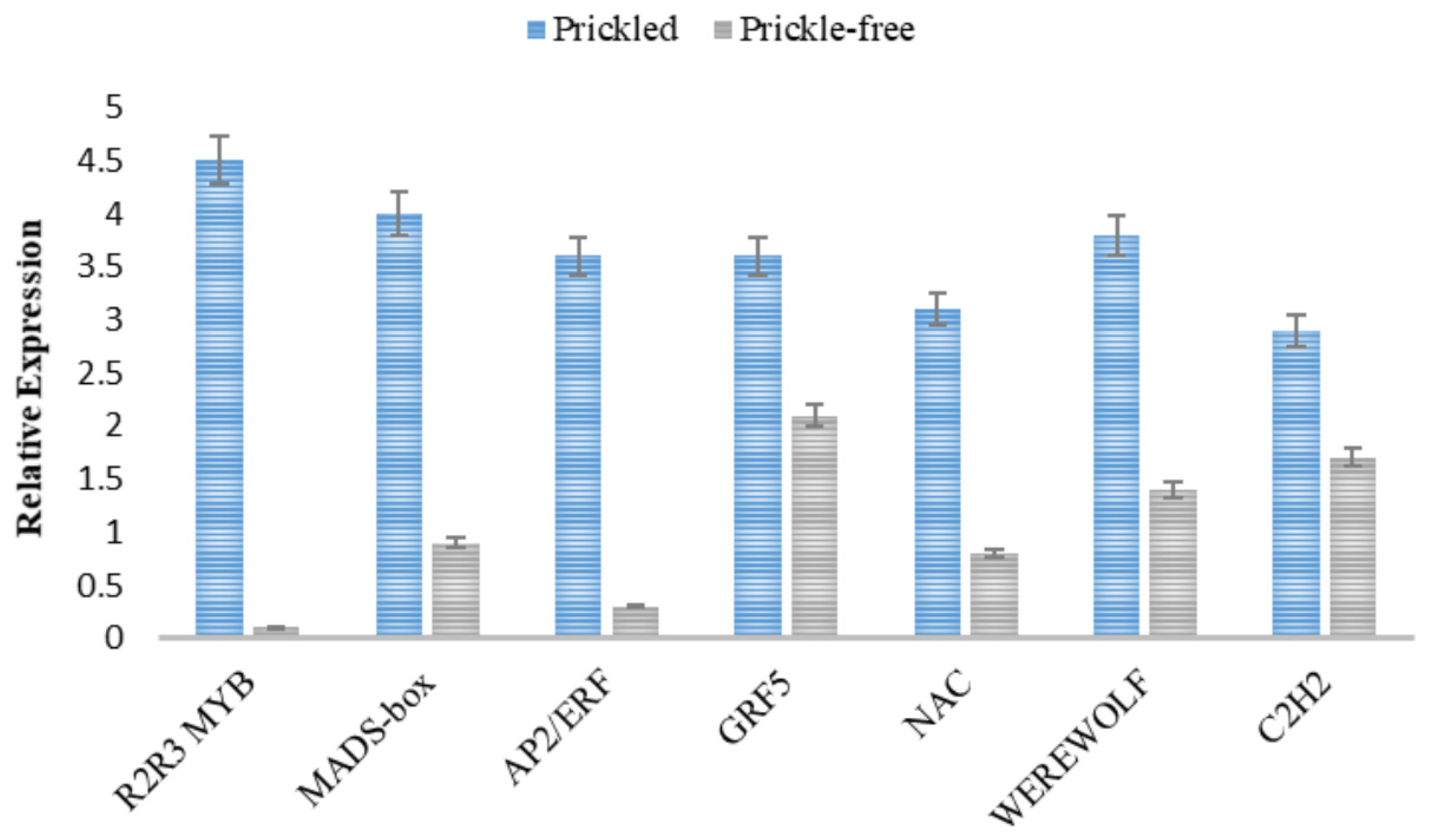
3.4. Validation of Transcriptome Data by qRT-PCR Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Doughari, J. An overview of plant immunity. J. Plant Pathol. Microbiol. 2015, 6, 10–4172. [Google Scholar]
- Johnson, H.B. Plant pubescence: An ecological perspective. Bot. Rev. 1975, 41, 233–258. [Google Scholar] [CrossRef]
- Schuepp, P. Tansley review no. 59: Leaf boundary layers. New Phytol. 1993, 125, 477–507. [Google Scholar] [CrossRef]
- Barton, K.E. Prickles, latex, and tolerance in the endemic Hawaiian prickly poppy (Argemone glauca): Variation between populations, across ontogeny, and in response to abiotic factors. Oecologia 2014, 174, 1273–1281. [Google Scholar] [CrossRef]
- Halpern, M.; Raats, D.; Lev-Yadun, S. The potential anti-herbivory role of microorganisms on plant thorns. Plant Signal. Behav. 2007, 2, 503–504. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, D.B.; Lloyd, A.M.; Marks, M.D. Progress in the molecular genetic analysis of trichome initiation and morphogenesis in Arabidopsis. Trends Plant Sci. 2000, 5, 214–219. [Google Scholar] [CrossRef]
- Bieniek, M.E.; Millington, W.F. Differentiation of lateral shoots as thorns in Ulex europaeus. Am. J. Bot. 1967, 54, 61–70. [Google Scholar] [CrossRef]
- Blaser, H.W. Morphology of the determinate thorn-shoots of Gleditsia. Amer. J. Bot. 1956, 43, 22–28. [Google Scholar] [CrossRef]
- Coyner, M.; Skirvin, R.M.; Norton, M.; Otterbacher, A. Thornlessness in blackberries: A review. Small Fruits Rev. 2005, 4, 83–106. [Google Scholar] [CrossRef]
- Esau, K. Anatomy of Seed Plants; John Wiley & Sons, Inc.: New York, NY, USA, 1977; pp. 455–500. [Google Scholar]
- Posluszny, U.; Fisher, J.B. Thorn and hook ontogeny in Artabotrys hexapetalus (Annonaceae). Am. J. Bot. 2000, 87, 1561–1570. [Google Scholar] [CrossRef]
- Clark, J.R.; Stafne, E.T.; Hall, H.K.; Finn, C.E. Blackberry breeding and genetics. Plant Breed. Rev. 2007, 29, 19. [Google Scholar]
- Finn, C.; Moore, P.; Kempler, C. Raspberry Cultivars: What’s New? What’s Succeeding? Where are the Breeding Programs Headed? Acta Hortic. 2008, 777, 33–40. [Google Scholar] [CrossRef]
- Clark, J.R.; Moore, J.N. ‘Natchez’thornless blackberry. HortScience 2008, 43, 1897–1899. [Google Scholar] [CrossRef] [Green Version]
- Asano, G.; Kubo, R.; Tanimoto, S. Growth, structure and lignin localization in rose prickle. Bull. Fac. Agric. 2008, 93, 117–125. [Google Scholar]
- Kellogg, A.A.; Branaman, T.J.; Jones, N.M.; Little, C.Z.; Swanson, J.D. Morphological studies of developing Rubus prickles suggest that they are modified glandular trichomes. Botany 2011, 89, 217–226. [Google Scholar] [CrossRef]
- Rajapakse, S.; Zhang, L.; Ballard, R.; Byrne, D. AFLP marker development in rose for genetic mapping: Comparison of three restriction enzyme pairs. Acta Hortic. 2001, 546, 619–627. [Google Scholar] [CrossRef]
