
A Two-Step PCR Protocol Enabling Flexible Primer Choice and High Sequencing Yield for Illumina MiSeq Meta-Barcoding



Abstract
:1. Introduction
2. Materials and Methods
Overview of Three-Step and Two-Step PCR Library Construction
3. Detailed Workflow of Two-Step PCR (2P) Protocol
3.1. Step 1: DNA Preparation
3.1.1. Sample Collection and Processing
3.1.2. DNA Extraction
3.1.3. Methods (Optional) to Prevent PCR Inhibitors in Extracted DNA Affecting the Following PCR Steps
3.2. Performing the First PCR Using Thermocycler Program “2P_1st”
3.2.1. PCR Reagent Preparation
3.2.2. PCR Amplification
3.3. First PCR Product Clean-Up
3.3.1. AMPure Bead Preparation
3.3.2. Mix AMPure Beads and PCR Products
3.3.3. Separate Solution versus AMPure Beads
3.3.4. Ethanol Wash
3.3.5. Beads and DNA Resuspension
3.3.6. AMpure Bead Separation and Removal
3.4. Performing the Second PCR Using Thermocycler Program “2P_2nd”
3.4.1. PCR Reagent Preparation
3.4.2. PCR Amplification
3.5. Second PCR Product Clean-Up
3.5.1. AMPure Beads Preparation
3.5.2. Mix AMPure Beads and PCR Products
3.5.3. Separate Solution versus AMPure Beads
3.5.4. Ethanol Wash
3.5.5. Beads and DNA Resuspension
3.5.6. AMpure Bead Separation and Removal
3.6. PCR Product Evaluation and Multiplex
3.6.1. Gel Preparation and Electrophoresis
3.6.2. Gel Examination
3.6.3. DNA Quantity and Purity Assessment
3.6.4. Sample Multiplexing
3.7. Submit Samples for Sequencing
3.7.1. Sample Submission to the Sequencing Facility
3.7.2. Submission Condition Inquiry
3.8. Hardware Requirements
- -
- Thermocycler: any thermocycler allowing temperature to decrease per cycle should work.
- -
- Magnetic stands: PCR purification step is essential to remove remaining dNTPs and primer dimers that might be present in PCR products. To perform purification with paramagnetic beads, a 96-well magnetic plate is essential.
- -
- Nucleotide spectrophotometer or fluorometer: to obtain an accurate concentration of DNA across the cleaned-up 2P_2nd product, a spectrophotometer or fluorometer is required. In addition to reporting the DNA concentration, the spectrophotometer also reveals common contaminations (e.g., ethanol and phenolic compounds) in the PCR product. The fluorometer, on the other hand, is believed to provide a more accurate estimation of DNA concentration.
- -
- Gel electrophoresis system: horizontal electrophoresis system.
