Multi-Trait Regressor Stacking Increased Genomic Prediction Accuracy of Sorghum Grain Composition

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
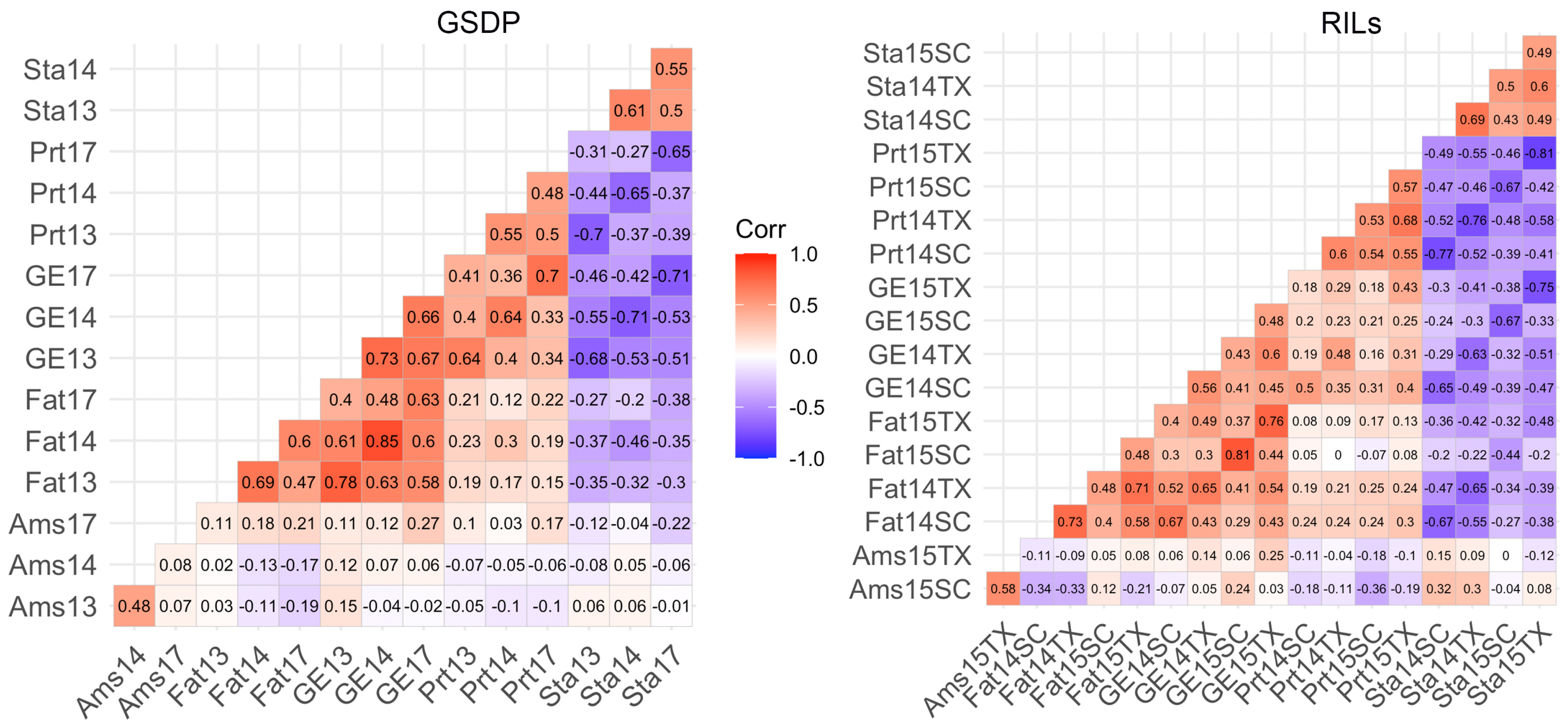
2.1. Phenotypic Variation
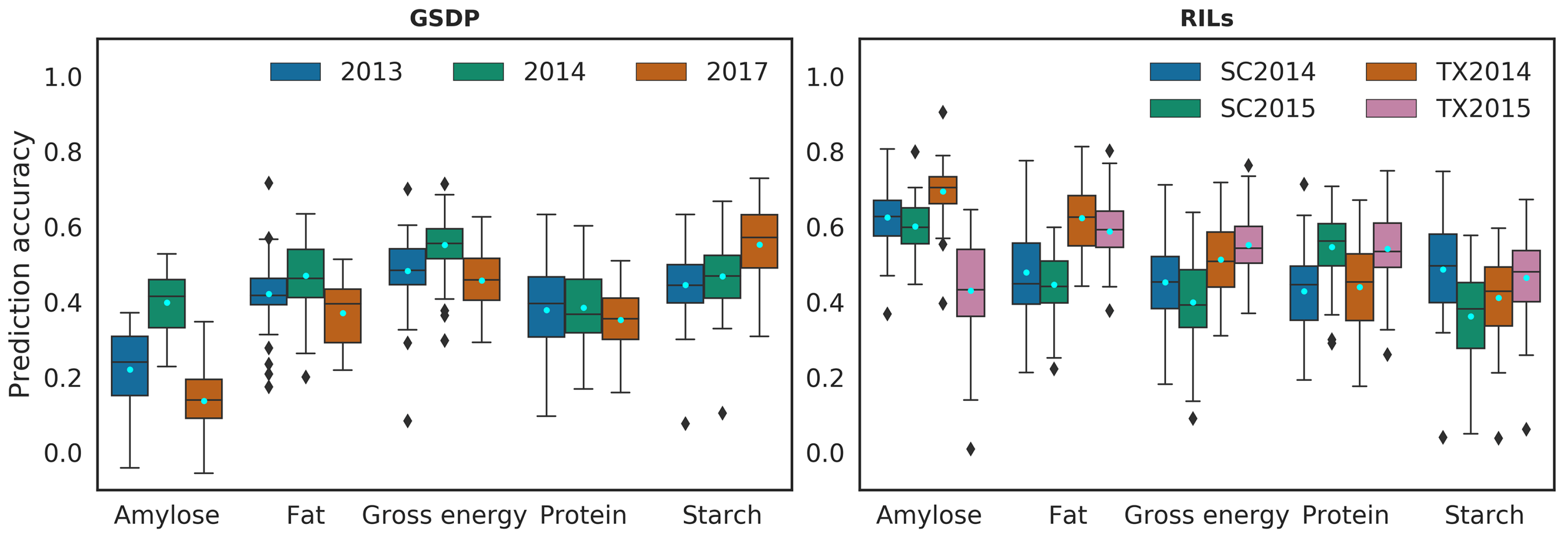
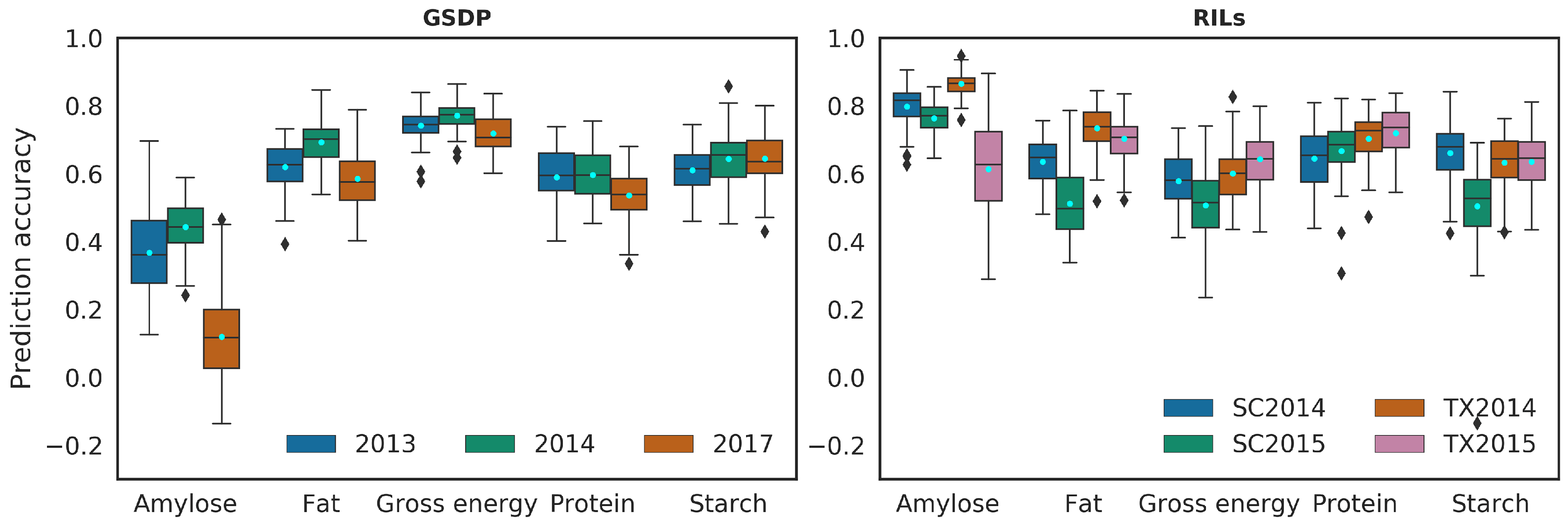
2.2. Prediction Performance in Single and Multiple Environment
2.3. Bayesian Multi-Output Regression Stacking
2.3.1. Five-Fold CV
2.3.2. Prediction of Whole Environment
3. Discussion
3.1. Trait Architecture and Prediction Accuracy
3.2. Multi-Trait Regressor Stacking
3.3. Application for Breeding
4. Materials and Methods
4.1. Plant Material
4.1.1. Grain Sorghum Diversity Panel
4.1.2. Recombinant Inbred Population
4.2. Phenotyping
4.3. Genotypic Data
4.4. Statistical Analysis
4.4.1. Single-Trait Single-Environment (STSE) Model:
4.4.2. Bayesian Multi-Environment (BME) GBLUP Model
4.4.3. Bayesian Multi-Output Regressor Stacking (BMORS)
4.4.4. Performance of Prediction Model:
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Abbreviations
| GSDP | Grain sorghum diversity panel |
| RIL | Recombinant inbred line |
| NIRS | Near infra-red spectroscopy |
| GP | Genomic prediction |
| GBLUP | Genomic best linear unbiased prediction |
| STSE | Single trait single environment |
| BME | Bayesian multi-environment |
| BMORS | Bayesian multi-output regressor stacking |
| MTME | Multi-trait multi-environment |
| CV | Cross validation |
| SC | South Carolina |
| TX | Texas |
| QTL | Quantitative trail loci |
| SNP | Single nucleotide polymorphism |
References
- Awika, J.M. Major cereal grains production and use around the world. In Advances in Cereal Science: Implications to Food Processing and Health Promotion; ACS Publications: Washington, DC, USA, 2011; pp. 1–13. [Google Scholar]
- Mace, E.S.; Tai, S.; Gilding, E.K.; Li, Y.; Prentis, P.J.; Bian, L.; Cruickshank, A. Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum. Nat. Commun. 2013, 4, 2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.R.; Schober, T.J.; Bean, S.R. Novel food and non-food uses for sorghum and millets. J. Cereal Sci. 2006, 44, 252–271. [Google Scholar] [CrossRef]