- Khadgi, A.; Weber, C.A. Morphological Characterization of Prickled and Prickle-free Rubus Using Scanning Electron Microscopy. HortScience 2020, 55, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.; Goel, R.; Bhardwaj, A.; Asif, M.H.; Sawant, S.V.; Misra, P. Transcriptome analysis provides insight into prickle development and its link to defense and secondary metabolism in Solanum viarum Dunal. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Breuer, C.; Kawamura, A.; Ichikawa, T.; Tominaga-Wada, R.; Wada, T.; Kondou, Y.; Muto, S.; Matsui, M.; Sugimoto, K. The trihelix transcription factor GTL1 regulates ploidy-dependent cell growth in the Arabidopsis trichome. Plant Cell 2009, 21, 2307–2322. [Google Scholar] [CrossRef] [Green Version]
- Marks, M.D.; Wenger, J.P.; Gilding, E.; Jilk, R.; Dixon, R.A. Transcriptome analysis of Arabidopsis wild-type and gl3–sst sim trichomes identifies four additional genes required for trichome development. Mol. Plant 2009, 2, 803–822. [Google Scholar] [CrossRef]
- Wagner, G.J. Secreting glandular trichomes: More than just hairs. Plant Phys. 1991, 96, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Pattanaik, S.; Patra, B.; Singh, S.K.; Yuan, L. An overview of the gene regulatory network controlling trichome development in the model plant, Arabidopsis. Front. Plant Sci. 2014, 5, 259. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.O.; Roeder, A.H. Themes and variations in cell type patterning in the plant epidermis. Curr. Opin. Genet. Dev. 2015, 32, 55–65. [Google Scholar] [CrossRef]
- Yang, C.; Ye, Z. Trichomes as models for studying plant cell differentiation. Cell. Mol. Life Sci. 2013, 70, 1937–1948. [Google Scholar] [CrossRef]
- Huchelmann, A.; Boutry, M.; Hachez, C. Plant glandular trichomes: Natural cell factories of high biotechnological interest. Plant Phys. 2017, 175, 6–22. [Google Scholar] [CrossRef] [Green Version]
- Hülskamp, M. Plant trichomes: A model for cell differentiation. Nat. Rev. Mol. Cell Biol. 2004, 5, 471–480. [Google Scholar] [CrossRef]
- Chen, C.; Yin, S.; Liu, X.; Liu, B.; Yang, S.; Xue, S.; Cai, Y.; Black, K.; Liu, H.; Dong, M. The WD-repeat protein CsTTG1 regulates fruit wart formation through interaction with the homeodomain-leucine zipper I protein Mict. Plant Phys. 2016, 171, 1156–1168. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, S.; Miao, H.; Wang, M.; Liu, P.; Wehner, T.C.; Gu, X. Molecular mapping and candidate gene analysis for numerous spines on the fruit of cucumber. J. Hered. 2016, 107, 471–477. [Google Scholar] [CrossRef]
- Zhou, N.; Tang, K.; Jeauffre, J.; Thouroude, T.; Arias, D.L.; Foucher, F.; Hibrand-Saint Oyant, L. Genetic determinism of prickles in rose. Theor. Appl. Genet. 2020, 133, 3017–3035. [Google Scholar] [CrossRef]
- Debener, T. Genetic analysis of horticulturally important morphological and physiological characters in diploid roses. Gartenbauwissenschaft 1999, 64, 14–19. [Google Scholar]
- Shupert, D.A.; Byrne, D.H.; Brent Pemberton, H. Inheritance of flower traits, leaflet number and prickles in roses. Acta Hortic. 2007, 751, 331–335. [Google Scholar] [CrossRef]
- Jennings, D. Balanced lethals and polymorphism in Rubus idaeus. Heredity 1967, 22, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Graham, J.; Smith, K.; MacKenzie, K.; Jorgenson, L.; Hackett, C.; Powell, W. The construction of a genetic linkage map of red raspberry (Rubus idaeus subsp. idaeus) based on AFLPs, genomic-SSR and EST-SSR markers. Theor. Appl. Genet. 2004, 109, 740–749. [Google Scholar] [CrossRef]