- -
- Gel imaging system: gel Documentation System
3.9. Synthetic Oligonucleotides
4. Results and Discussion
4.1. Evaluation of PCR Efficiency by Assessing the Amplicon Length
4.2. The Effect of PCR Library Protocols (2P, 3P, and 3P+Cleanup) on the Quantity and Quality of Sequence Reads
4.3. Across Run Comparison for the Proportion of Undetermined Barcoded Sequences Generated with 2P vs. 3P Approaches
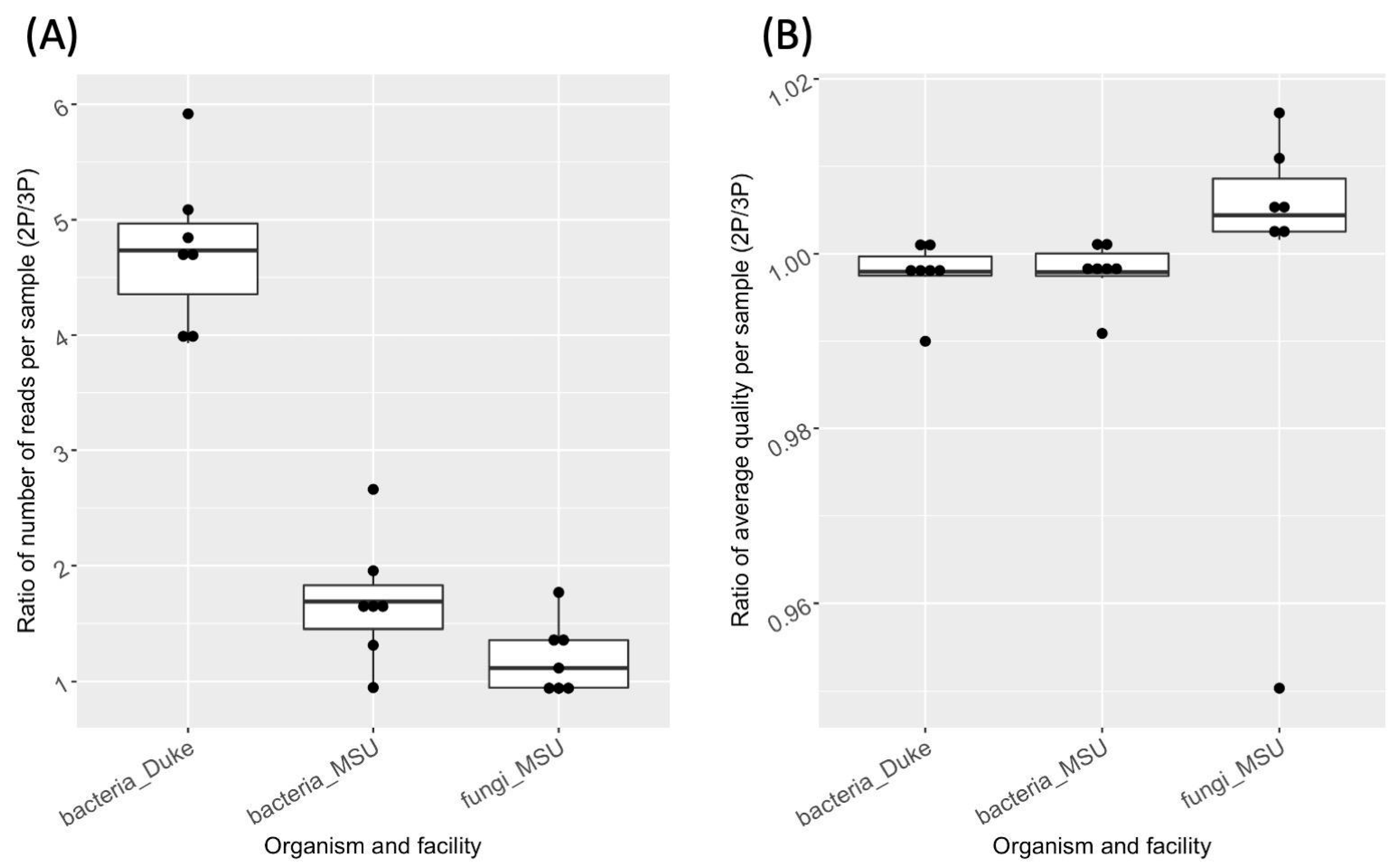
4.4. Evaluation of 2P and 3P Performance for Libraries Targeting Different Taxonomic Groups
4.5. 2P and 3P Comparison between Sequencing Facilities
4.6. Touchdown Technique in Improving Multi-Step PCR for Next-Generation Amplicon Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schnell, I.B.; Bohmann, K.; Gilbert, M. Tag jumps illuminated—reducing sequence-to-sample misidentifications in metabarcoding studies. Mol. Ecol. Resour. 2015, 15, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.P.; Peay, K.G. Sequence Depth, Not PCR Replication, Improves Ecological Inference from Next Generation DNA Sequencing. PLoS ONE 2014, 9, e90234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Truong, C.; Gabbarini, L.A.; Corrales, A.; Mujic, A.B.; Escobar, J.M.; Moretto, A.; Smith, M.E. Ectomycorrhizal fungi and soil enzymes exhibit contrasting patterns along elevation gradients in southern Patagonia. New Phytol. 2019, 222, 1936–1950. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Liao, H.-L.; Arnold, A.E.; Bonito, G.; Lutzoni, F. RNA-based analyses reveal fungal communities structured by a senescence gradient in the moss Dicranum scoparium and the presence of putative multi-trophic fungi. New Phytol. 2018, 218, 1597–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Benucci, G.M.N.; Burnard, D.; Shepherd, L.D.; Bonito, G.; Munkacsi, A.B. Evidence for Co-evolutionary History of Early Diverging Lycopodiaceae Plants With Fungi. Front. Microbiol. 2020, 10, 2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beule, L.; Chen, K.-H.; Hsu, C.-M.; Mackowiak, C.; Dubeux, J.C.B.; Blount, A.; Liao, H.-L.; Dubeux, J.C.B., Jr. Soil bacterial and fungal communities of six bahiagrass cultivars. PeerJ 2019, 7, e7014. [Google Scholar] [CrossRef] [PubMed]