- Taylor, J. Food product development using sorghum and millets: Opportunities and challenges. Qual. Assur. Saf. Crop. Foods 2012, 4, 151. [Google Scholar] [CrossRef]
- Zhu, F. Structure, physicochemical properties, modifications, and uses of sorghum starch. Compr. Rev. Food Sci. Food Saf. 2014, 13, 597–610. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Bernardo, R.; Yu, J. Prospects for genomewide selection for quantitative traits in maize. Crop. Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef] [Green Version]
- Habier, D.; Fernando, R.L.; Dekkers, J.C. The impact of genetic relationship information on genome-assisted breeding values. Genetics 2007, 177, 2389–2397. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Dekkers, J.C.; Fernando, R.L.; Jannink, J.L. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: A barley case study. Genetics 2009, 182, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Combs, E.; Bernardo, R. Accuracy of genomewide selection for different traits with constant population size, heritability, and number of markers. Plant Genome 2013, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Dreisigacker, S. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgueño, J.; de los Campos, G.; Weigel, K.; Crossa, J. Genomic prediction of breeding values when modeling genotype × environment interaction using pedigree and dense molecular markers. Crop. Sci. 2012, 52, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Jarquín, D.; Crossa, J.; Lacaze, X.; Du Cheyron, P.; Daucourt, J.; Lorgeou, J.; Burgueño, J. A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor. Appl. Genet. 2014, 127, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Jannink, J.L. Multiple-trait genomic selection methods increase genetic value prediction accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Zhao, F.; Wang, Y.; Zhang, Y.; Du, L.; Su, G. Comparison of single-trait and multiple-trait genomic prediction models. BMC Genet. 2014, 15, 30. [Google Scholar] [CrossRef] [Green Version]
