Genetic Variation and Sequence Diversity of Starch Biosynthesis and Sucrose Metabolism Genes in Sweet Potato

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Sample Preparation and DNA Extraction
2.3. Genetic Diversity Analysis, Population Structure Analysis, and Starch Properties Evaluation
2.4. Candidate Gene Selection
2.5. Sequence Analysis and Primer Design
2.6. Gene Cloning and Reference Sequence Determination
2.7. DNA Equivalent Pooling
2.8. Amplicon Sequencing
2.9. Genotype Calling and Variation Filtering
2.10. Total Polymorphism Rate Calculation and Non-Synonymous SNP (nsSNP) Detection
2.11. Marker Development and Identification
2.12. ILP Marker Development and Identification
3. Results
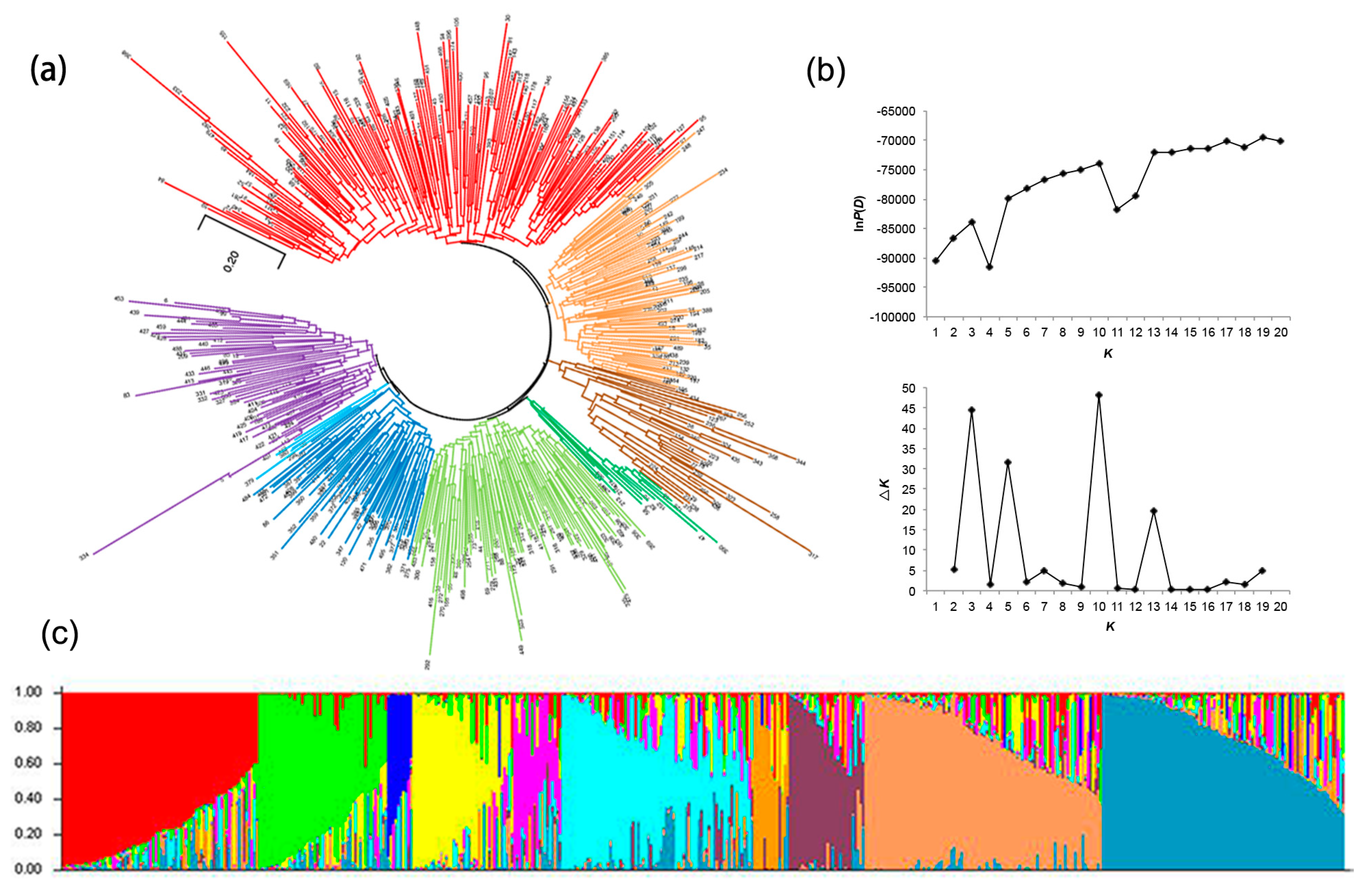
3.1. The Sweet Potato Germplasms Exhibited High Genetic and Phenotypic Diversity
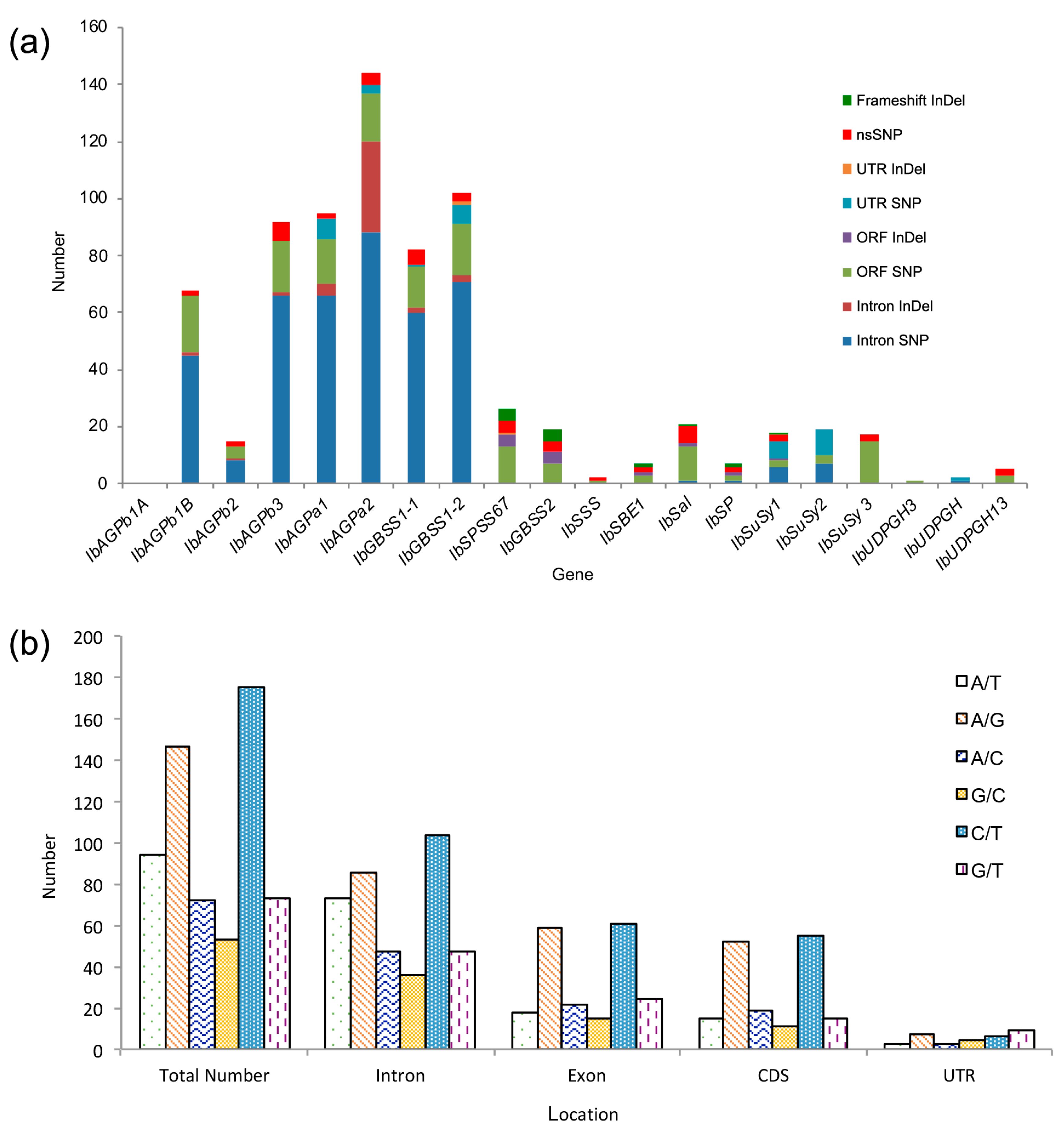
3.2. Twenty Candidate Genes Were Captured for Variation Detection
3.3. Number of Reads and Average Coverage Obtained from NGS
3.4. Detection of SNPs and InDels
3.5. Non-Synonymous Substitutions were Identified in Starch Biosynthesis and Metabolism Genes
3.6. Development of CAPS Markers and Verification of SNPs
3.7. Frameshift InDels were Detected
3.8. Two Gene Forms were Detected in IbAGPb3
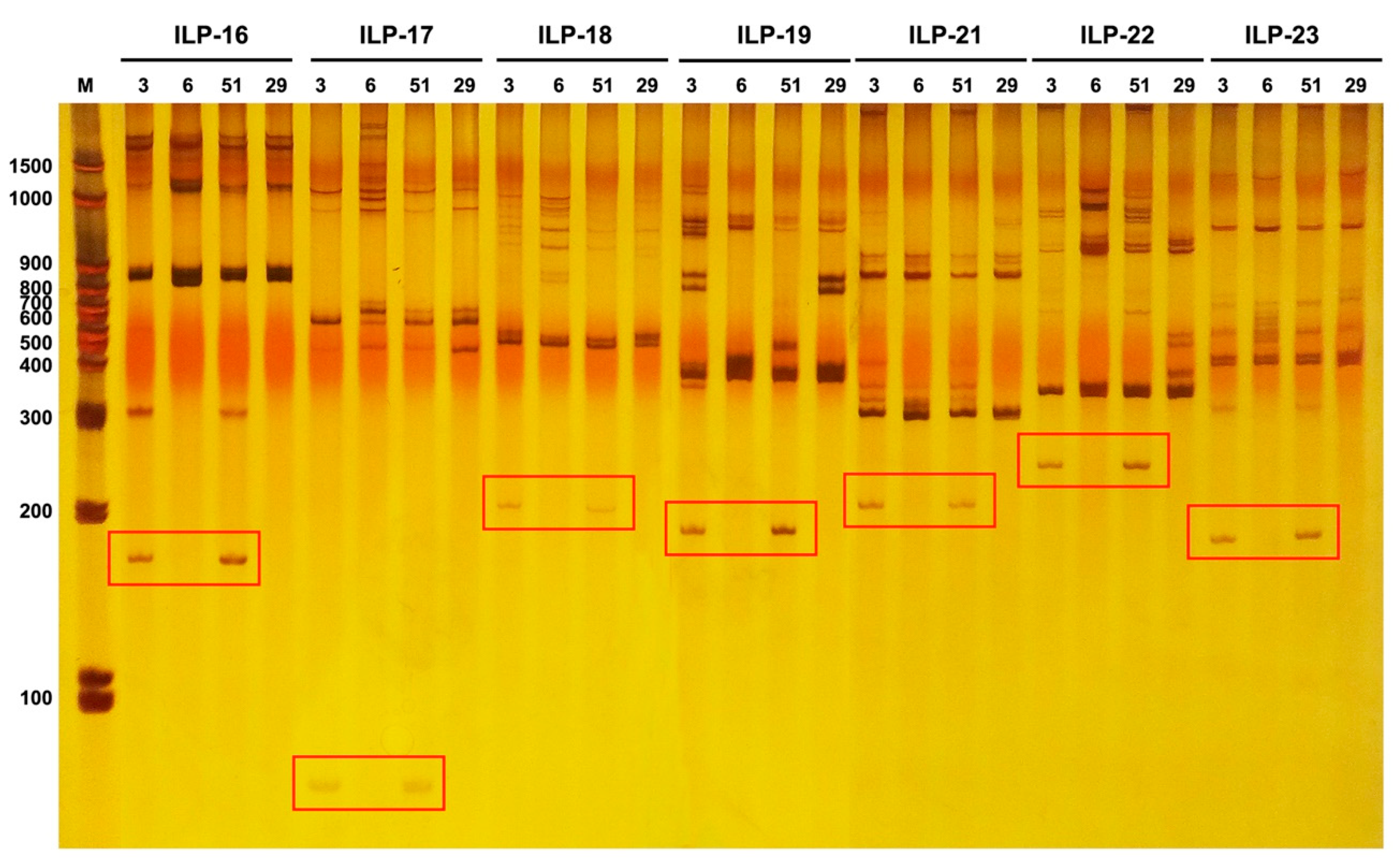
3.9. Intron Loss in the IbGBSS1-1 Genes
4. Discussion
4.1. Effective Strategies for Capturing and Identifying Allelic Variations in Hexaploid Sweet Potato
4.2. Characteristics of Gene Sequence Variation in Target Genes
4.3. CAPS Markers are An Effective Tool for SNP Genotyping in Sweet Potato
4.4. Creation of Intron-Loss Alleles Might Be a Characteristic Mechanism of Regulating Gene Expression in Sweet Potato
4.5. The Impact of Genetic Variations to Phenotype in Allohexaploid Sweet Potato
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kharabian-Masouleh, A.; Waters, D.L.E.; Reinke, R.F.; Henry, R.J. Discovery of polymorphisms in starch-related genes in rice germplasm by amplification of pooled DNA and deeply parallel sequencing. Plant Biotech. J. 2011, 9, 1074–1085. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.; Duran, C.; Berkman, P.J.; Lorenc, M.T.; Stiller, J.; Manoli, S.; Hayden, M.J.; Forrest, K.L.; Fleury, D.; Bumann, U.; et al. Single nucleotide polymorphism discovery from wheat next-generation sequence data. Plant Biotechnol. J. 2012, 10, 743–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhng, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaswall, K.; Sharma, H.; Bhandawat, A.; Sagar, R.; Yadav, V.K.; Sharma, V.; Mahajan, V.; Roy, J.; Singh, M. Development of intron length polymorphic (ILP) markers in onion (Allium cepa L.), and their cross-species transferability in garlic (A. sativum L.) and wild relatives. Genet. Resour. Crop. Evol. 2019, 66, 1379–1388. [Google Scholar] [CrossRef]