- Longley, R.; Noel, Z.A.; Benucci, G.M.N.; Chilvers, M.I.; Trail, F.; Bonito, G. Crop Management Impacts the Soybean (Glycine max) Microbiome. Front. Microbiol. 2020, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hsu, C.; Dubeux, J.C.B., Jr.; Mackowiak, C.; Blount, A.; Han, X.; Liao, H. Effects of rhizoma peanut cultivars (Arachis glabrata Benth.) on the soil bacterial diversity and predicted function in nitrogen fixation. Ecol. Evol. 2019, 9, 12676–12687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.-L.; Chen, Y.; Bruns, T.D.; Peay, K.G.; Taylor, J.W.; Branco, S.; Talbot, J.M.; Vilgalys, R. Metatranscriptomic analysis of 567 ectomycorrhizal roots reveal genes associated with Piloderma-Pinus symbiosis: Improved methodologies for assessing gene 568 expression in situ. Environ. Microbiol. 2014, 16, 3730–3742. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org (accessed on 1 November 2018).
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Stortchevoi, A.; Kamelamela, N.; Levine, S.S. SPRI Beads-based Size Selection in the Range of 2–10kb. J. Biomol. Tech. JBT 2020, 31, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Lucena-Aguilar, G.; Sánchez-López, A.M.; Barberá;n-Aceituno, C.; Carrillo-Ávila, J.A.; López-Guerrero, J.A.; Aguilar-Quesada, R. DNA source selection for downstream applications based on DNA quality indicators analysis. Biopreserv. Biobank. 2016, 14, 264–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.L.; Walters, W.A.; Lennon, N.J.; Bochicchio, J.; Krohn, A.; Caporaso, J.G.; Pennanen, T. Accurate Estimation of Fungal Diversity and Abundance through Improved Lineage-Specific Primers Optimized for Illumina Amplicon Sequencing. Appl. Environ. Microbiol. 2016, 82, 7217–7226. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press: Cambridge, MA, USA, 1990; pp. 315–322. [Google Scholar]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a Prokaryotic Universal Primer for Simultaneous Analysis of Bacteria and Archaea Using Next-Generation Sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Don, R.H.; Cox, P.T.; Wainwright, B.J.; Baker, K.; Mattick, J.S. ‘Touchdown’ PCR to circumvent spurious priming during gene 561 amplification. Nucleic Acids Res. 1991, 19, 4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protocol | 2P (This Study) | 3P (Chen et al., 2018) | 3P+Cleanup |
|---|---|---|---|
| Task Summary (Description) | Step No. | Step Included or Not | Step Included or Not |
| DNA extraction | Step 1 | Yes | Yes |
| PCR amplification (Amplify with FGRSP/RGRSP primers) | NA | Yes | Yes |
| PCR amplification (Amplify with FGRSP_F1-F6/RGRSP_F1-F6) | Step 2 | Yes | Yes |
| Beads clean-up (1st PCR product clean-up) | Step 3 | NA | Yes |