- Lado, B.; Vázquez, D.; Quincke, M.; Silva, P.; Aguilar, I.; Gutiérrez, L. Resource allocation optimization with multi-trait genomic prediction for bread wheat (Triticum aestivum L.) baking quality. Theor. Appl. Genet. 2018, 131, 2719–2731. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Toledo, F.H.; Pérez-Hernández, O.; Eskridge, K.M.; Rutkoski, J. A genomic Bayesian multi-trait and multi-environment model. G3 Genes Genomes Genet. 2016, 6, 2725–2744. [Google Scholar] [CrossRef] [Green Version]
- Montesinos-López, O.A.; Montesinos-López, A.; Luna-Vázquez, F.J.; Toledo, F.H.; Pérez-Rodríguez, P.; Lillemo, M.; Crossa, J. An R package for Bayesian analysis of multi-environment and multi-trait multi-environment data for genome-based prediction. G3 Genes Genomes Genet. 2019, 9, 1355–1369. [Google Scholar] [CrossRef] [Green Version]
- Spyromitros-Xioufis, E.; Tsoumakas, G.; Groves, W.; Vlahavas, I. Multi-target regression via input space expansion: Treating targets as inputs. Mach. Learn. 2016, 104, 55–98. [Google Scholar]
- Spyromitros-Xioufis, E.; Tsoumakas, G.; Groves, W.; Vlahavas, I. Multi-label classification methods for multi-target regression. arXiv 2012, arXiv:12116581. [Google Scholar]
- Heffner, E.L.; Jannink, J.L.; Iwata, H.; Souza, E.; Sorrells, M.E. Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop. Sci. 2011, 51, 2597–2606. [Google Scholar] [CrossRef] [Green Version]
- Battenfield, S.D.; Guzmán, C.; Gaynor, R.C.; Singh, R.P.; Peña, R.J.; Dreisigacker, S.; Poland, J.A. Genomic selection for processing and end-use quality traits in the CIMMYT spring bread wheat breeding program. Plant Genome 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haile, J.K.; N’Diaye, A.; Clarke, F.; Clarke, J.; Knox, R.; Rutkoski, J.; Pozniak, C.J. Genomic selection for grain yield and quality traits in durum wheat. Mol. Breed. 2018, 38, 75. [Google Scholar] [CrossRef]
