
De Novo SNP Discovery and Genotyping of Iranian Pimpinella Species Using ddRAD Sequencing

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and DNA Extraction
2.2. ddRAD Library Preparation
2.3. Sequence Quality Analysis and Filtering
2.4. De Novo Assembly, Read Alignment and SNP Identification
2.5. Phylogenetic Tree Construction of Eight Pimpinella Species and Principal Component Analysis
2.6. Functional Analysis of SNP-Associated Contig
3. Results
3.1. Genotyping-by-Sequencing Library Construction and Sequencing
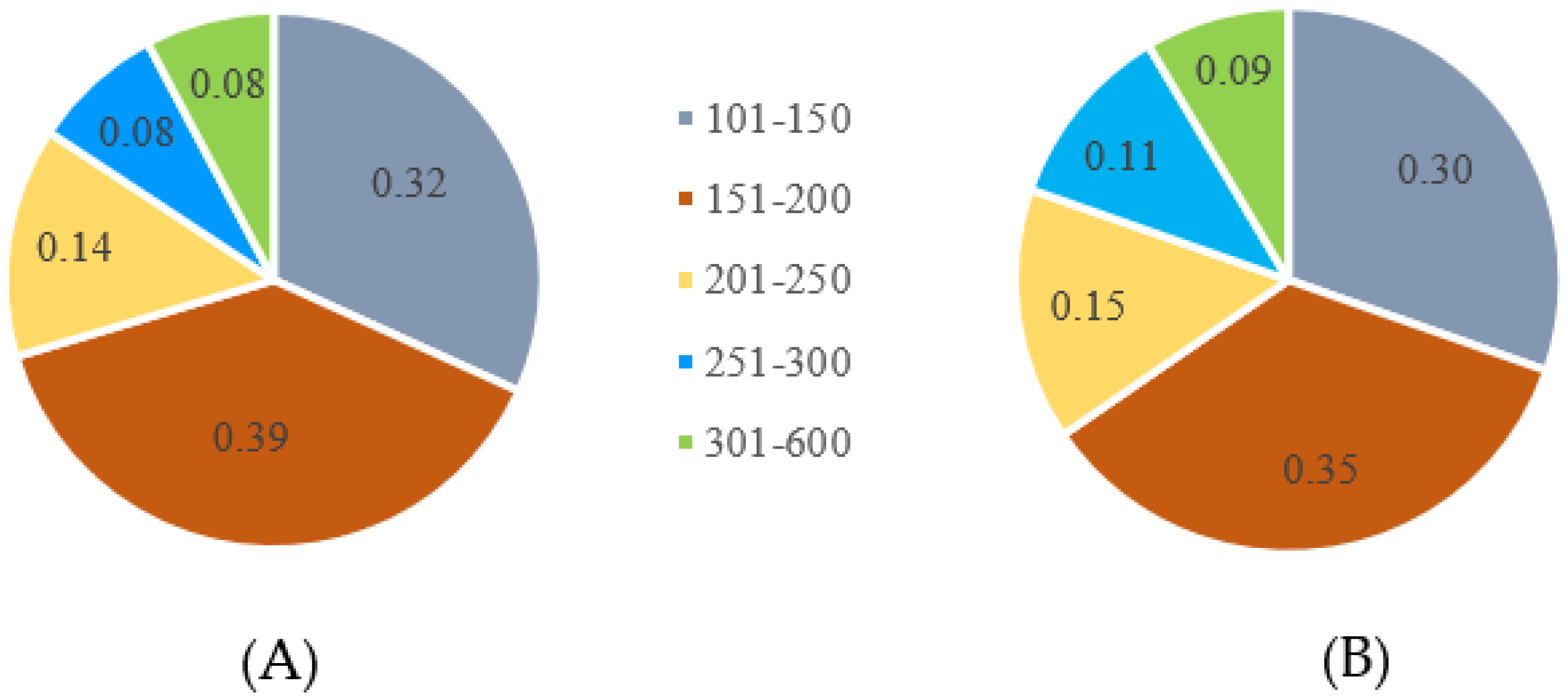
3.2. SNP Calling and Filtering
3.3. Population Structure and Genetic Relationship Analysis
3.4. Functional Analysis of SNP-Associated Scaffolds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iovene, M.; Grzebelus, E.; Carputo, D.; Jiang, J.; Simon, P.W. Major cytogenetic landmarks and karyotype analysis in Daucus carota and other Apiaceae. Am. J. Bot. 2008, 95, 793–804. [Google Scholar] [CrossRef]
- Pourgholami, M.; Majzoob, S.; Javadi, M.; Kamalinejad, M.; Fanaee, G.; Sayyah, M. The fruit essential oil of Pimpinella anisum exerts anticonvulsant effects in mice. J. Ethnopharmacol. 1999, 66, 211–215. [Google Scholar] [CrossRef]
- Özcan, M.M.; Chalchat, J.C. Chemical composition and antifungal effect of anise (Pimpinella anisum L.) fruit oil at ripening stage. Ann. Microbiol. 2006, 56, 353–358. [Google Scholar] [CrossRef]
- Fujimatu, E.; Ishikawa, T.; Kitajima, J. Aromatic compound glucosides, alkyl glucoside and glucide from the fruit of anise. Phytochemistry 2003, 63, 609–616. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; de Andrade, C.R.; Cassano, A.O.; De Souza, F.A.; Ambrosio, S.R.; da Costa, F.B.; de Oliveira, A.M. Antispasmodic and relaxant effects of the hidroalcoholic extract of Pimpinella anisum (Apiaceae) on rat anococcygeus smooth muscle. J. Ethnopharmacol. 2007, 110, 23–29. [Google Scholar] [CrossRef]
- Güvenalp, Z.; Ozbek, H.; Yuzbasioglu, M.; Kuruuzum-Uz, A.; Demirezer, L.O. Flavonoid Quantification and Antioxidant Activities of Some Pimpinella species. Rev. Anal. Chem. 2010, 29, 233–240. [Google Scholar] [CrossRef]
- Kuruüzüm-Uz, A.; Güvenalp, Z.; Yuzbasioglu, M.; Özbek, H.; Kazaz, C.; Demirezer, L. Flavonoids from Pimpinella kotschyana. Planta Medica 2010, 76, 274. [Google Scholar] [CrossRef]
- Mozaffarian, V. Studies on the flora of Iran, new species and new records. Pak. J. Bot. 2002, 34, 391–396. [Google Scholar]
- Awad, N.M.; Turky, A.S.; Mazhar, A. Effects of bio-and chemical nitrogenous fertilizers on yield of anise Pimpinella anisum and biological activities of soil irrigated with agricultural drainage water. Egypt. J. Soil Sci. 2005, 45, 265. [Google Scholar]