| PCR amplification (Amplify with PCR_F, PCR_R_bc (x)) | Step 4 | Yes | Yes |
| Beads clean-up (2nd PCR product clean-up) | Step 5 | Yes | Yes |
| PCR product evaluation and multiplex | Step 6 | Yes | Yes |
| Sequencing | Step 7 | Yes | Yes |
| Two-Step PCR (2P) (This Study) | Three-Step PCR (3P) (Chen et al., 2018) | |||||||
|---|---|---|---|---|---|---|---|---|
| Primer Sets | Cycle Number | Product Name | Annealing T (°C) | DNA Input (μL) | Cycle Number | Product Name | Annealing T. (°C) | DNA Input (μL) |
| FGRSP, RGRSP | N/A | N/A | N/A | N/A | 10 | 3P_1st | Constant | 0.5 |
| FGRSP_F1-F6, RGRSP_F1-F6 | 15 | 2P_1st | Touchdown | 1 | 10 | 3P_2nd | Constant | 2.5 |
| PCR_F, PCR_R_bc (x) | 15 | 2P_2nd | Touchdown | 1.6 | 10 | 3P_3rd | Constant | 10 |
| 2P_1st | 2P_2nd | ||||
|---|---|---|---|---|---|
| Reagent | Volume (μL) | Volume (μL) | Reagent | Volume (μL) | Volume (μL) |
| 10× PCR buffer | 1.25 | 10× PCR buffer | 2.5 | ||
| MgCl2 (50 mM) | 0.375 | MgCl2 (50 mM) | 0.75 | ||
| FGRSP_F1-F6 (10 μM) | 0.25 | PCR_F primer (10 μM) | 0.5 | ||
| RGRSP_F1-F6 (10 μM) | 0.25 | dNTP 10 mM | 0.5 | ||
| dNTP 10 mM | 0.25 | Taq (5 U/μL) | 0.08 | ||
| Taq (5 U/μL) | 0.05 | Water | 18.57 | ||
| Water | 9.075 | Master mix: 22.9 | |||
| Master mix: 11.5 | Product from C_2P_1st | 1.6 | Add individually | ||
| DNA (1:50×) * | 1 | Add individually | Barcode_primer | 0.5 | Add individually |
| Total = 12.5 | Total = 25 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.-H.; Longley, R.; Bonito, G.; Liao, H.-L. A Two-Step PCR Protocol Enabling Flexible Primer Choice and High Sequencing Yield for Illumina MiSeq Meta-Barcoding. Agronomy 2021, 11, 1274. https://doi.org/10.3390/agronomy11071274
Chen K-H, Longley R, Bonito G, Liao H-L. A Two-Step PCR Protocol Enabling Flexible Primer Choice and High Sequencing Yield for Illumina MiSeq Meta-Barcoding. Agronomy. 2021; 11(7):1274. https://doi.org/10.3390/agronomy11071274
Chicago/Turabian StyleChen, Ko-Hsuan, Reid Longley, Gregory Bonito, and Hui-Ling Liao. 2021. "A Two-Step PCR Protocol Enabling Flexible Primer Choice and High Sequencing Yield for Illumina MiSeq Meta-Barcoding" Agronomy 11, no. 7: 1274. https://doi.org/10.3390/agronomy11071274
APA StyleChen, K. -H., Longley, R., Bonito, G., & Liao, H. -L. (2021). A Two-Step PCR Protocol Enabling Flexible Primer Choice and High Sequencing Yield for Illumina MiSeq Meta-Barcoding. Agronomy, 11(7), 1274. https://doi.org/10.3390/agronomy11071274