- Schulthess, A.W.; Wang, Y.; Miedaner, T.; Wilde, P.; Reif, J.C.; Zhao, Y. Multiple-trait-and selection indices-genomic predictions for grain yield and protein content in rye for feeding purposes. Theor. Appl. Genet. 2016, 129, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tucker, D.M.; Basten, C.J.; Gandhi, H.; Ersoz, E.; Guo, B.; Gay, G. The impact of population structure on genomic prediction in stratified populations. Theor. Appl. Genet. 2014, 127, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Duhnen, A.; Gras, A.; Teyssèdre, S.; Romestant, M.; Claustres, B.; Daydé, J.; Mangin, B. Genomic selection for yield and seed protein content in Soybean: A study of breeding program data and assessment of prediction accuracy. Crop. Sci. 2017, 57, 1325–1337. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.; Panozzo, J.; Walker, C.; Choy, A.; Kant, S.; Wong, D.; Spangenberg, G.C. Accelerating wheat breeding for end-use quality with multi-trait genomic predictions incorporating near infrared and nuclear magnetic resonance-derived phenotypes. Theor. Appl. Genet. 2017, 130, 2505–2519. [Google Scholar] [CrossRef]
- Rapp, M.; Lein, V.; Lacoudre, F.; Lafferty, J.; Müller, E.; Vida, G.; Leiser, W.L. Simultaneous improvement of grain yield and protein content in durum wheat by different phenotypic indices and genomic selection. Theor. Appl. Genet. 2018, 131, 1315–1329. [Google Scholar] [CrossRef]
- Boyles, R.E.; Pfeiffer, B.K.; Cooper, E.A.; Rauh, B.L.; Zielinski, K.J.; Myers, M.T.; Kresovich, S. Genetic dissection of sorghum grain quality traits using diverse and segregating populations. Theor. Appl. Genet. 2017, 130, 697–716. [Google Scholar] [CrossRef] [Green Version]
- Velazco, J.G.; Jordan, D.R.; Mace, E.S.; Hunt, C.H.; Malosetti, M.; Van Eeuwijk, F.A. Genomic prediction of grain yield and drought-adaptation capacity in sorghum is enhanced by multi-trait analysis. Front. Plant Sci. 2019, 10, 997. [Google Scholar] [CrossRef] [Green Version]
- Murray, S.C.; Sharma, A.; Rooney, W.L.; Klein, P.E.; Mullet, J.E.; Mitchell, S.E.; Kresovich, S. Genetic improvement of sorghum as a biofuel feedstock: I. QTL for stem sugar and grain nonstructural carbohydrates. Crop. Sci. 2008, 48, 2165–2179. [Google Scholar] [CrossRef]