- Keep, E.; Knight, V.H.; Parker, J.H. Rubus coreanus as donor of resistance to cane diseases and mildew in red raspberry breeding. Euphytica 1977, 26, 505–510. [Google Scholar] [CrossRef]
- Molina-Bravo, R.; Fernandez, G.E.; Sosinski, B.R. Quantitative trait locus analysis of tolerance to temperature fluctuations in winter, fruit characteristics, flower color, and prickle-free canes in raspberry. Mol. Breed. 2014, 33, 267–280. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Oshima, Y.; Mitsuda, N. The MIXTA-like Transcription factor MYB16 is a major regulator of cuticle formation in vegetative organs. Plant Signal. Behav. 2013, 8, e26826. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2Go Tutorial; Bioinformatics and Genomics Department Prince Felipe Research Center: Valencia, Spain, 2009. [Google Scholar]
- Seo, E.; Choi, D. Functional studies of transcription factors involved in plant defenses in the genomics era. Brief. Funct. Genom. 2015, 14, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Schilmiller, A.L.; Last, R.L.; Pichersky, E. Harnessing plant trichome biochemistry for the production of useful compounds. Plant J. 2008, 54, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.; Wang, E.; Shepherd, R. New approaches for studying and exploiting an old protuberance, the plant trichome. Ann. Bot. 2004, 93, 3. [Google Scholar] [CrossRef] [Green Version]
- Balcke, G.U.; Bennewitz, S.; Bergau, N.; Athmer, B.; Henning, A.; Majovsky, P.; Jiménez-Gómez, J.M.; Hoehenwarter, W.; Tissier, A. Multi-omics of tomato glandular trichomes reveals distinct features of central carbon metabolism supporting high productivity of specialized metabolites. Plant Cell 2017, 29, 960–983. [Google Scholar] [CrossRef] [Green Version]
- Mazid, M.; Khan, T.; Mohammad, F. Role of secondary metabolites in defense mechanisms of plants. Biol. Med. 2011, 3, 232–249. [Google Scholar]
- Zhao, M.; Morohashi, K.; Hatlestad, G.; Grotewold, E.; Lloyd, A. The TTG1-bHLH-MYB complex controls trichome cell fate and patterning through direct targeting of regulatory loci. Development 2008, 135, 1991–1999. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.C.; Brown, M.L.; Schiefelbein, J. How do cells know what they want to be when they grow up? Lessons from epidermal patterning in Arabidopsis. Ann. Rev. Plant Biol. 2003, 54, 403–430. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; He, H.; Li, Y.; Ai, Q.; Yu, D. MYB82 functions in regulation of trichome development in Arabidopsis. J. Exp. Bot. 2014, 65, 3215–3223. [Google Scholar] [CrossRef] [Green Version]
- Payne, C.T.; Zhang, F.; Lloyd, A.M. GL3 encodes a bHLH protein that regulates trichome development in Arabidopsis through interaction with GL1 and TTG1. Genetics 2000, 156, 1349–1362. [Google Scholar]
- Rerie, W.G.; Feldmann, K.A.; Marks, M.D. The GLABRA2 gene encodes a homeo domain protein required for normal trichome development in Arabidopsis. Genes Dev. 1994, 8, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Schnittger, A.; Folkers, U.; Schwab, B.; Jürgens, G.; Hülskamp, M. Generation of a spacing pattern: The role of TRIPTYCHON in trichome patterning in Arabidopsis. Plant Cell 1999, 11, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Tachibana, T.; Shimura, Y.; Okada, K. Epidermal cell differentiation in Arabidopsis determined by a Myb homolog, CPC. Science 1997, 277, 1113–1116. [Google Scholar] [CrossRef]