- Zheljazkov, V.D.; Callahan, A.; Cantrell, C.L. Yield and Oil Composition of 38 Basil (Ocimum basilicum L.) Accessions Grown in Mississippi. J. Agric. Food Chem. 2008, 56, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Giachino, R.R.A. Investigation of the genetic variation of anise (Pimpinella anisum L.) using RAPD and ISSR markers. Genet. Resour. Crop. Evol. 2019, 67, 763–780. [Google Scholar] [CrossRef]
- Nurcahyanti, A.D.R.; Nasser, I.J.; Sporer, F.; Graf, J.; Bermawie, N.; Reichling, J.; Wink, M. Chemical Composition of the Essential Oil from Aerial Parts of Javanian Pimpinella pruatjan Molk. and Its Molecular Phylogeny. Diversity 2016, 8, 15. [Google Scholar] [CrossRef] [Green Version]
- Tabanca, N.; Douglas, A.W.; Bedir, E.; Dayan, F.E.; Kirimer, N.; Baser, K.H.C.; Aytac, Z.; Khan, I.A.; Scheffler, B.E. Patterns of essential oil relationships in Pimpinella (Umbelliferae) based on phylogenetic relationships using nuclear and chloroplast sequences. Plant Genet. Resour. 2005, 3, 149–169. [Google Scholar] [CrossRef]
- Spalik, K.; Downie, S.R. Intercontinental disjunctions in Cryptotaenia (Apiaceae, Oenantheae): An appraisal using molecular data. J. Biogeogr. 2007, 34, 2039–2054. [Google Scholar] [CrossRef]
- Wang, Z.X.; Downie, S.R.; Tan, J.B.; Liao, C.Y.; Yu, Y.; He, X.J. Molecular phylogenetics of Pimpinella and allied genera (Apiaceae), with emphasis on Chinese native species, inferred from nrDNA ITS and cpDNA intron sequence data. Nord. J. Bot. 2014, 32, 642–657. [Google Scholar] [CrossRef]
- Deschamps, S.; Llaca, V.; May, G.D. Genotyping-by-Sequencing in Plants. Biology 2012, 1, 460–483. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Purushotham, P.M.; Han, K.; Lee, H.-R.; Nah, G.; Kang, B.-C. Development of a Genetic Map for Onion (Allium cepa L.) Using Reference-Free Genotyping-by-Sequencing and SNP Assays. Front. Plant Sci. 2017, 8, 1606. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Natarajan, S.; Biswas, M.K.; Shirasawa, K.; Isobe, S.; Kim, H.-T.; Park, J.-I.; Seong, C.-N.; Nou, I.-S. SNP discovery of Korean short day onion inbred lines using double digest restriction site-associated DNA sequencing. PLoS ONE 2018, 13, e0201229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, C.; Liu, Y.; VanBuren, R.; Yao, X.; Zhong, C.; Huang, H. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015, 22, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Alipour, H.; Bihamta, M.R.; Mohammadi, V.; Peyghambari, S.A.; Bai, G.; Zhang, G. Genotyping-by-Sequencing (GBS) Revealed Molecular Genetic Diversity of Iranian Wheat Landraces and Cultivars. Front. Plant Sci. 2017, 8, 1293. [Google Scholar] [CrossRef]