- Sukumaran, S.; Xiang, W.; Bean, S.R.; Pedersen, J.F.; Kresovich, S.; Tuinstra, M.R.; Yu, J. Association mapping for grain quality in a diverse sorghum collection. Plant Genome 2012, 5, 126–135. [Google Scholar] [CrossRef]
- Jannink, J.L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Briefings Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, S.; Boyles, R.; Cooper, E.; Brenton, Z.; Myers, M.; Kresovich, S. Impact of sorghum racial structure and diversity on genomic prediction of grain yield components. Crop. Sci. 2020, 60, 132–148. [Google Scholar] [CrossRef] [Green Version]
- Bhatta, M.; Gutierrez, L.; Cammarota, L.; Cardozo, F.; Germán, S.; Gómez-Guerrero, B.; Castro, A.J. Multi-trait Genomic Prediction Model Increased the Predictive Ability for Agronomic and Malting Quality Traits in Barley (Hordeum vulgare L.). G3 Genes Genomes Genet. 2020, 10, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casa, A.M.; Pressoir, G.; Brown, P.J.; Mitchell, S.E.; Rooney, W.L.; Tuinstra, M.R.; Kresovich, S. Community resources and strategies for association mapping in sorghum. Crop. Sci. 2008, 48, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Boyles, R.E.; Cooper, E.A.; Myers, M.T.; Brenton, Z.; Rauh, B.L.; Morris, G.P.; Kresovich, S. Genome-wide association studies of grain yield components in diverse sorghum germplasm. Plant Genome 2016, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kays, S.E.; Barton, F.E. Rapid prediction of gross energy and utilizable energy in cereal food products using near-infrared reflectance spectroscopy. J. Agric. Food Chem. 2002, 50, 1284–1289. [Google Scholar] [CrossRef]
- De Alencar Figueiredo, L.F.; Sine, B.; Chantereau, J.; Mestres, C.; Fliedel, G.; Rami, J.F.; Courtois, B. Variability of grain quality in sorghum: Association with polymorphism in Sh2, Bt2, SssI, Ae1, Wx and O2. Theor. Appl. Genet. 2010, 121, 1171–1185. [Google Scholar] [CrossRef]