- Li, W.; Zhu, Z.; Chern, M.; Yin, J.; Yang, C.; Ran, L.; Cheng, M.; He, M.; Wang, K.; Wang, J.; et al. A natural allele of a transcription factor in rice confers broad-spectrum blast resistance. Cell 2017, 170, 114–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Chen, B.; Xu, K.; Wu, J.; Song, W.; Bancroft, I.; Harper, A.L.; Trick, M.; Liu, S.; Gao, G.; et al. Genome-wide association study dissects the genetic architecture of seed weight and seed quality in rapeseed (Brassica napus L.). DNA Res. 2014, 21, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef]
- Burri, B.J. Evaluating sweet potato as an intervention food to prevent vitamin A deficiency. Compr. Rev. Food Sci. Food Saf. 2011, 10, 118–130. [Google Scholar] [CrossRef]
- Mitra, S. Nutritional status of orange-fleshed sweet potatoes in alleviating Vitamin A malnutrition through a food-based approach. J. Nutr. Food Sci. 2012, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wu, Z.; Tang, D.; Luo, K.; Lu, H.; Liu, Y.; Dong, J.; Wang, X.; Lv, C.; Wang, J.; et al. Comparative transcriptome analysis reveals critical function of sucrose metabolism related-enzymes in starch accumulation in the storage root of sweet potato. Front. Plant Sci. 2017, 8, 914. [Google Scholar] [CrossRef]
- Nedunchezhiyan, M.; Byju, G.; Jata, S.K. Sweet Potato Agronomy. Fruit Veg. Cereal. Sci. Biotech. 2012, 6, 1–10. [Google Scholar]
- Srichuwong, S.; Orikasa, T.; Matsuki, J.; Shiina, T.; Kobayashi, T.; Tokuyasu, K. Sweet potato having a low temperature-gelatinizing starch as a promising feedstock for bioethanol production. Biomass Bioenergy 2012, 39, 120–127. [Google Scholar] [CrossRef]
- Koçar, G.; Civaş, N. An overview of biofuels from energy crops: Current status and future prospects. Renew. Sustain. Energy Rev. 2013, 28, 900–916. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, Z.; Tang, D.; Lv, C.; Luo, K.; Zhao, Y.; Liu, X.; Huang, Y.; Wang, J. Development and identification of SSR markers associated with starch properties and β-Carotene content in the storage root of sweet potato (Ipomoea batatas L.). Front. Plant Sci. 2016, 7, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Yang, J.; Hong, Y.; Liu, G.; Zheng, J.; Gu, Z.; Zhang, P. Impact of amylose content on starch physicochemical properties in transgenic sweet potato. Carbohydr. Polym. 2015, 122, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; He, S.; Zhao, N.; Zhai, H.; Liu, Q. A sucrose non-fermenting-1-related protein kinase-1 gene, IbSnRK1, improves starch content, composition, granule size, degree of crystallinity and gelatinization in transgenic sweet potato. Plant Biotechnol. J. 2019, 17, 21–32. [Google Scholar] [CrossRef]