- Julio, H.-E.; Vikram, P.; Singh, R.P.; Kilian, A.; Carling, J.; Song, J.; Burgueno-Ferreira, J.A.; Bhavani, S.; Huerta-Espino, J.; Payne, T.; et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genom. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, D.; Thudi, M.; Kale, S.; Azam, S.; Roorkiwal, M.; Gaur, P.M.; Kishor, P.K.; Nguyen, H.; Sutton, T.; Varshney, R.K. Genotyping-by-sequencing based intra-specific genetic map refines a ‘‘QTL-hotspot” region for drought tolerance in chickpea. Mol. Genet. Genom. 2015, 290, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Iquira, E.; Humira, S.; François, B. Association mapping of QTLs for sclerotinia stem rot resistance in a collection of soybean plant introductions using a genotyping by sequencing (GBS) approach. BMC Plant Biol. 2015, 15, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Arbizu, C.I.; Ellison, S.L.; Senalik, D.; Simon, P.W.; Spooner, D.M. Genotyping-by-sequencing provides the discriminating power to investigate the subspecies of Daucus carota (Apiaceae). BMC Evol. Biol. 2016, 16, 234. [Google Scholar] [CrossRef] [Green Version]
- Davik, J.; Sargent, D.J.; Brurberg, M.B.; Lien, S.; Kent, M.; Alsheikh, M. A ddRAD Based Linkage Map of the Cultivated Strawberry, Fragaria xananassa. PLoS ONE 2015, 10, e0137746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RAD seq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Hojati, M.; Modarres-Sanavy, S.A.M.; Karimi, M.; Ghanati, F. Responses of growth and antioxidant systems in Carthamustinctorius L. under water deficit stress. Acta Physiol. Plant. 2011, 33, 105–112. [Google Scholar] [CrossRef]
- Severn-Ellis, A.A.; Scheben, A.; Neik, T.X.; Saad, N.S.M.; Pradhan, A.; Batley, J. Genotyping for Species Identification and Diversity Assessment Using Double-Digest Restriction Site-Associated DNA Sequencing (ddRAD-Seq), Legume Genomics; Springer: Berlin/Heidelberg, Germany, 2020; pp. 159–187. [Google Scholar]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. A Quality Control Tool for High Throughput Sequencing Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 16 May 2010).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. bioRxiv 2021, in press. [Google Scholar]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Thamilarasan, S.K.; Natarajan, S.; Park, J.-I.; Chung, M.-Y.; Nou, I.-S. De Novo Assembly and Transcriptome Analysis of Bulb Onion (Allium cepa L.) during Cold Acclimation Using Contrasting Genotypes. PLoS ONE 2016, 11, e0161987. [Google Scholar] [CrossRef]
- Novaes, E.; Drost, D.R.; Farmerie, W.G.; Pappas, G.J.; Grattapaglia, D.; Sederoff, R.R.; Kirst, M. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genom. 2008, 9, 354. [Google Scholar] [CrossRef] [Green Version]
- Izzatullayeva, V.; Akparov, Z.; Babayeva, S.; Ojaghi, J.; Abbasov, M. Efficiency of using RAPD and ISSR markers in evaluation of genetic diversity in sugar beet. Turk. J. Boil. 2014, 38, 429–438. [Google Scholar] [CrossRef]