- Wang, S.; Kwak, S.-H.; Zeng, Q.; Ellis, B.E.; Chen, X.-Y.; Schiefelbein, J.; Chen, J.-G. TRICHOMELESS1 regulates trichome patterning by suppressing GLABRA1 in Arabidopsis. Development 2007, 134, 3873–3882. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.; Xia, K.; Chen, J.G.; Wang, S. Functional characterization of TRICHOMELESS2, a new single-repeat R3 MYB transcription factor in the regulation of trichome patterning in Arabidopsis. BMC Plant Biol. 2011, 11, 176. [Google Scholar] [CrossRef] [Green Version]
- Tominaga-Wada, R.; Nukumizu, Y.; Sato, S.; Wada, T. Control of plant trichome and root-hair development by a tomato (Solanum lycopersicum) R3 MYB transcription factor. PLoS ONE 2013, 8, e54019. [Google Scholar] [CrossRef] [Green Version]
- Esch, J.J.; Chen, M.A.; Hillestad, M.; Marks, M.D. Comparison of TRY and the closely related At1g01380 gene in controlling Arabidopsis trichome patterning. Plant J. 2004, 40, 860–869. [Google Scholar] [CrossRef]
- Kirik, V.; Simon, M.; Huelskamp, M.; Schiefelbein, J. The ENHANCER OF TRY AND CPC1 gene acts redundantly with TRIPTYCHON and CAPRICE in trichome and root hair cell patterning in Arabidopsis. Dev. Biol. 2004, 268, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Kirik, V.; Simon, M.; Wester, K.; Schiefelbein, J.; Hulskamp, M. ENHANCER of TRY and CPC 2 (ETC2) reveals redundancy in the region-specific control of trichome development of Arabidopsis. Plant Mol. Biol. 2004, 55, 389–398. [Google Scholar] [CrossRef]
- Simon, M.; Lee, M.M.; Lin, Y.; Gish, L.; Schiefelbein, J. Distinct and overlapping roles of single-repeat MYB genes in root epidermal patterning. Dev. Biol. 2007, 311, 566–578. [Google Scholar] [CrossRef]
- Esch, J.J.; Chen, M.; Sanders, M.; Hillestad, M.; Ndkium, S.; Idelkope, B.; Neizer, J.; Marks, M.D. A contradictory GLABRA3 allele helps define gene interactions controlling trichome development in Arabidopsis. Development 2003, 130, 5885–5894. [Google Scholar] [CrossRef] [Green Version]
- Hülskamp, M.; Miséra, S.; Jürgens, G. Genetic dissection of trichome cell development in Arabidopsis. Cell 1994, 76, 555–566. [Google Scholar] [CrossRef]
- Ishida, T.; Kurata, T.; Okada, K.; Wada, T. A genetic regulatory network in the development of trichomes and root hairs. Annu. Rev. Plant Biol. 2008, 59, 365–386. [Google Scholar] [CrossRef]
- Pesch, M.; Hülskamp, M. Creating a two-dimensional pattern de novo during Arabidopsis trichome and root hair initiation. Curr. Opin. Genet. Dev. 2004, 14, 422–427. [Google Scholar] [CrossRef]
- Schellmann, S.; Schnittger, A.; Kirik, V.; Wada, T.; Okada, K.; Beermann, A.; Thumfahrt, J.; Jürgens, G.; Hülskamp, M. TRIPTYCHON and CAPRICE mediate lateral inhibition during trichome and root hair patterning in Arabidopsis. EMBO J. 2002, 21, 5036–5046. [Google Scholar] [CrossRef] [Green Version]