- Tumwegamire, S.; Kapinga, R.; Rubaihayo, P.R.; Labonte, D.R.; Grüneberg, W.J.; Burgos, G.; Felde, T.Z.; Carpio, R.; Pawelzik, E.; Mwanga, R.O.M. Evaluation of dry Matter, protein, starch, sucrose, β-carotene, iron, zinc, calcium, and magnesium in East African sweetpotato [Ipomoea batatas (L.) Lam] germplasm. HortScience 2011, 46, 348–357. [Google Scholar] [CrossRef]
- Lai, Y.C.; Wang, S.Y.; Gao, H.Y.; Nguyen, K.M.; Nguyen, C.H.; Shih, M.C.; Lin, K.H. Physicochemical properties of starches and expression and activity of starch biosynthesis-related genes in sweet potatoes. Food Chem. 2016, 199, 556–564. [Google Scholar] [CrossRef]
- Kharabian-Masouleh, A.; Waters, D.L.E.; Reinke, R.F.; Ward, R.; Henry, R.J. SNP in starch biosynthesis genes associated with nutritional and functional properties of rice. Sci. Rep. 2012, 2, 557. [Google Scholar] [CrossRef]
- Cook, J.P.; McMullen, M.D.; Holland, J.B.; Tian, F.; Bradbury, P.; Ross-Ibarra, J.; Buckler, E.S.; Flint-Garcia, S.A. Genetic architecture of maize kernel composition in the nested association mapping and inbred association panels. Plant Physiology 2012, 158, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, L.; Nader-Nieto, A.C.; Schönhals, E.M.; Walkemeier, B.; Gebhardt, C. SNPs in genes functional in starch-sugar interconversion associate with natural variation of tuber starch and sugar content of potato (Solanum tuberosum L.). G3 Genes Genomes Genet. 2014, 4, 1797–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Otani, M.; Hamada, T.; Kim, S.H. Increase of amylose content of sweetpotato starch by RNA interference of the starch branching enzyme II gene (IbSBEII). Plant Biotech. 2006, 23, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Otani, M.; Hamada, T.; Katayama, K.; Kitahara, K.; Kim, S.H.; Takahata, Y.; Kim, S.-H.; Yasuhiro Takahata, Y.; Suganuma, T.; Shimada, T. Inhibition of the gene expression for granule-bound starch synthase I by RNA interference in sweet potato plants. Plant Cell Rep. 2007, 26, 1801–1807. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Zhang, H.; Zhai, H.; Liu, Q.; He, S. A soluble starch synthase I gene, IbSSI, alters the content, composition, granule size and structure of starch in transgenic sweet potato. Sci. Rep. 2017, 7, 2315. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.X.; Wu, Y.L.; Zhang, Y.D.; Yang, J.; Fan, W.J.; Zhang, H.; Zhao, S.S.; Yuan, L.; Zhang, P. CRISPR/Cas9-based mutagenesis of starch biosynthetic genes in sweet potato (Ipomoea Batatas) for the improvement of starch quality. Int. J. Mol. Sci. 2019, 20, 4702. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef]