- Morris, G.P.; Ramu, P.; Deshpande, S.P.; Hash, C.T.; Shah, T.; Upadhyaya, H.D.; Riera-Lizarazu, O.; Brown, P.J.; Acharya, C.B.; Mitchell, S.E.; et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. USA 2013, 110, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. 2019. Available online: https://www.R-project.org/ (accessed on 26 April 2019).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kassambara, A.; Mundt, F. Factoextra: Extract and visualize the results of multivariate data analyses. R Package Version 2017, 1, 337–354. [Google Scholar]
- Kruijer, W.; Boer, M.P.; Malosetti, M.; Flood, P.J.; Engel, B.; Kooke, R.; van Eeuwijk, F.A. Marker-based estimation of heritability in immortal populations. Genetics 2015, 199, 379–398. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, O.A.; Montesinos-López, A.; Crossa, J.; Cuevas, J.; Montesinos-López, J.C.; Gutiérrez, Z.S.; Singh, R. A Bayesian genomic multi-output regressor stacking model for predicting multi-trait multi-environment plant breeding data. G3 Genes Genomes Genet. 2019, 9, 3381–3393. [Google Scholar] [CrossRef] [Green Version]
- Godbole, S.; Sarawagi, S. Discriminative methods for multi-labeled classification. In Proceedings of the Pacific-Asia Conference on Knowledge Discovery and Data Mining, Sydney, Australia, 26–28 May 2004; Springer: Berlin/Heidelberg, Germany, 2004; pp. 22–30. [Google Scholar]
- Pérez, P.; de Los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
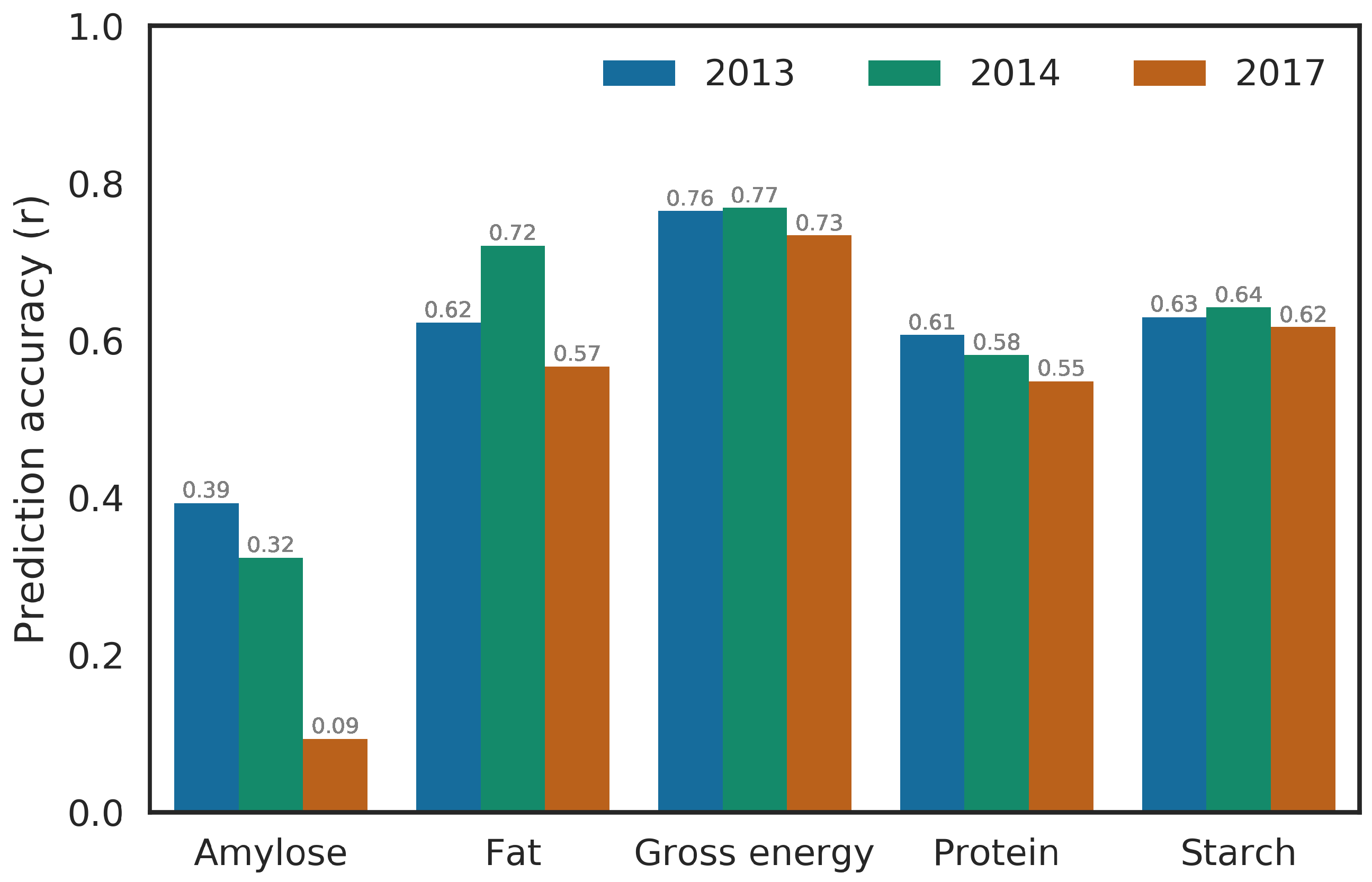{kind=link}
| Trait | NIRS | GSDP | RILs | |||
|---|---|---|---|---|---|---|
| SECV | Mean ± SD | Mean ± SD | ||||
| Amylose | 0.60 | 2.24 | 13.87 ± 2.98 | 0.24 | 11.49 ± 4.32 | 0.77 |
| Fat | 0.41 | 0.53 | 2.53 ± 0.57 | 0.54 | 3.07 ± 0.67 | 0.76 |
| Gross energy | 0.71 | 25.80 | 4108.33 ± 55.15 | 0.59 | 4124.56 ± 41.74 | 0.69 |
| Protein | 0.96 | 0.27 | 12.02 ± 1.45 | 0.39 | 11.43 ± 1.03 | 0.83 |
| Starch | 0.89 | 0.75 | 68.30 ± 2.44 | 0.44 | 68.37 ± 1.87 | 0.79 |
| Trait | 2013 | 2014 | 2017 | |||
|---|---|---|---|---|---|---|
| BME | BMORS | BME | BMORS | BME | BMORS | |
| Amylose | −11 | 66 | −5 | 11 | −13 | −13 |
| Fat | −24 | 47 | −12 | 47 | −27 | 58 |
| Gross energy | 3 | 54 | −2 | 40 | 1 | 57 |
| Protein | −3 | 56 | −1 | 55 | −8 | 52 |