- Schiefelbein, J. Cell-fate specification in the epidermis: A common patterning mechanism in the root and shoot. Curr. Opin. Plant Biol. 2003, 6, 74–78. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Kirik, V.; Lee, M.M.; Wester, K.; Herrmann, U.; Zheng, Z.; Oppenheimer, D.; Schiefelbein, J.; Hulskamp, M. Functional diversification of MYB23 and GL1 genes in trichome morphogenesis and initiation. Development 2005, 132, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Liang, Z.; Zhao, S.; Nan, M.G.; Tran, L.S.P.; Lu, K.; Huang, Y.B.; Li, J.N. The evolutionary history of R2R3-MYB proteins across 50 eukaryotes: New insights into subfamily classification and expansion. Sci. Rep. 2015, 5, 11037. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.M.; Schiefelbein, J. Developmentally distinct MYB genes encode functionally equivalent proteins in Arabidopsis. Development 2001, 128, 1539–1546. [Google Scholar]
- Noda, K.I.; Glover, B.J.; Linstead, P.; Martin, C. Flower colour intensity depends on specialized cell shape controlled by a Myb-related transcription factor. Nature 1994, 369, 661–664. [Google Scholar] [CrossRef]
- Baumann, K.; Perez-Rodriguez, M.; Bradley, D.; Venail, J.; Bailey, P.; Jin, H.; Koes, R.; Roberts, K.; Martin, C. Control of cell and petal morphogenesis by R2R3 MYB transcription factors. Development 2007, 134, 1691–1701. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Yang, S.S.; Liang, Z.; Feng, B.R.; Liu, L.; Huang, Y.B.; Tang, Y.X. Genome-wide analysis of the MYB transcription factor superfamily in soybean. BMC Plant Biol. 2012, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Glover, B.J.; Perez-Rodriguez, M.; Martin, C. Development of several epidermal cell types can be specified by the same MYB-related plant transcription factor. Development 1998, 125, 3497–3508. [Google Scholar]
- Jaffé, F.W.; Tattersall, A.; Glover, B.J. A truncated MYB transcription factor from Antirrhinum majus regulates epidermal cell outgrowth. J. Exp. Bot. 2007, 58, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Jakoby, M.J.; Falkenhan, D.; Mader, M.T.; Brininstool, G.; Wischnitzki, E.; Platz, N.; Hudson, A.; Hülskamp, M.; Larkin, J.; Schnittger, A. Transcriptional profiling of mature Arabidopsis trichomes reveals that NOECK encodes the MIXTA-like transcriptional regulator MYB106. Plant Phys. 2008, 148, 1583–1602. [Google Scholar] [CrossRef] [Green Version]
- Machado, A.; Wu, Y.; Yang, Y.; Llewellyn, D.J.; Dennis, E.S. The MYB transcription factor GhMYB25 regulates early fibre and trichome development. Plant J. 2009, 59, 52–62. [Google Scholar] [CrossRef]
- Perez-Rodriguez, M.; Jaffe, F.W.; Butelli, E.; Glover, B.J.; Martin, C. Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development 2005, 132, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Walford, S.A.; Wu, Y.; Llewellyn, D.J.; Dennis, E.S. GhMYB25-like: A key factor in early cotton fibre development. Plant J. 2011, 65, 785–797. [Google Scholar] [CrossRef]
- Yang, S.; Cai, Y.; Liu, X.; Dong, M.; Zhang, Y.; Chen, S.; Zhang, W.; Li, Y.; Tang, M.; Zhai, X. A CsMYB6-CsTRY module regulates fruit trichome initiation in cucumber. J. Exp. Bot. 2018, 69, 1887–1902. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Zhang, C.; Li, J.; Wang, L.; Ren, Z. Genome-wide identification and characterization of R2R3MYB family in Cucumis sativus. PLoS ONE 2012, 7, e47576. [Google Scholar] [CrossRef] [Green Version]