- Su, W.J.; Zhao, N.; Lei, J.; Wang, L.J.; Chai, S.S.; Yang, X.S. SNP sites developed by specific length amplification fragment sequencing (SLAF-seq) in Sweetpotato. Sci. Agric. Sinica 2016, 49, 27–34. [Google Scholar]
- Kou, M.; Xu, J.; Li, Q.; Liu, Y.; Wang, X.; Tang, W.; Yan, H.; Zhang, Y.G.; Ma, D.F. Development of SNP markers using RNA-seq technology and tetra-primer ARMS-PCR in sweetpotato. J. Integr. Agric. 2017, 16, 464–470. [Google Scholar] [CrossRef]
- Shirasawa, K.; Tanaka, M.; Takahata, Y.; Ma, D.F.; Cao, Q.H.; Liu, Q.C.; Zhai, H.; Kwak, S.-S.; Jeong, J.C.; Cheol, J.; et al. A high-density SNP genetic map consisting of a complete set of homologous groups in autohexaploid sweetpotato (Ipomoea batatas). Sci. Rep. 2017, 7, 44207. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Moeinzadeh, M.H.; Kuhl, H.; Helmuth, J.; Xiao, P.; Haas, S.; Liu, G.; Zheng, J.L.; Sun, Z.; Weijuan Fan, W.J.; et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants 2017, 3, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Francki, M.G.; Forster, J.W. Identification, characterization and interpretation of single-nucleotide sequence variation in allopolyploid crop species. Plant Biotech. J. 2012, 10, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wu, Z.D.; Li, Y.H.; Zhang, H.; Wang, L.P.; Zhou, Q.L.; Tang, D.B.; Fu, Y.F.; He, F.F.; Jiang, Y.C.; et al. ISSR-based molecular characterization of an elite germplasm collection of sweet potato (Ipomoea batatas L.) in China. J. Integr. Agric. 2014, 13, 2346–2361. [Google Scholar] [CrossRef] [Green Version]
- Luo, K.; Lu, H.X.; Wu, Z.D.; Wu, X.L.; Yin, W.; Tang, D.B.; Wang, J.C.; Zhang, K. Genetic diversity and population structure analysis of main sweet potato breeding parents in southwest China. Sci. Agric. Sinica 2016, 49, 593–608. [Google Scholar]
- Kim, S.H.; Hamada, T. Rapid and reliable method of extracting DNA and RNA from sweetpotato, Ipomoea batatas (L). Lam. Biotechnol. Lett. 2005, 27, 1841–1845. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System, Version 2.1. Exeter Software; Setauket: New York, NY, USA, 2002. [Google Scholar]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenyetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Notes, A. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Hubisz, M.J.; Falush, D.; Stephens, M.; Pritchard, J.K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 2009, 9, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef] [PubMed]
