
Transcriptomic Analysis of Short-Term Salt-Stress Response in Mega Hybrid Rice Seedlings

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth Condition and Salinity Stress Treatment
2.2. Phenotype Evaluation of Seedlings under Salt Stress
2.3. RNA Extraction and cDNA Library Preparation and Sequencing
2.4. Data Filtering and Assembly
2.5. Analysis of Differentially Expressed Genes and Functional Annotation
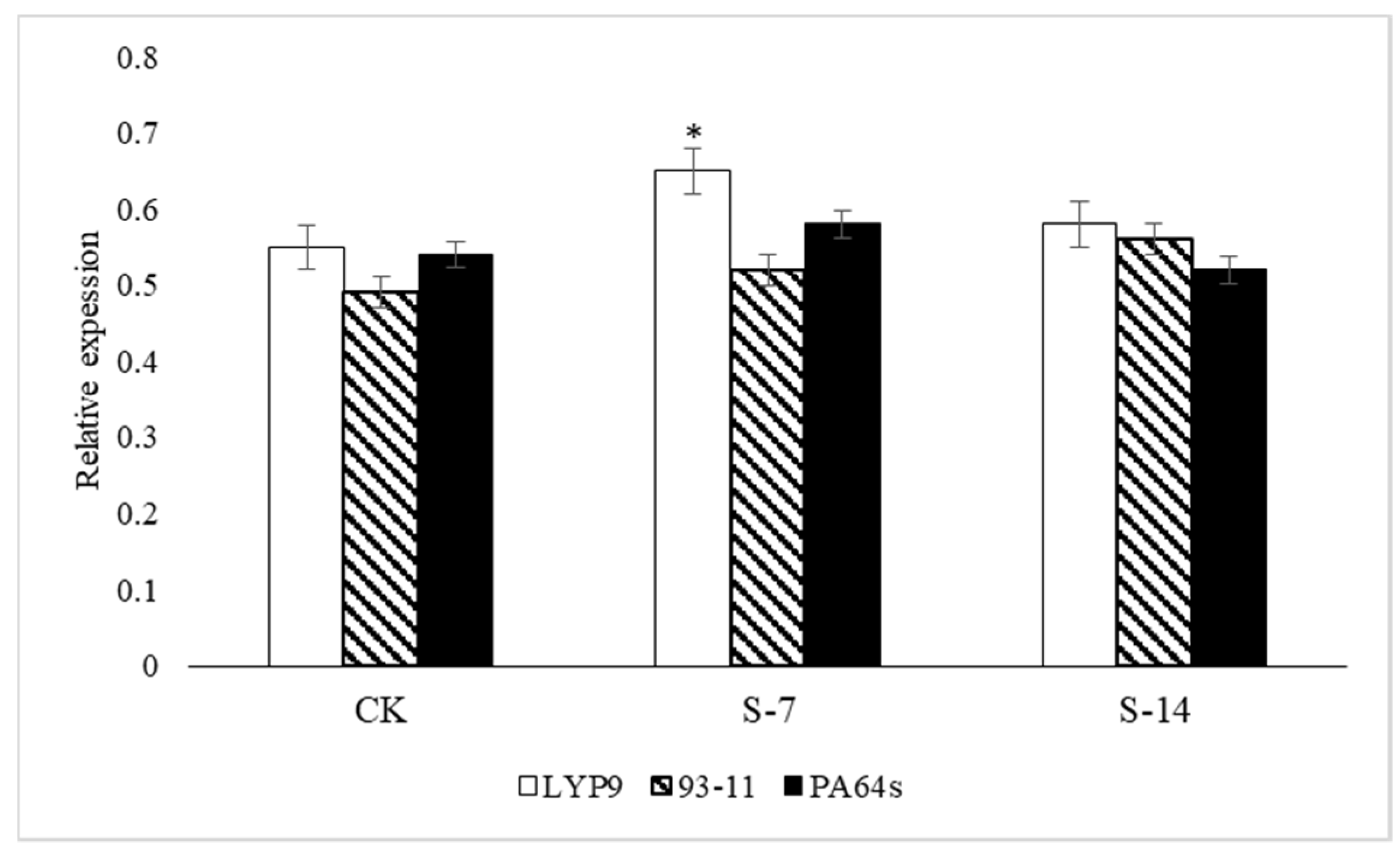
2.6. RT-qPCR Analysis
3. Results
3.1. Transcriptome Sequencing of the Hybrid LYP9 and Its Parents under Two Salinity Stresses
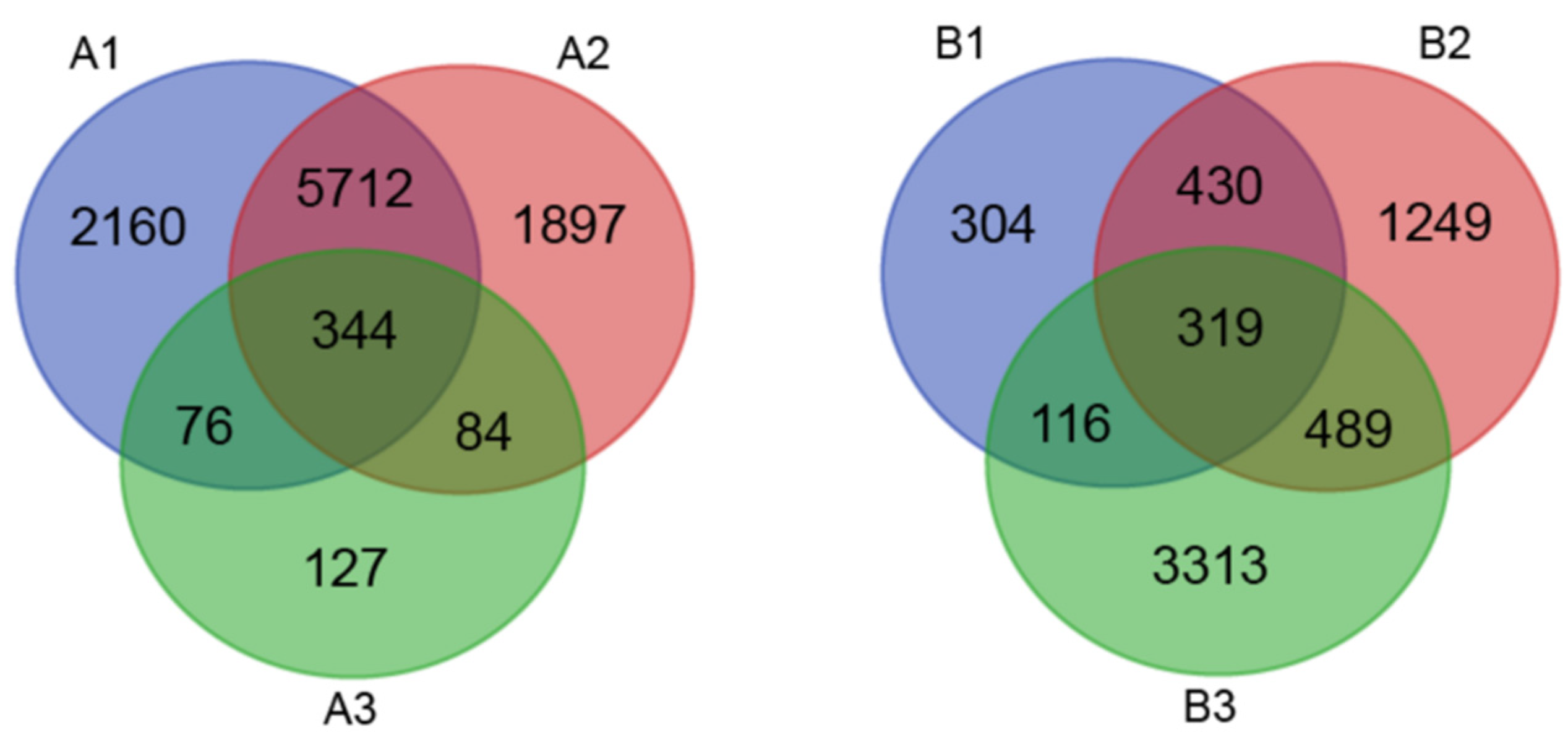
3.2. Identification of Salt Stress Response Differentially Expressed Genes (DEGs) by RNA-Seq