- Matus, J.; Aquea, F.; Acre-Johnson, P. Analysis of the grape MYB R2R3 subfamily reveals expanded wine quality-related clades and conserved gene structure organization across Vitis and Arabidopsis genomes. BMC Plant Biol. 2008, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, O.; Nahal, H.; Foong, J.; Provart, N.J.; Campbell, M.M. Expansion and diversification of the Populus R2R3-MYB family of transcription factors. Plant Phys. 2009, 149, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Oshima, Y.; Shikata, M.; Koyama, T.; Ohtsubo, N.; Mitsuda, N.; Ohme-Takagi, M. MIXTA-like transcription factors and WAX INDUCER1/SHINE1 coordinately regulate cuticle development in Arabidopsis and Torenia fournieri. Plant Cell 2013, 25, 1609–1624. [Google Scholar] [CrossRef] [Green Version]
- González, M.; Carrasco, B.; Salazar, E. Genome-wide identification and characterization of R2R3MYB family in Rosaceae. Genomics Data 2016, 9, 50–57. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhou, Q.; Zhang, W.; Fu, Y.; Huang, H. ASYMMETRIC LEAVES1, an Arabidopsis gene that is involved in the control of cell differentiation in leaves. Planta 2002, 214, 694–702. [Google Scholar] [CrossRef]
- Riechmann, J.L.; Meyerowitz, E.M. The AP2/EREBP family of plant transcription factors. Biol. Chem. 1998, 379, 633–646. [Google Scholar]
- Licausi, F.; Giorgi, F.M.; Zenoni, S.; Osti, F.; Pezzotti, M.; Perata, P. Genomic and transcriptomic analysis of the AP2/ERF superfamily in Vitis vinifera. BMC Genom. 2010, 11, 719. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Phys. 2006, 140, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Sharoni, A.M.; Nuruzzaman, M.; Satoh, K.; Shimizu, T.; Kondoh, H.; Sasaya, T.; Choi, I.R.; Omura, T.; Kikuchi, S. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Phys. 2011, 52, 344–360. [Google Scholar] [CrossRef]
- Tsuwamoto, R.; Yokoi, S.; Takahata, Y. Arabidopsis EMBRYOMAKER encoding an AP2 domain transcription factor plays a key role in developmental change from vegetative to embryonic phase. Plant Mol. Biol. 2010, 73, 481–492. [Google Scholar] [CrossRef]
- Moose, S.P.; Sisco, P.H. Glossy15, an APETALA2-like gene from maize that regulates leaf epidermal cell identity. Genes Dev. 1996, 10, 3018–3027. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Gao, D.; Xiong, Y.; Tang, X.; Xiao, X.; Wang, C.; Yu, S. Hairy leaf 6, an AP2/ERF transcription factor, interacts with OsWOX3B and regulates trichome formation in rice. Mol. Plant 2017, 10, 1417–1433. [Google Scholar] [CrossRef] [Green Version]
- Castelán-Muñoz, N.; Herrera, J.; Cajero-Sánchez, W.; Arrizubieta, M.; Trejo, C.; Garcia-Ponce, B.; Sánchez, M.D.L.P.; Álvarez-Buylla, E.R.; Garay-Arroyo, A. MADS-box genes are key components of genetic regulatory networks involved in abiotic stress and plastic developmental responses in plants. Front. Plant Sci. 2019, 10, 853. [Google Scholar] [CrossRef] [Green Version]
- Bowman, J.L.; Baum, S.F.; Eshed, Y.; Putterill, J.; Alvarez, J. 4 Molecular Genetics of Gynoecium Development in Arabidopsis. Curr. Top. Dev. Biol. 1999, 45, 155–205. [Google Scholar]
- Weigel, D. The genetics of flower development: From floral induction to ovule morphogenesis. Ann. Rev. Genet. 1995, 29, 19–39. [Google Scholar] [CrossRef]