- Isobe, S.; Shirasawa, K.; Hirakawa, H. Current status in whole genome sequencing and analysis of Ipomoea spp. Plant Cell Rep. 2019, 38, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingman, M.; Gyllensten, U. SNP frequency estimation using massively parallel sequencing of pooled DNA. Eur. J. Hum. Genet. 2009, 17, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Madelaine, R.; Notwell, J.H.; Skariah, G.; Halluin, C.; Chen, C.C.; Bejerano, G.; Mourrain, P. A screen for deeply conserved non-coding GWAS SNPs uncovers a MIR-9-2 functional mutation associated to retinal vasculature defects in human. Nucleic Acids Res. 2018, 46, 3517–3531. [Google Scholar] [CrossRef] [Green Version]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef] [Green Version]
- Hyma, K.E.; Barba, P.; Wang, M.; Londo, J.P.; Acharya, C.B.; Mitchell, S.E.; Sun, Q.; Reisch, B.; Cadle-Davidson, L. Heterozygous mapping strategy (HetMappS) for high resolution genotyping-by-sequencing markers: A case study in grapevine. PLoS ONE 2015, 10, 0134880. [Google Scholar] [CrossRef] [Green Version]
- Mugford, S.T.; Fernandez, O.; Brinton, J.; Flis, A.; Krohn, N.; Encke, B.; Regina Feil, R.; Sulpice, R.; Lunn, J.E.; Stitt, M.; et al. Regulatory properties of ADP glucose pyrophosphorylase are required for adjustment of leaf starch synthesis in different photoperiods. Plant Physiol. 2014, 166, 1733–1747. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.J.; Nevo, E. Exploring natural selection to guide breeding for agriculture. Plant Biotechnol. J. 2014, 12, 655–662. [Google Scholar] [CrossRef]
- Shavrukov, Y.N. CAPS markers in plant biology. Russ. J. Genet. Appl. Res. 2016, 6, 279–287. [Google Scholar] [CrossRef]
- van Wesemael, J.; Hueber, Y.; Kissel, E.; Campos, N.; Swennen, R.; Carpentier, S. Homeolog expression analysis in an allotriploid non-model crop via integration of transcriptomics and proteomics. Sci. Rep. 2018, 8, 1353. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.K.; Sonah, H.; Singh, N.K. Intron gain, a dominant evolutionary process supporting high levels of gene expression in rice. J. Plant Biochem. Biotechnol. 2016, 25, 142–146. [Google Scholar] [CrossRef]
- Hir, H.L.; Nott, A.; Moore, M.J. How introns influence and enhance eukaryotic gene expression. TRENDS Biochem. Sci. 2003, 28, 215–220. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Mourier, T.; Penny, D. The biology of intron gain and loss. TRENDS Genet. 2006, 22, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Mourier, T.; Jeffares, D.C. Eukaryotic Intron Loss. Science 2003, 300, 1393. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Soriano, S.F.; Cao, Q.H.; Yang, X.S.; Lu, G.Q. Hexaploid sweetpotato (Ipomoea batatas (L.) Lam.) may not be a true type to either auto- or allopolyploid. PLoS ONE 2020, 15, e0229624. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.H.; Yu, M.N.; Liu, M.; Zhang, R.; Sun, W.; Qian, M.C.; Duan, H.C.; Chang, W.; Ma, J.Q.; Qu, C.M.; et al. Genome-wide identification, evolutionary and expression analyses of the GALACTINOL SYNTHASE gene family in rapeseed and tobacco. Int. J. Mol. Sci. 2017, 18, 2768. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Steinmetz, L.M.; Gu, X.; Scharfe, C.; Davis, R.W.; Li, W.-H. Role of duplicate genes in genetic robustness against null mutations. Nature 2003, 421, 63–66. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Gene | Position | Nucleotide Substitution | Amino acid Change | Polarity Changed | Amino Acid Change in Conserved Domain |
|---|---|---|---|---|---|---|
| 1 | IbAGPb1B | 2,992 bp/457 aa | C/G | His to Gln | No | |
| 2 | 3,304 bp/507 aa | T/C | Val to Ala | No | ||
| 3 | IbAGPb2 | 1,572 bp/205 aa | G/A | Asp to Asn | Yes | Yes |
| 4 | 2,454 bp/340 aa | A/G | Try to His | No | Yes | |
| 5 | IbAGPb3 | 2 bp/1 aa | T/A | Start codon | ||
| 6 | 29 bp/10 aa | C/G | Ala to Gly | No | ||
| 7 | 160 bp/54 aa | G/A | Gly to Ser | Yes | ||
| 8 | 163 bp/55 aa | G/A | Thr to Ala | Yes | ||
| 9 | 166 bp/56 aa | A/G | Lys to Glu | No | ||
| 10 | 1,526 bp/294 aa | C/A | Pro to Gln | Yes | Yes | |
| 11 | 2,511 bp/445 aa | T/A | Phe to Try | Yes | ||
| 12 | IbAGPa1 | 125 bp/19 aa | G/T | Glu to Thr | Yes | |
| 13 | 2,263 bp/310 aa | C/T | Ala to Val | No | ||
| 14 | IbAGPa2 | 2,140 bp/286 aa | C/A | Phe to Leu | No | Yes |
| 15 | 2,538 bp/342 aa | A/C | Gln to Pro | Yes | Yes | |
| 16 | 2,717 bp/376 aa | G/C | Phe to Leu | No | ||
| 17 | 2,971 bp/423 aa | G/C | Val to Leu | Noa | ||
| 18 | IbGBSS1-1 | 2,296 bp/217 aa | A/C | Lys to Asn | Yes | |
| 19 | 4,276 bp | G/T | pre-termination | |||
| 20 | 4,280 bp/585 aa | T/G | Val to Gly | Yes | ||
| 21 | 4,283 bp/586 aa | C/A | Cys to Asp | Yes | ||
| 22 | 4,311 pb/595 aa | C/G | Asp to Glu | No | ||
| 23 | IbGBSS1-2 | 1,372 bp/137aa | A/G | Ile to Val | No | Yes |
| 24 | 1,892 bp/216aa | A/G | Ser to Gly | No | Yes | |
| 25 | 2,736 bp/376 aa | T/G | Val to Gly | Yes | ||
| 26 | IbSPSS67 | 198 bp/59 aa | T/C | Leu to Pro | No | |
| 27 | 421 bp/133 aaD | A/T | Glu to Asp | No | Yes | |
| 28 | 441 bp/140 aa | C/T | Pro to Leu | No | Yes | |
| 29 | 1,779 bp/586 aa | C/A | Ala to Asp | No | ||
| 30 | IbGBSS2 | 779 bp/209 aa | T/C | Phe to Leu | No | Yes |
| 31 | 797–798 bp/238 aa | AA/GG | Asn to Gly | Yes | Yes | |
| 32 | 915 bp /277 aa | T/A | Val to Glu | No | Yes | |
| 33 | 1,985 bp/634 aa | A/G | Ser to Gly | Yes | ||
| 34 | IbSSS | 691 bp/111 aa | G/A | Glu to Lys | No | |
| 35 | IbSBE1 | 658 bp | G/T | per-termination | ||
| 36 | 1,654 bp/493 aa | T/C | Ser to Pro | Yes | Yes | |
| 37 | IbSal | 139 bp/42 aa | A/G | Lys to Arg | No | |
| 38 | 453 bp/147 aa | C/A | Gln to Lys | Yes | Yes | |
| 39 | 641–642 bp/210 aa | TA/GT | Thr to Ser | No | ||
| 40 | 898 bp/295 aa | A/G | Lys to Arg | No | Yes | |
| 41 | 1467 bp | G/T | per-termination | |||
| 42 | 1472 bp/486 aa | G/T | Trp to Cys | No | Yes | |