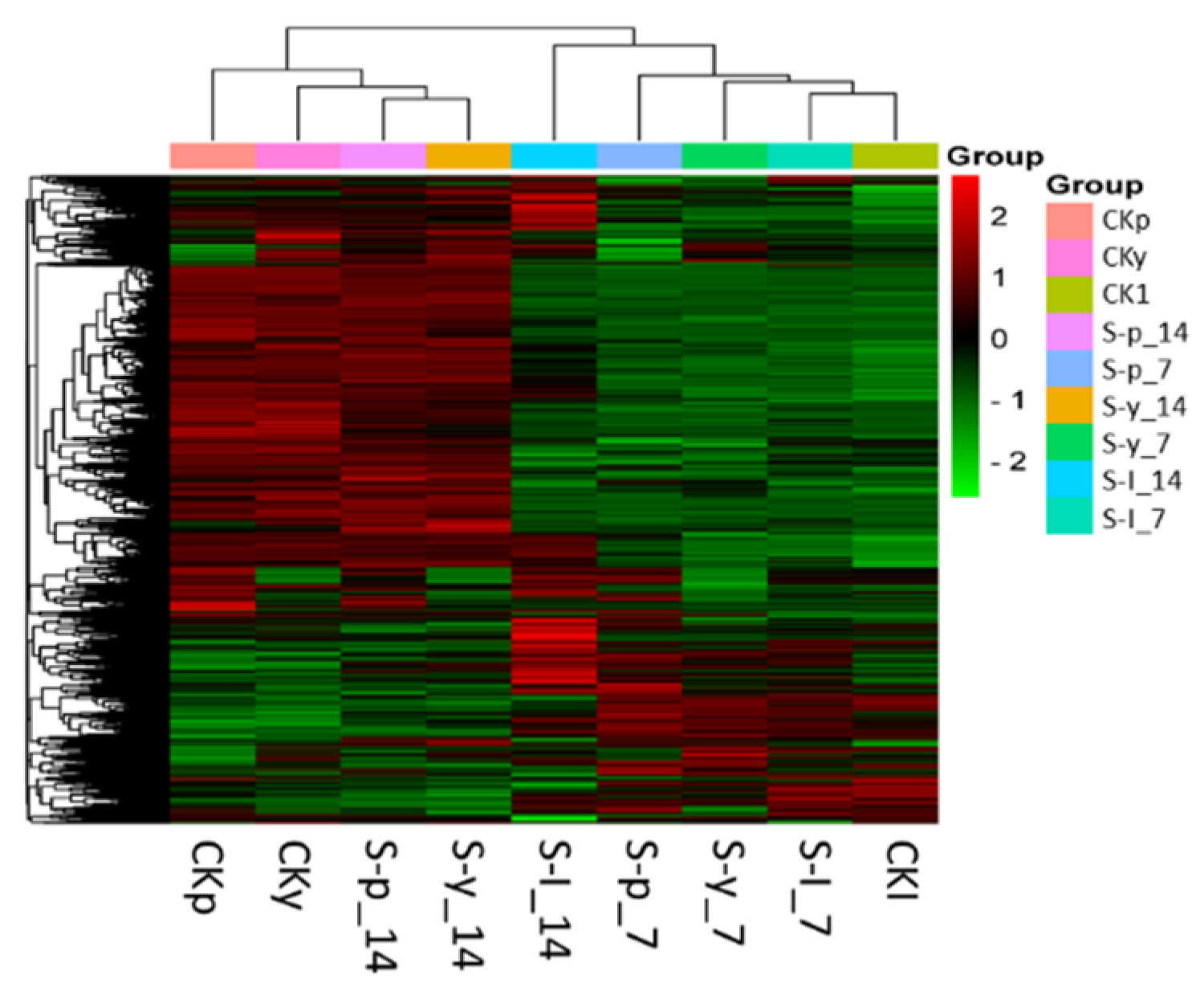
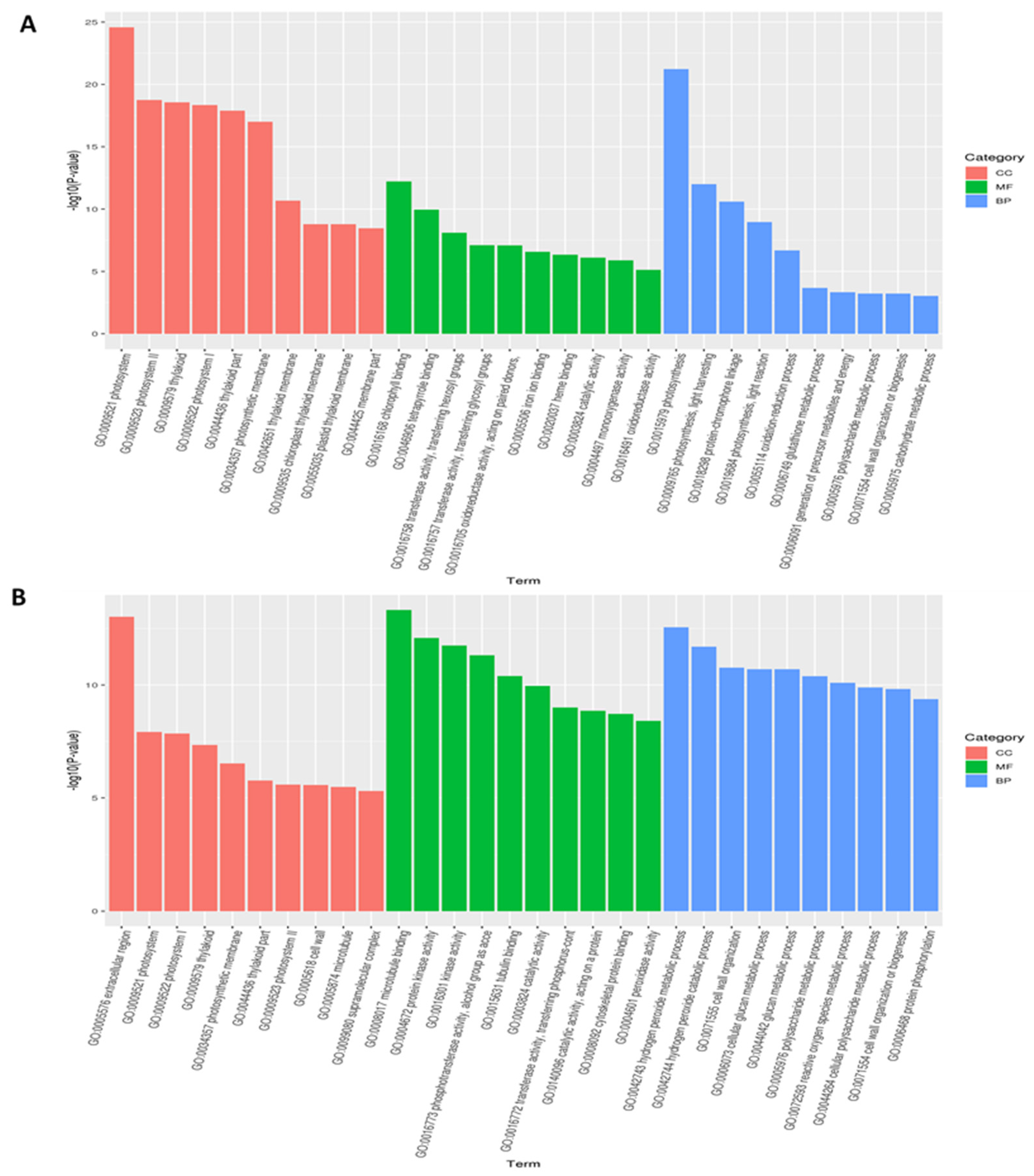
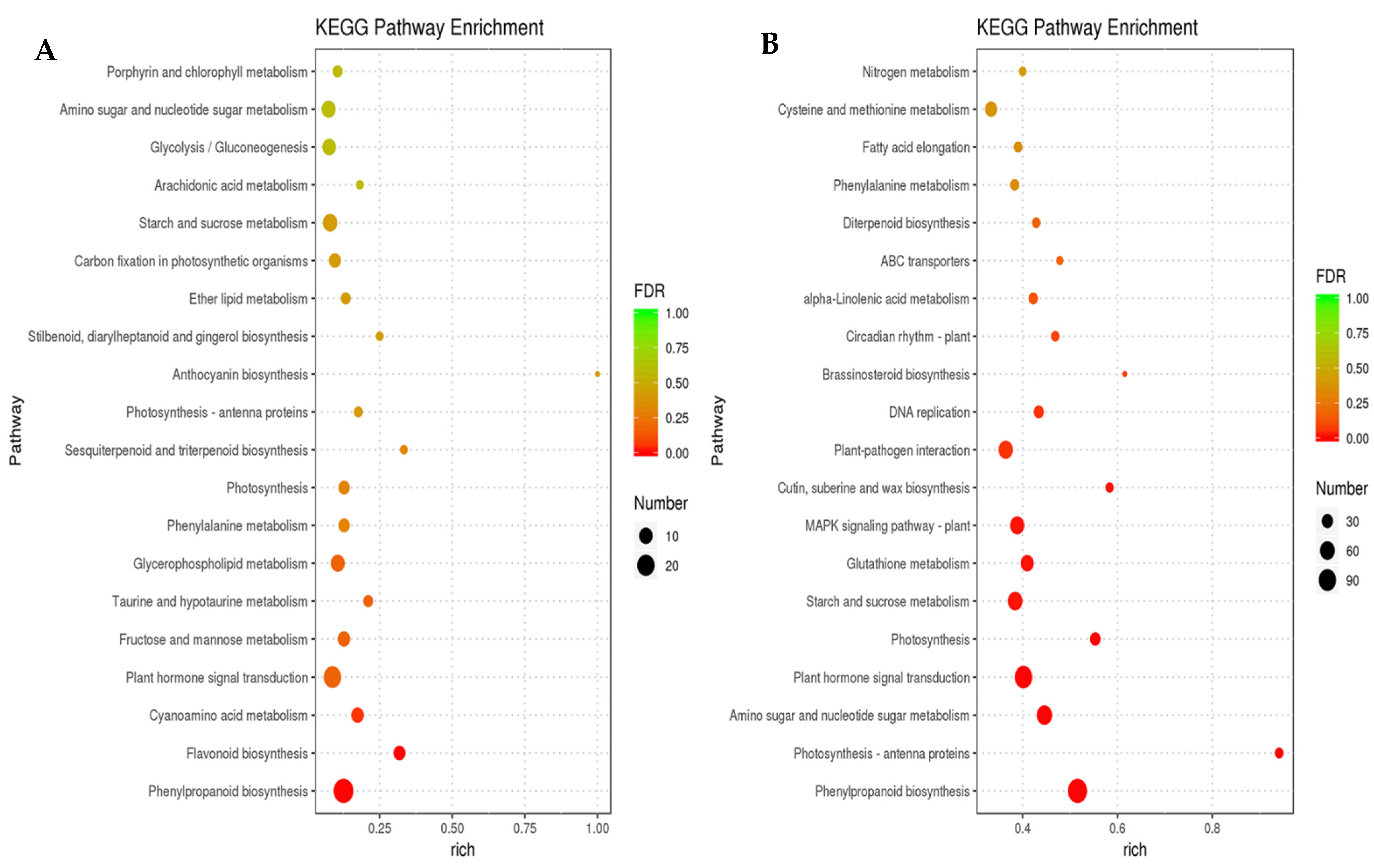
3.3. Functional Classification of Common DEGs
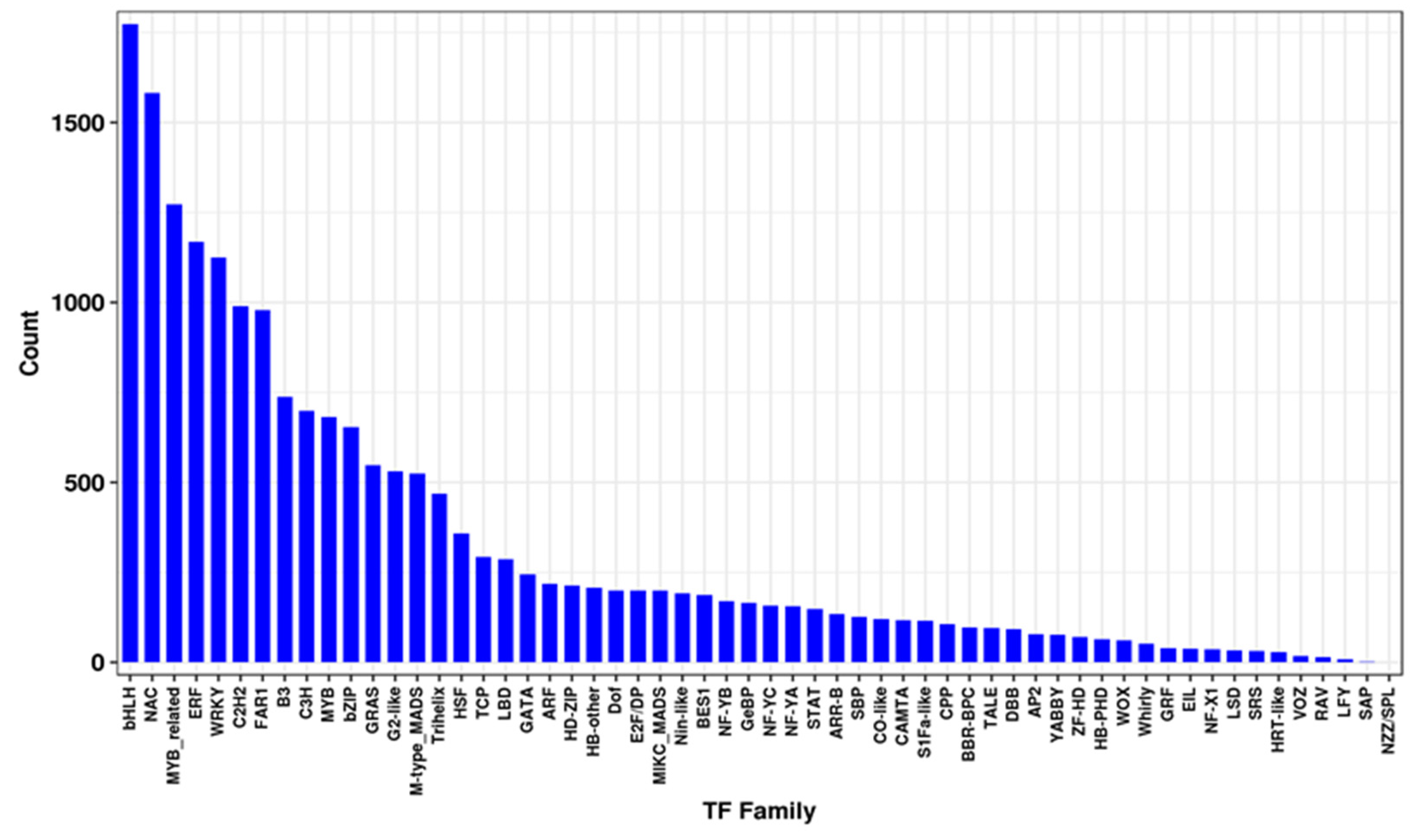
3.4. Identification of Transcription Factors That Respond to Salt Stress
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Yang, S.; Gong, J.; Zhao, Q.; Feng, Q.; Zhan, Q.; Zhao, Y.; Li, W.; Cheng, B.; Xia, J.; et al. Genomic architecture of heterosis for yield traits in rice. Nature 2016, 537, 629–633. [Google Scholar] [CrossRef]
- Kaeppler, S. Heterosis: Many Genes, Many Mechanisms-End the Search for an Undiscovered Unifying Theory. ISRN Bot. 2012, 2012, 682824. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Liu, C.; Zhang, Y.; Cui, Y.; Hu, H.; Jahan, N.; Lv, Y.; Qian, Q.; Guo, L. A 3-bp deletion of WLS5 gene leads to weak growth and early leaf senescence in rice. Rice 2019, 12, 26. [Google Scholar] [CrossRef]
- Bao, J.; Lee, S.; Chen, C.; Zhang, X.; Zhang, Y.; Liu, S.; Clark, T.; Wang, J.; Cao, M.; Yang, H.; et al. Serial Analysis of Gene Expression Study of a Hybrid Rice Strain (LYP9) and Its Parental Cultivars. Plant Physiol. 2005, 138, 1216. [Google Scholar] [CrossRef] [Green Version]
- Krieger, U.; Lippman, Z.B.; Zamir, D. The flowering gene SINGLE FLOWER TRUSS drives heterosis for yield in tomato. Nat. Genet. 2010, 42, 459–463. [Google Scholar] [CrossRef]
- Shang, L.; Wang, Y.; Cai, S.; Wang, X.; Li, Y.; Abduweli, A.; Hua, J. Partial Dominance, Overdominance, Epistasis and QTL by Environment Interactions Contribute to Heterosis in Two Upland Cotton Hybrids. G3 2015, 6, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Swanson-Wagner, R.A.; DeCook, R.; Jia, Y.; Bancroft, T.; Ji, T.; Zhao, X.; Nettleton, D.; Schnable, P.S. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 2009, 326, 1118–1120. [Google Scholar] [CrossRef]
- Paschold, A.; Jia, Y.; Marcon, C.; Lund, S.; Larson, N.B.; Yeh, C.-T.; Ossowski, S.; Lanz, C.; Nettleton, D.; Schnable, P.S.; et al. Complementation contributes to transcriptome complexity in maize (Zea mays L.) hybrids relative to their inbred parents. Genome Res. 2012, 22, 2445–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.; Messing, J. Gene