| Starch | 4 | 37 | −2 | 38 | 1 | 17 |
| Average | −6 | 52 | −4 | 38 | −9 | 34 |
| Trait | SC2014 | SC2015 | TX2014 | TX2015 | ||||
|---|---|---|---|---|---|---|---|---|
| BME | BMORS | BME | BMORS | BME | BMORS | BME | BMORS | |
| Amylose | 2 | 28 | 0 | 28 | −1 | 25 | 12 | 43 |
| Fat | 5 | 33 | 1 | 15 | 2 | 18 | 2 | 20 |
| Gross energy | 7 | 28 | −3 | 27 | 1 | 18 | 3 | 17 |
| Protein | 10 | 51 | 1 | 23 | 5 | 60 | −4 | 33 |
| Starch | 5 | 36 | −4 | 40 | 7 | 54 | 4 | 37 |
| Average | 6 | 35 | −1 | 27 | 3 | 35 | 3 | 30 |
| Trait | Year × Location | Location | Year | |||||
|---|---|---|---|---|---|---|---|---|
| SC2014 | SC2015 | TX2014 | TX2015 | SC | TX | 2014 | 2015 | |
| Amylose | 0.79 | 0.80 | 0.88 | 0.60 | 0.76 | 0.74 | 0.74 | 0.65 |
| Fat | 0.69 | 0.49 | 0.78 | 0.74 | 0.60 | 0.71 | 0.64 | 0.60 |
| Gross energy | 0.56 | 0.49 | 0.62 | 0.66 | 0.48 | 0.58 | 0.56 | 0.56 |
| Protein | 0.65 | 0.66 | 0.66 | 0.70 | 0.59 | 0.58 | 0.61 | 0.66 |
| Starch | 0.64 | 0.52 | 0.68 | 0.60 | 0.55 | 0.56 | 0.56 | 0.55 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapkota, S.; Boatwright, J.L.; Jordan, K.; Boyles, R.; Kresovich, S. Multi-Trait Regressor Stacking Increased Genomic Prediction Accuracy of Sorghum Grain Composition. Agronomy 2020, 10, 1221. https://doi.org/10.3390/agronomy10091221
Sapkota S, Boatwright JL, Jordan K, Boyles R, Kresovich S. Multi-Trait Regressor Stacking Increased Genomic Prediction Accuracy of Sorghum Grain Composition. Agronomy. 2020; 10(9):1221. https://doi.org/10.3390/agronomy10091221
Chicago/Turabian StyleSapkota, Sirjan, J. Lucas Boatwright, Kathleen Jordan, Richard Boyles, and Stephen Kresovich. 2020. "Multi-Trait Regressor Stacking Increased Genomic Prediction Accuracy of Sorghum Grain Composition" Agronomy 10, no. 9: 1221. https://doi.org/10.3390/agronomy10091221
APA StyleSapkota, S., Boatwright, J. L., Jordan, K., Boyles, R., & Kresovich, S. (2020). Multi-Trait Regressor Stacking Increased Genomic Prediction Accuracy of Sorghum Grain Composition. Agronomy, 10(9), 1221. https://doi.org/10.3390/agronomy10091221