| 43 | IbSP | 3,584 bp/458 aa | T/C | Ser to Pro | Yes | |
| 44 | 3,743 bp/511 aa | A/G | Lys to Glu | No | ||
| 45 | IbSuSy1 | 94 bp/9 aa | C/A | Thr to Asn | No | |
| 46 | 110 bp/12 aa | A/C | Gln to Pro | Yes | ||
| 47 | IbSuSy3 | 911–912 bp/289 aa | GG/CC | Gly to Ala | No | Yes |
| 48 | 1,280–1,281 bp/412 aa | GT/AC | Ser to Asn | No | Yes | |
| 49 | IbUDPGH13 | 140 bp/38 aa | G/A | Arg to Gln | Yes | Yes |
| 50 | 1,318 bp/431 aa | A/T | Lys to stop codon |
| Marker | Gene | Position | Base Change | Restriction Endonuclease | Primer Position | Primer Sequence |
|---|---|---|---|---|---|---|
| CAPS1 | IbAGPa1 | 2263 | C/T | BtgZI | 1875 | CGCTGGAGATCACCTATACCGAATGG |
| 2511 | CAGTGAGACTTCACATAGAGCTACTG | |||||
| CAPS2 | IbAGPa2 | 2538 | A/C | HindIII | 2242 | CCCTGGAGCCAATGACTTTGGAAGTG |
| 2794 | CACTGTCCGTGACATCAGCATCAAGC | |||||
| CAPS3 | IbAGPa2 | 2971 | G/C | DdeI | 2730 | GCTCCAATCTACACTCAGCCTCGATA |
| 3274 | GCCAATGCCAATTGGGATGCTGCCC | |||||
| CAPS4 | IbGBSS1-1 | 4283 | C/A | BseYI | 3878 | GTTGCTTGTGCTCAGTGTGAAACTG |
| 4327 | CAAGTGGTGCAATTTCGTCTCCTTC | |||||
| CAPS5 | IbSP | 3584 | T/C | RsaI | 3407 | TGGAGTTATGAGCTGATGGAGAAGC |
| 3937 | ATGAATCTCGGCAACTCCATTTACA | |||||
| CAPS6 | IbSuSy1 | 110 | A/C | EcoRII | 6 | TGTGACACCCGGGGAGCCTTCGTTCA |
| 470 | GCCACGTGTACCTATTTCAAGCAAAC |
| No. | Gene | Position (bp) | Deletion(D)/Insertion(I) | Deletion/Insertion nucleotide | Influence on protein |
|---|---|---|---|---|---|
| 1 | IbSPSS67 | 188–189 | I | C | Reading frame shift, and translation pre-terminate 64 bp downstream of the insertion site |
| 2 | 234 | D | A | Reading frame shift, and translation pre-terminate 32 bp downstream of the deletion site | |
| 3 | 437–438 | I | C | Glu to Ala (139 aa), reading frame shift and translation pre-terminate 22 bp downstream of the insertion site | |
| 4 | 1776 | D | T | Reading frame shift, amino acid changed and ORF become longer | |
| 5 | IbGBSS2 | 681–682 | I | A | Met to Ile (176 aa), and reading frame shift, and translation pre-terminate 30 bp downstream of the insertion site |
| 6 | 873 | D | A | Asp to Ala (240 aa), reading frame shift and translation pre-terminate 5 bp downstream of the deletion site | |
| 7 | 914 | D | G | Val to Trp (254 aa), and reading frame shift, translation pre-terminate 51 bp downstream of the deletion site | |
| 8 | 915 | D | T | Val to Gly (254 aa), and reading frame shift and translation pre-terminate 50 bp downstream of the deletion site | |
| 9 | IbSBE1 | 1835 | D | C | Ala to Val (553 aa), translation pre-terminate 32 bp downstream of the deletion site |
| 10 | IbSal | 1981–1982 | I | G | His to Gln, reading frame shift and translation pre-termination |