expression of a gene family in maize based on noncollinear haplotypes. Proc. Natl. Acad. Sci. USA 2003, 100, 9055–9060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jiang, H.; Wang, H.; Cui, J.; Wang, J.; Hu, J.; Guo, L.; Qian, Q.; Xue, D. Transcriptome Analysis of Rice Seedling Roots in Response to Potassium Deficiency. Sci. Rep. 2017, 7, 5523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y.; et al. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695. [Google Scholar] [CrossRef] [Green Version]
- Long, L.; Yang, W.; Liao, P.; Guo, Y.; Kumar, A.; Gao, W. Transcriptome analysis reveals differentially expressed ERF transcription factors associated with salt response in cotton. Plant Sci. 2019, 281, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, M.; Lu, Q.; Ge, Q.; Rashid, M.H.o.; Liu, A.; Gong, J.; Shang, H.; Gong, W.; Li, J.; et al. Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and Chromosome Segment Substitution Lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wei, Y.; Nettleton, D.; Brummer, E.C. Comparative gene expression profiles between heterotic and non-heterotic hybrids of tetraploid Medicago sativa. BMC Plant Biol. 2009, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- FAO Soils Portal. Available online: http://www.fao.org/soils-portal/soil-management/management-of-some-problem-soils/salt-affected-soils/more-information-on-salt-affected-soils/en/ (accessed on 21 June 2021).