- Colombo, L.; Franken, J.; Koetje, E.; van Went, J.; Dons, H.J.; Angenent, G.C.; van Tunen, A.J. The petunia MADS box gene FBP11 determines ovule identity. Plant Cell 1995, 7, 1859–1868. [Google Scholar]
- Ma, H.; Yanofsky, M.F.; Meyerowitz, E.M. AGL1-AGL6, an Arabidopsis gene family with similarity to floral homeotic and transcription factor genes. Genes Dev. 1991, 5, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Rounsley, S.D.; Ditta, G.S.; Yanofsky, M.F. Diverse roles for MADS box genes in Arabidopsis development. Plant Cell 1995, 7, 1259–1269. [Google Scholar]
- Zhang, H.; Forde, B.G. An Arabidopsis MADS box gene that controls nutrient-induced changes in root architecture. Science 1998, 279, 407–409. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Liljegren, S.J.; Pelaz, S.; Gold, S.E.; Burgeff, C.; Ditta, G.S.; Vergara-Silva, F.; Yanofsky, M.F. MADS-box gene evolution beyond flowers: Expression in pollen, endosperm, guard cells, roots and trichomes. Plant J. 2000, 24, 457–466. [Google Scholar] [CrossRef]
- Tweneboah, S.; Oh, S.K. Biological roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in solanaceous crops. J. Plant Biotech. 2017, 44, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Bartholomew, E.; Black, K.; Dong, M.; Zhang, Y.; Yang, S.; Cai, Y.; Xue, S.; Weng, Y.; Ren, H. Comprehensive analysis of NAC transcription factors and their expression during fruit spine development in cucumber (Cucumis sativus L.). Hort. Res. 2018, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Primers Used for Transcriptome Data Validation | |||
|---|---|---|---|
| Primer Sequence 5′ to 3′ | |||
| S.N. | Gene IDs | Forward | Reverse |
| 1 | 19386_g | CCCTCATAATCTCCACAGGTTT | ATTCCAGCCACTGCCATAATA |
| 2 | 3610_g | TCGTGGTGCATCAGCTTTAG | CTCCATCTTCCTGCCCATATTT |
| 3 | 4624_g | GAGGAGATTGGGATGGATGTT | CAGATGCTCCAATGCTGAAAG |
| 4 | 9394_g | CTTCTGTGATCGAATTGGGTTTG | CAGCACCACCACCTTGATAA |
| 5 | 29335_g | GCAGCTAAGGACATGGAGAAAG | GGGATATGATGATGCTGGGTTTAG |
| 6 | 21030_g | GTCAGTGACTGGTACAGGTATTT | CGATCCCTACTTTCCACCATAC |
| 7 | 18962_g | CGCATCCGGTCTTACCATTTA | TAGGCAGCATTACCGAAACTC |
| 8 | 14085_g | GCCTCTCTGTATTTCCCTATGC | GCGGAGGTTGATCGATTCTT |
| 9 | 9950_g | CTCGATACCGAACCTCCAAAG | CTCCGCAAACCCTAGCTAAA |
| 10 | 5631_g | TCATCACCGAGTCCAAACAC | GCACGGGTTTGATGAATTGG |
| Selected Primers Used for Expression Analysis for Transcription Factors | ||||
|---|---|---|---|---|
| Primer Sequence 5′ to 3′ | ||||
| S.N. | Gene IDs | Gene Names | Forward | Reverse |
| 1 | 8958_g | R2R3-MYB | GCGGAGGACGGTTTGATTAG | CCACAGAAACCCTCCATGATATT |
| 2 | 3714_g | MADS-box | CAACAGCAGCAAACGAATATGA | GGTGATTGGACTCGAGGATTAC |
| 3 | 9441_g | NAC | ACGTGCTGATAACCCAGATG | CAACTCCACCAGTAGCCAAA |
| 4 | 5478_g | C2H2 | CAGTTTGCAGTGCTGTGATTAT | GCAAACTGCCCTGACAAATC |
| 5 | 13766_g | WEREWOLF | AGTTTGTGGAGCCTGATAATGA | GTGGGAAGAGTGTTAGGCTTAG |
| 6 | 19810_g | AP2/ERF | GAGGTGATAATCGGAAGCAAGA | GACCAGAAGAGCATCCCATATC |
| 7 | 8771_g | GRF5 | AGGGACGAGACGACCATATTA | GACGCCTTCTTTCTTTCTTTCTTC |
| Gene IDs | Log2-Fold Change | p-Value (Adjusted) | Description |
|---|---|---|---|
| 4820_g | −7.73 | 1.40 × 10−22 | Myb-related protein |
| 26462_g | −7.45 | 4.64 × 10−17 | GDSL esterase/lipase At45670 |
| 4747_g | −7.10 | 2.15 × 10−18 | Phylloplanin-like |