- Tripathi, N.; Saini, N.; Mehto, V.; Kumar, S.; Tiwari, S. Assessment of genetic diversity among Withania somnifera collected from central India using RAPD and ISSR analysis. Med. Aromat. Plant Sci. Biotechnol 2012, 6, 33–39. [Google Scholar]
- Marakli, S. Transferability of Barley Retrotransposons (Sukkula and Nikita) to Investigate Genetic Structure of Pimpinella anisum L. Marmara Fen Bilim. Derg. 2018, 30, 217–220. [Google Scholar] [CrossRef]
- Fereidounfar, S.; Ghahremaninejad, F.; Khajehpiri, M. Phylogeny of the Southwest Asian Pimpinella and related genera based on nuclear and plastid sequences. Genet. Mol. Res. 2016, 15, 1–17. [Google Scholar] [CrossRef]
- Taranto, F.; D’Agostino, N.; Greco, B.; Cardi, T.; Tripodi, P. Genome-wide SNP discovery and population structure analysis in pepper (Capsicum annuum) using genotyping by sequencing. BMC Genom. 2016, 17, 943. [Google Scholar] [CrossRef] [Green Version]
- Paris, J.R.; Stevens, J.R.; Catchen, J.M. Lost in parameter space: A road map for stacks. Methods Ecol. Evol. 2017, 8, 1360–1373. [Google Scholar] [CrossRef]
- Baldwin, S.; Pither-Joyce, M.; Wright, K.; Chen, L.; McCallum, J. Development of robust genomic simple sequence repeat markers for estimation of genetic diversity within and among bulb onion (Allium cepa L.) populations. Mol. Breed. 2012, 30, 1401–1411. [Google Scholar] [CrossRef]
- Duangjit, J.; Bohanec, B.; Chan, A.; Town, C.; Havey, M.J. Transcriptome sequencing to produce SNP-based genetic maps of onion. Theor. Appl. Genet. 2013, 126, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Fuerst, P. Population Bottlenecks and Nonequilibrium Models in Population Genetics. II. Number of Alleles in a Small Population that was Formed by a Recent Bottleneck. Genetics 1985, 111, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Campagna, L.; Gronau, I.; Silveira, L.F.; Siepel, A.; Lovette, I.J. Distinguishing noise from signal in patterns of genomic divergence in a highly polymorphic avian radiation. Mol. Ecol. 2015, 24, 4238–4251. [Google Scholar] [CrossRef] [PubMed]
- Ravinet, M.; Westram, A.; Johannesson, K.; Butlin, R.; André, C.; Panova, M. Shared and nonshared genomic divergence in parallel ecotypes of L ittorina saxatilis at a local scale. Mol. Ecol. 2016, 25, 287–305. [Google Scholar] [CrossRef]
- Bus, A.; Hecht, J.; Huettel, B.; Reinhardt, R.; Stich, B. High-throughput polymorphism detection and genotyping in Brassica napus using next-generation RAD sequencing. BMC Genom. 2012, 13, 281. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, S.; Kim, H.-T.; Thamilarasan, S.K.; Veerappan, K.; Park, J.-I.; Nou, I.-S. Whole Genome Re-Sequencing and Characterization of Powdery Mildew Disease-Associated Allelic Variation in Melon. PLoS ONE 2016, 11, e0157524. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, K.; Kuwata, C.; Watanabe, M.; Fukami, M.; Hirakawa, H.; Isobe, S. Target Amplicon Sequencing for Genotyping Genome-Wide Single Nucleotide Polymorphisms Identified by Whole-Genome Resequencing in Peanut. Plant Genome 2016, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Li, Z.; Wu, J.; Xu, Y.; Yang, X.; Fan, L.; Fang, R.; Zhou, X. Analysis of genetic variation and diversity of Rice stripe virus populations through high-throughput sequencing. Front. Plant Sci. 2015, 6, 176. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Cornuet, J.M.; Luikart, G. Description and Power Analysis of Two Tests for Detecting Recent Population Bottlenecks From Allele Frequency Data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Prober, S.M.; Brown, A. Conservation of the Grassy White Box Woodlands: Population Genetics and Fragmentation of Eucalyptus albens. Conserv. Biol. 1994, 8, 1003–1013. [Google Scholar] [CrossRef]
- Glémin, S.; Bazin, E.; Charlesworth, D. Impact of mating systems on patterns of sequence polymorphism in flowering plants. In Proceedings of the Royal Society of London. Series B: Biological Sciences, The Royal Society, London, UK, 14 September 2006; pp. 3011–3019. [Google Scholar]
- Zhao, K.; Tung, C.-W.; Eizenga, G.C.; Wright, M.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.R.; Mezey, J.G.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef]
- Rubin, B.E.R.; Ree, R.H.; Moreau, C.S. Inferring Phylogenies from RAD Sequence Data. PLoS ONE 2012, 7, e33394. [Google Scholar] [CrossRef] [Green Version]
- Eivazi, A.; Naghavi, M.R.; Hajheidari, M.; Pirseyedi, S.; Ghaffari, M.; Mohammadi, S.; Majidi, I.; Salekdeh, G.; Mardi, M. Assessing wheat (Triticum aestivum L.) genetic diversity using quality traits, amplified fragment length polymorphisms, simple sequence repeats and proteome analysis. Ann. Appl. Biol. 2007, 152, 81–91. [Google Scholar] [CrossRef]
- Gupta, P.; Idris, A.; Mantri, S.; Asif, M.H.; Yadav, H.K.; Roy, J.K.; Tuli, R.; Mohanty, C.S.; Sawant, S.V. Discovery and use of single nucleotide polymorphic (SNP) markers in Jatropha curcas L. Mol. Breed. 2012, 30, 1325–1335. [Google Scholar] [CrossRef]
- Lo, M.-T.; Hinds, D.; Tung, D.A.H.J.Y.; Franz, C.; Fan, C.-C.; Wang, Y.; Smeland, O.B.; Schork, C.-C.F.A.; Holland, D.; Kauppi, K.; et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat. Genet. 2017, 49, 152–156. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela-Muñoz, V.; Gallardo-Escárate, C. TLR and IMD signaling pathways from Caligus rogercresseyi (Crustacea: Copepoda): In silico gene expression and SNPs discovery. Fish Shellfish. Immunol. 2014, 36, 428–434. [Google Scholar] [CrossRef]