- Jahan, N.; Zhang, Y.; Lv, Y.; Song, M.; Zhao, C.; Hu, H.; Cui, Y.; Wang, Z.; Yang, S.; Zhang, A.; et al. QTL analysis for rice salinity tolerance and fine mapping of a candidate locus qSL7 for shoot length under salt stress. Plant Growth Regul. 2020, 90, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome Analysis of Salt Stress Responsiveness in the Seedlings of Dongxiang Wild Rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, P.; Mohanty, B.; Wijaya, E.; Lee, D.-Y.; Lim, T.-M.; Lin, Q.; Xu, J.; Loh, C.-S.; Kumar, P.P. Transcriptomics analysis of salt stress tolerance in the roots of the mangrove Avicennia officinalis. Sci. Rep. 2017, 7, 10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.R.; Bagali, P.G.; Hittalmani, S.; Shashidhar, H.E. Molecular mapping of quantitative trait loci associated with seedling tolerance to salt stress in rice (Oryza sativa L.). Curr. Sci. 2000, 78, 162–164. [Google Scholar]
- Yamaguchi, T.; Blumwald, E. Developing salt-tolerant crop plants: Challenges and opportunities. Trends Plant Sci. 2005, 10, 615–620. [Google Scholar] [CrossRef]
- Zhang, Y.; Lv, Y.; Jahan, N.; Chen, G.; Ren, D.; Guo, L. Sensing of Abiotic Stress and Ionic Stress Responses in Plants. Int. J. Mol. Sci. 2018, 19, 3298. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, A.; Zhang, W. Efficient acquisition of iron confers greater tolerance to saline-alkaline stress in rice (Oryza sativa L.). J. Exp. Bot. 2016, 67, 6431–6444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Xing, F.; Xu, C.; Zhang, Q.; Che, J.; Wang, X.; Song, J.; Li, X.; Xiao, J.; Chen, L.-L.; et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc. Natl. Acad. Sci. USA 2019, 116, 5653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Kim, D.; Ju, H.; Kwon, T.; Oh, C.; Ahn, S. Mapping QTLs for salt tolerance in an introgression line population between Japonica cultivars in rice. J. Crop Sci. Biotechnol. 2009, 12, 121. [Google Scholar] [CrossRef]
- Ma, T.; Wu, W.; Wang, Y. Transcriptome analysis of rice root responses to potassium deficiency. BMC Plant Biol. 2012, 12, 161. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Zhang, H.; Tian, Y.; Li, F.; Zhang, Z.; Lu, X.; Chen, X.; Huang, R. Expression of TERF1 in rice regulates expression of stress-responsive genes and enhances tolerance to drought and high-salinity. Plant Cell Rep. 2008, 27, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, A.; Zhang, W. Comparative studies on tolerance of rice genotypes differing in their tolerance to moderate salt stress. BMC Plant Biol. 2017, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Gundel, P.E.; Garibaldi, L.A.; Martínez-Ghersa, M.A.; Ghersa, C.M. Neotyphodium endophyte transmission to Lolium multiflorum seeds depends on the host plant fitness. Environ. Exp. Bot. 2011, 71, 359–366. [Google Scholar] [CrossRef]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Huang, C.h.; Peng, F.; Xue, X.; Wang, T. Effect of salt stress on photosynthesis and related physiological characteristics of Lycium ruthenicum Murr. Acta Agric. Scand. Sect. B Soil Plant Sci. 2017, 67, 680–692. [Google Scholar] [CrossRef]
- Wang, N.; Qian, Z.; Luo, M.; Fan, S.; Zhang, X.; Zhang, L. Identification of Salt Stress Responding Genes Using Transcriptome Analysis in Green Alga Chlamydomonas reinhardtii. Int. J. Mol. Sci. 2018, 19, 3359. [Google Scholar] [CrossRef] [Green Version]
- Moolna, A.; Bowsher, C.G. The physiological importance of photosynthetic ferredoxin NADP+ oxidoreductase (FNR) isoforms in wheat. J. Exp. Bot. 2010, 61, 2669–2681. [Google Scholar] [CrossRef]
- Jaradat, M.; Feurtado, J.; Huang, D.; Lu, Y.; Cutler, A.J. Multiple roles of the transcription factor AtMYBR1/AtMYB44 in ABA signaling, stress responses, and leaf senescence. BMC Plant Biol. 2013, 13, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, D.; Lüking, O.; Yang, O. Plant tolerance to drought and salinity: Stress regulating transcription factors and their functional significance in the cellular transcriptional network. Plant Cell Rep. 2011, 30, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.-H.; Li, C.-W.; Su, R.-C.; Cheng, C.-P.; Sanjaya; Tsai, Y.-C.; Chan, M.-T. A tomato bZIP transcription factor, SlAREB, is involved in water deficit and salt stress response. Planta 2010, 231, 1459–1473. [Google Scholar] [CrossRef]