| 4737_g | −6.53 | 1.83 × 10−13 | Phylloplanin |
| 28832_g | −6.20 | 3.55 × 10−17 | Putative proteinase inhibitor I13, potato inhibitor I |
| 8958_g | −6.03 | 5.87 × 10−5 | Transcription Factor Myb16-Like (Rosa chinensis) |
| 26472_g | −5.67 | 1.23 × 10−15 | Transcription factor MYB8-like |
| 4741_g | −5.63 | 3.02 × 10−28 | Very-long-chain (3R)-3-hydroxyacyl-CoA dehydratase PASTICCINO 2A-like |
| 13982_g | −5.29 | 8.87 × 10−16 | Major latex-like protein |
| 13766_g | −5.13 | 3.46 × 10−22 | Transcription factor WER-like |
| 23514_g | −5.10 | 5.62 × 10−7 | Ethylene-responsive transcription factor ERF109-like |
| 3903_g | −4.76 | 8.02 × 10−15 | NAC domain-containing protein 79-like |
| 19874_g | −4.60 | 2.25 × 10−11 | Rosa chinensis proline-rich 33-kDa extensin-related protein-like |
| 3948_g | −3.26 | 5.15 × 10−8 | Rosa chinensis uncharacterized LOC112189886 |
| 26292_g | −3.02 | 1.38 × 10−62 | Uncharacterized protein LOC112197621 (Rosa chinensis) |
| 23873_g | 21.66 | 4.26 × 10−11 | Putative spindle and kinetochore-associated protein |
| 22675_g | 8.48 | 6.66 × 10−9 | Transcription factor MYB36 (Rosa chinensis) |
| 5242_g | 7.11 | 7.16 × 10−8 | Uncharacterized protein LOC112181570 |
| 13029_g | 6.61 | 2.31 × 10−8 | Uncharacterized protein LOC112167160 (Rosa chinensis) |
| 14750_g | 6.26 | 2.31 × 10−10 | Protein SRG1-like (Rosa chinensis) |
| 12450_g | 6.23 | 6.32 × 10−6 | Putative jacalin-like lectin domain-containing protein (Rosa chinensis) |
| 1056_g | 6.00 | 2.47 × 10−9 | 1-aminocyclopropane-1-carboxylate oxidase 5-like |
| 5349_g | 5.75 | 2.68 × 10−6 | Transcription factor RAX2-like |
| 20962_g | 5.74 | 1.31 × 10−7 | Peroxidase 27-like |
| 18908_g | 5.55 | 3.99 × 10−5 | Probable beta-1,3-galactosyltransferase 8 isoform X2 |
| 11647_g | 5.397 | 1.11 × 10−7 | Probable E3 ubiquitin-protein ligase ATL44 |
| 9170_g | 5.05 | 1.08 × 10−11 | Hypothetical protein |
| 2126_g | 4.95 | 4.48 × 10−5 | Hypothetical protein RchiOBHm_Chr1g0330981 (Rosa chinensis) |
| 19143_g | 4.95 | 4.24 × 10−13 | Transcription factor bHLH94-like (Fragaria vesca subsp. vesca) |
| 23421_g | 4.93 | 4.84 × 10−5 | Putative plant lipid transfer protein/Par allergen (Rosa chinensis) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khadgi, A.; Weber, C.A. RNA-Seq Analysis of Prickled and Prickle-Free Epidermis Provides Insight into the Genetics of Prickle Development in Red Raspberry (Rubus ideaus L.). Agronomy 2020, 10, 1904. https://doi.org/10.3390/agronomy10121904
Khadgi A, Weber CA. RNA-Seq Analysis of Prickled and Prickle-Free Epidermis Provides Insight into the Genetics of Prickle Development in Red Raspberry (Rubus ideaus L.). Agronomy. 2020; 10(12):1904. https://doi.org/10.3390/agronomy10121904
Chicago/Turabian StyleKhadgi, Archana, and Courtney A. Weber. 2020. "RNA-Seq Analysis of Prickled and Prickle-Free Epidermis Provides Insight into the Genetics of Prickle Development in Red Raspberry (Rubus ideaus L.)" Agronomy 10, no. 12: 1904. https://doi.org/10.3390/agronomy10121904
APA StyleKhadgi, A., & Weber, C. A. (2020). RNA-Seq Analysis of Prickled and Prickle-Free Epidermis Provides Insight into the Genetics of Prickle Development in Red Raspberry (Rubus ideaus L.). Agronomy, 10(12), 1904. https://doi.org/10.3390/agronomy10121904