| 11 | IbSP | 3580–3581 | I | T | Amino acid changed from the insertion site and translation pre-terminate 26 bp downstream of the insertion site |
| 12 | IbSuSy1 | 96 | I | T | End coding and translation pre-termination |
| No. | Primer Name | Primer Sequence | Primer Length | Tm (°C) | Intron No. Detected | Start/Stop Site in Reference Sequence | Length of PCR Products without and with Intron Loss |
|---|---|---|---|---|---|---|---|
| 1 | FIbILP-16 | CGTGCTTCCACACTCTTGCAGTAGCTG | 27 | 69.16 | 1 | 412 | 662/166 |
| RIbILP-16 | CCCCACTTTTGATTCTCCAGAAGTGGCA | 27 | 67.54 | 1074 | |||
| 2 | FIbILP-17 | GGACTTGGAGATGTTCTTGGAGGATTGCC | 29 | 68.87 | 2 | 1294 | 444/77 |
| RIbILP-17 | CGGGGACACACTGTCATAACTCTATGCC | 28 | 69.01 | 1737 | |||
| 3 | FIbILP-18 | GTACAAAGATGCTTGGGATACCTGTGTG | 28 | 66.08 | 3 and 4 | 1747 | 392/196 |
| RIbILP-18 | GTCCTTGTAATCTTTCCCAGCCTTGGG | 27 | 67.64 | 2139 | |||
| 4 | FIbILP-19 | CAACCAGTTGCGGTTCAGTTTGTTGTGCC | 29 | 68.87 | 5 and 6 | 2139 | 357/173 |
| RIbILP-19 | TCATGTAGATTCCTCTCGACTGGTACATGG | 30 | 67.37 | 2496 | |||
| 5 | FIbILP-21 | CACGGGTACTTGTAATGGAATGGATACCCA | 30 | 67.37 | 9 | 2985 | 287/206 |
| RIbILP-21 | TGTCTGAGCCTTTCTGCTCTTCAAGTCTG | 29 | 67.45 | 3272 | |||
| 6 | FIbILP-22 | GCAGAAAGGCTCAGACATTCTTTATGCTGC | 30 | 67.37 | 10 | 3256 | 325/235 |
| RIbILP-22 | GAGACCACACGGCTCAAATCTGCTCG | 26 | 69.32 | 3581 | |||
| 7 | FIbILP-23 | GAGCCGTGTGGTCTCTTTCAGTTGCA | 26 | 67.75 | 11 and 12 | 3567 | 371/178 |
| RIbILP-23 | CAGTGGTTATCACCTTCAGCACGTCCT | 27 | 67.64 | 3938 | |||
| 8 | FIbILP-24 | CACTGAAATGATCAAGAACTGCATGTCAC | 29 | 64.62 | 13 | 3976 | 351/137 |
| RIbILP-24 | CAAGTGGTGCAATTTCGTCTCCTTCAA | 27 | 64.6 | 4327 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Luo, K.; Li, S.; Peng, D.; Tang, D.; Lu, H.; Zhao, Y.; Lv, C.; Wang, J. Genetic Variation and Sequence Diversity of Starch Biosynthesis and Sucrose Metabolism Genes in Sweet Potato. Agronomy 2020, 10, 627. https://doi.org/10.3390/agronomy10050627
Zhang K, Luo K, Li S, Peng D, Tang D, Lu H, Zhao Y, Lv C, Wang J. Genetic Variation and Sequence Diversity of Starch Biosynthesis and Sucrose Metabolism Genes in Sweet Potato. Agronomy. 2020; 10(5):627. https://doi.org/10.3390/agronomy10050627
Chicago/Turabian StyleZhang, Kai, Kai Luo, Shixi Li, Deliang Peng, Daobin Tang, Huixiang Lu, Yong Zhao, Changwen Lv, and Jichun Wang. 2020. "Genetic Variation and Sequence Diversity of Starch Biosynthesis and Sucrose Metabolism Genes in Sweet Potato" Agronomy 10, no. 5: 627. https://doi.org/10.3390/agronomy10050627
APA StyleZhang, K., Luo, K., Li, S., Peng, D., Tang, D., Lu, H., Zhao, Y., Lv, C., & Wang, J. (2020). Genetic Variation and Sequence Diversity of Starch Biosynthesis and Sucrose Metabolism Genes in Sweet Potato. Agronomy, 10(5), 627. https://doi.org/10.3390/agronomy10050627