- Blumwald, E. Sodium transport and salt tolerance in plants. Curr. Opin. Cell Biol. 2000, 12, 431–434. [Google Scholar] [CrossRef]
- Chen, H.-C.; Cheng, W.-H.; Hong, C.-Y.; Chang, Y.-S.; Chang, M.-C. The transcription factor OsbHLH035 mediates seed germination and enables seedling recovery from salt stress through ABA-dependent and ABA-independent pathways, respectively. Rice 2018, 11, 50. [Google Scholar] [CrossRef]
- Toda, Y.; Tanaka, M.; Ogawa, D.; Kurata, K.; Kurotani, K.-i.; Habu, Y.; Ando, T.; Sugimoto, K.; Mitsuda, N.; Katoh, E.; et al. RICE SALT SENSITIVE3 Forms a Ternary Complex with JAZ and Class-C bHLH Factors and Regulates Jasmonate-Induced Gene Expression and Root Cell Elongation. Plant Cell 2013, 25, 1709. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data (bp) | Clean Data (bp) | Q20 (%) | Q30 (%) | Total Mapped |
|---|---|---|---|---|---|
| CKl_1 | 7,923,985,022 | 7,518,704,344 | 97.32 | 93.36 | 46,372,093 (93.13%) |
| CKl_2 | 6,480,020,342 | 6,146,625,630 | 97.58 | 93.36 | 37,908,455 (93.13%) |
| CKy_1 | 7,446,201,694 | 7,064,073,846 | 97.44 | 93.61 | 44,311,077 (94.72%) |
| CKy_2 | 7,585,693,380 | 7,207,629,244 | 97.58 | 93.89 | 44,852,039 (93.97%) |
| CKp_1 | 7,190,156,100 | 6,797,538,000 | 97.47 | 93.56 | 43,303,322 (95.56%) |
| CKp_2 | 7,312,592,400 | 6,912,223,200 | 97.51 | 93.64 | 43,166,663 (93.67%) |
| S-l_7_1 | 7,253,315,100 | 6,839,539,800 | 97.53 | 93.67 | 42,824,053 (93.92%) |
| S-l_7_2 | 7,295,699,100 | 6,890,722,800 | 97.35 | 93.24 | 43,026,950 (93.66%) |
| S-y_7_1 | 7,274,557,500 | 6,870,173,400 | 97.54 | 93.66 | 43,685,297 (95.38%) |
| S-y_7_2 | 8,194,719,300 | 7,745,438,400 | 97.38 | 93.35 | 49,557,272 (95.97%) |
| S-p_7_1 | 7,487,282,700 | 7,087,584,600 | 97.56 | 93.74 | 45,026,497 (95.29%) |
| S-p_7_2 | 7,109,772,900 | 6,710,789,400 | 97.51 | 93.64 | 42,096,782 (94.10%) |
| S-l_14_1 | 8,286,700,612 | 7,863,880,378 | 97.30 | 93.34 | 48,575,755 (93.27%) |
| S-l_14_2 | 6,374,416,680 | 6,056,998,372 | 97.24 | 93.25 | 36,757,809 (91.64%) |
| S-y_14_1 | 7,336,768,974 | 6,948,243,860 | 97.49 | 93.72 | 43,152,182 (93.78%) |
| S-y_14_2 | 7,461,388,972 | 7,071,076,924 | 97.46 | 93.64 | 44,220,646 (94.43%) |
| S-p_14_1 | 8,189,553,600 | 7,718,297,100 | 98.01 | 94.62 | 48,529,365 (94.31%) |
| S-p_14_2 | 6,494,686,800 | 6,111,858,300 | 97.97 | 94.51 | 38,808,204 (95.24%) |
| Traits | DEGpp | DEGhp | ||||||
|---|---|---|---|---|---|---|---|---|
| L/Y | L/P | DEGhpu | DEGo | H2P | B2P | L2P | ||
| CK | 1727 | 9724 | 9827 | 10,139 | 470 | 3430 | 1523 | 6505 |
| 7-day | 1729 | 1397 | 1952 | 1592 | 142 | 1412 | 811 | 501 |
| 14-day | 342 | 7581 | 5473 | 7993 | 65 | 3251 | 192 | 4834 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahan, N.; Lv, Y.; Song, M.; Zhang, Y.; Shang, L.; Lu, Y.; Ye, G.; Qian, Q.; Gao, Z.; Guo, L. Transcriptomic Analysis of Short-Term Salt-Stress Response in Mega Hybrid Rice Seedlings. Agronomy 2021, 11, 1328. https://doi.org/10.3390/agronomy11071328
Jahan N, Lv Y, Song M, Zhang Y, Shang L, Lu Y, Ye G, Qian Q, Gao Z, Guo L. Transcriptomic Analysis of Short-Term Salt-Stress Response in Mega Hybrid Rice Seedlings. Agronomy. 2021; 11(7):1328. https://doi.org/10.3390/agronomy11071328
Chicago/Turabian StyleJahan, Noushin, Yang Lv, Mengqiu Song, Yu Zhang, Lianguang Shang, Ying Lu, Guoyou Ye, Qian Qian, Zhenyu Gao, and Longbiao Guo. 2021. "Transcriptomic Analysis of Short-Term Salt-Stress Response in Mega Hybrid Rice Seedlings" Agronomy 11, no. 7: 1328. https://doi.org/10.3390/agronomy11071328
APA StyleJahan, N., Lv, Y., Song, M., Zhang, Y., Shang, L., Lu, Y., Ye, G., Qian, Q., Gao, Z., & Guo, L. (2021). Transcriptomic Analysis of Short-Term Salt-Stress Response in Mega Hybrid Rice Seedlings. Agronomy, 11(7), 1328. https://doi.org/10.3390/agronomy11071328