- Khodadadi, M.; Fotokian, M.H.; Miransari, M. Genetic diversity of wheat (Triticum aestivum L.) genotypes based on cluster and principal component analyses for breeding strategies. Aust. J. Crop. Sci. 2011, 5, 17–24. [Google Scholar]
- Guan, X.; Nah, G.; Song, Q.; Udall, J.A.; Stelly, D.M.; Chen, Z.J. Transcriptome analysis of extant cotton progenitors revealed tetraploidization and identified genome-specific single nucleotide polymorphism in diploid and allotetraploid cotton. BMC Res. Notes 2014, 7, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravelombola, W.; Shi, A.; Weng, Y.; Mou, B.; Motes, D.; Clark, J.; Chen, P.; Srivastava, V.; Qin, J.; Dong, L.; et al. Association analysis of salt tolerance in cowpea (Vigna unguiculata (L.) Walp) at germination and seedling stages. Theor. Appl. Genet. 2017, 131, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Egea, L.A.; Mérida-García, R.; Kilian, A.; Hernandez, P.; Dorado, G. Assessment of Genetic Diversity and Structure of Large Garlic (Allium sativum) Germplasm Bank, by Diversity Arrays Technology “Genotyping-by-Sequencing” Platform (DArTseq). Front. Genet. 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | KEGG Pathway | Number of Sequences | Enzyme |
|---|---|---|---|
| map03030 | DNA replication | 1 | DNA ligase [EC:6.5.1.1] |
| map03020 | RNA polymerase | 1 | RNA nucleotidyltransferase [EC:2.7.7.6] |
| map00270 | Cysteine and methionine metabolism | 1 | Adenosylhomocysteinase [EC:3.3.1.1] |
| map00190 | Oxidative phosphorylation | 1 | Ubiquinone reductase [EC:7.1.1.2], ATP synthase [EC:3.6.3.14] |
| map00195 | Photosynthesis | 1 | Photosystem II [EC:1.10.3.9] |
| map00143 | Metabolic pathways | 1 | NADH dehydrogenase [EC:7.1.1.2] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehravi, S.; Ranjbar, G.A.; Mirzaghaderi, G.; Severn-Ellis, A.A.; Scheben, A.; Edwards, D.; Batley, J. De Novo SNP Discovery and Genotyping of Iranian Pimpinella Species Using ddRAD Sequencing. Agronomy 2021, 11, 1342. https://doi.org/10.3390/agronomy11071342
Mehravi S, Ranjbar GA, Mirzaghaderi G, Severn-Ellis AA, Scheben A, Edwards D, Batley J. De Novo SNP Discovery and Genotyping of Iranian Pimpinella Species Using ddRAD Sequencing. Agronomy. 2021; 11(7):1342. https://doi.org/10.3390/agronomy11071342
Chicago/Turabian StyleMehravi, Shaghayegh, Gholam Ali Ranjbar, Ghader Mirzaghaderi, Anita Alice Severn-Ellis, Armin Scheben, David Edwards, and Jacqueline Batley. 2021. "De Novo SNP Discovery and Genotyping of Iranian Pimpinella Species Using ddRAD Sequencing" Agronomy 11, no. 7: 1342. https://doi.org/10.3390/agronomy11071342
APA StyleMehravi, S., Ranjbar, G. A., Mirzaghaderi, G., Severn-Ellis, A. A., Scheben, A., Edwards, D., & Batley, J. (2021). De Novo SNP Discovery and Genotyping of Iranian Pimpinella Species Using ddRAD Sequencing. Agronomy, 11(7), 1342. https://doi.org/10.3390/agronomy11071342