
Temperature Dependences of IR Spectra of Humic Substances of Brown Coal


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. FTIR Measurements
3. Results
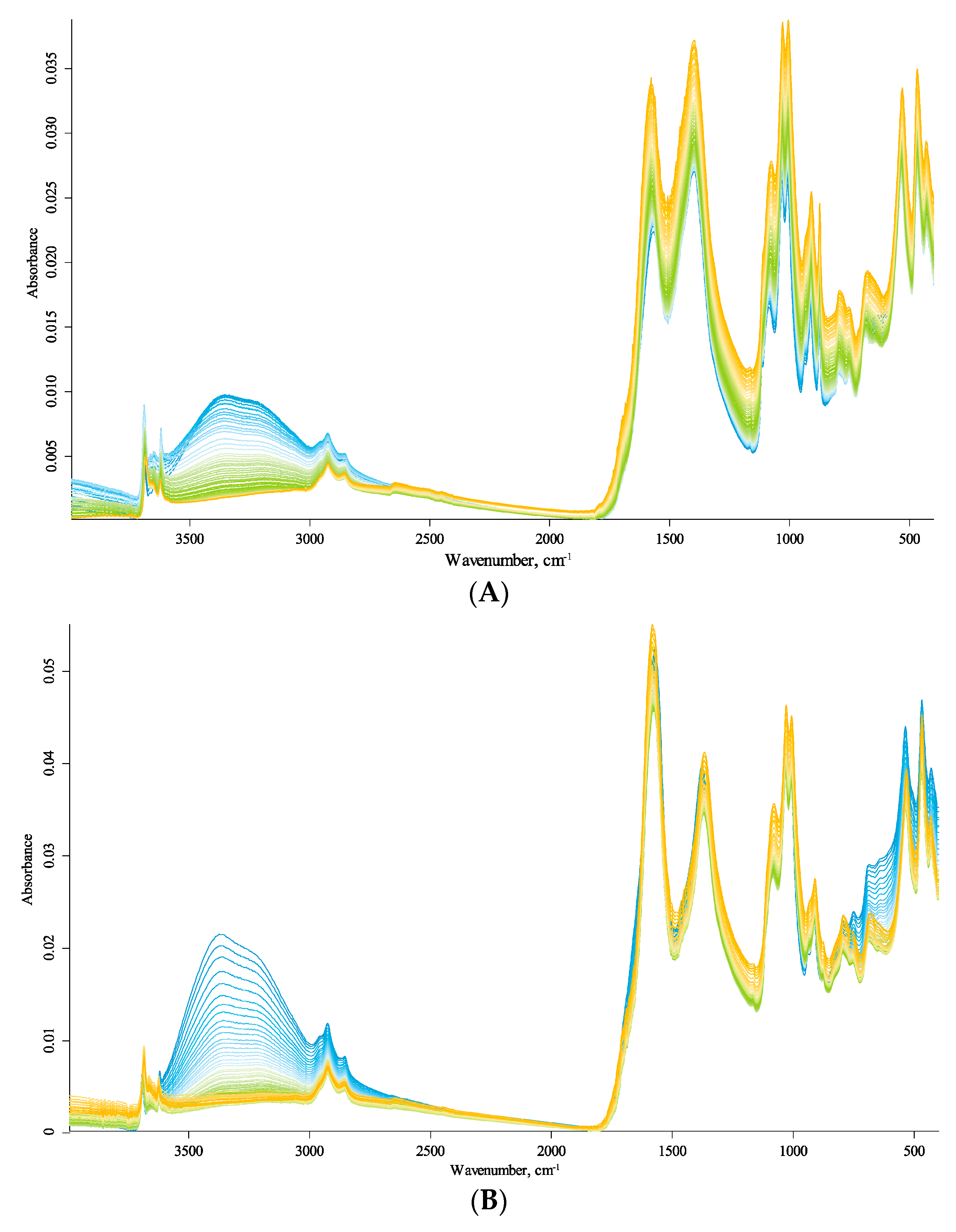
3.1. Post-Registration Processing of IR Spectra
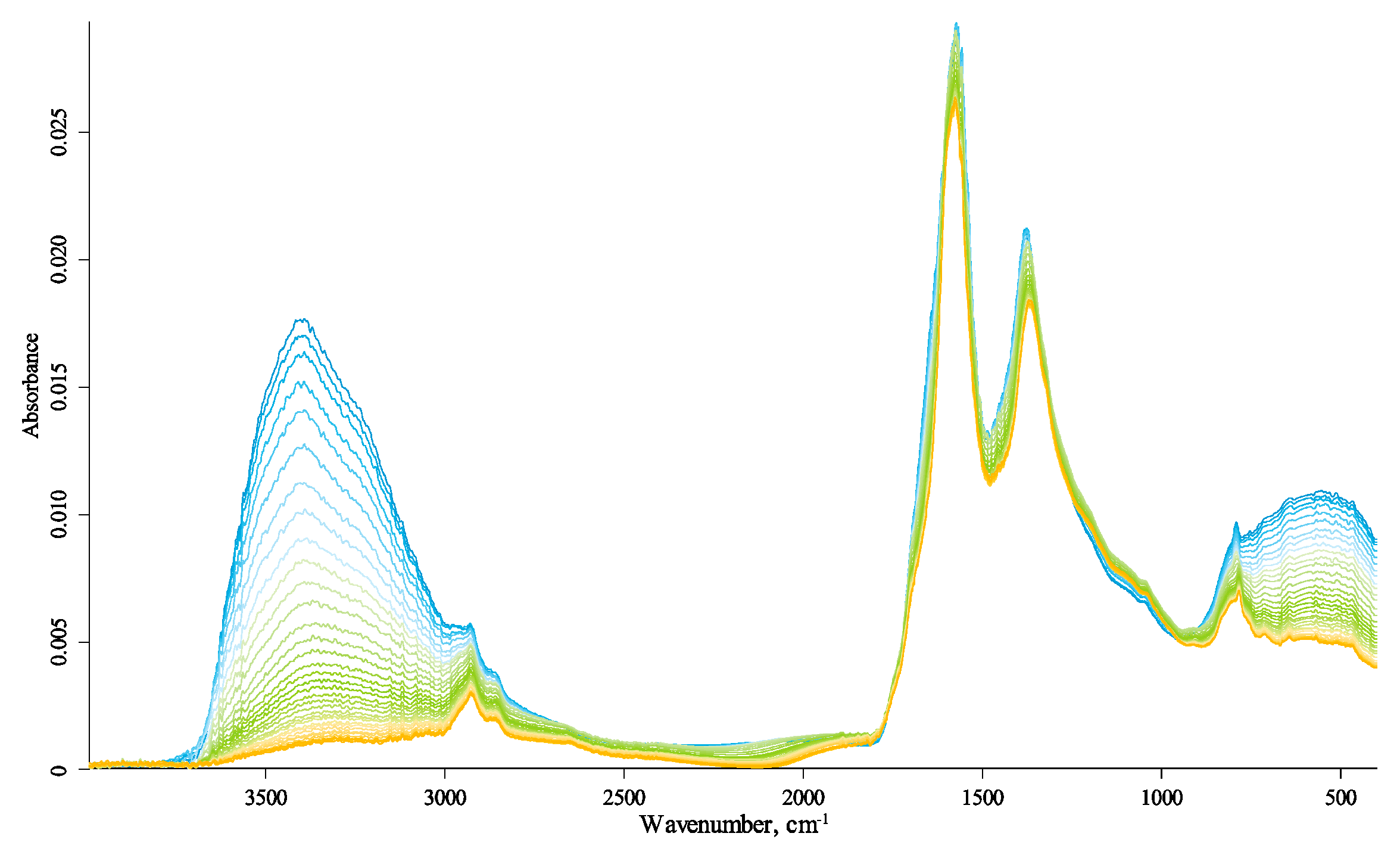
3.2. General Description of IR Spectra
3.3. Primary Band Assignment
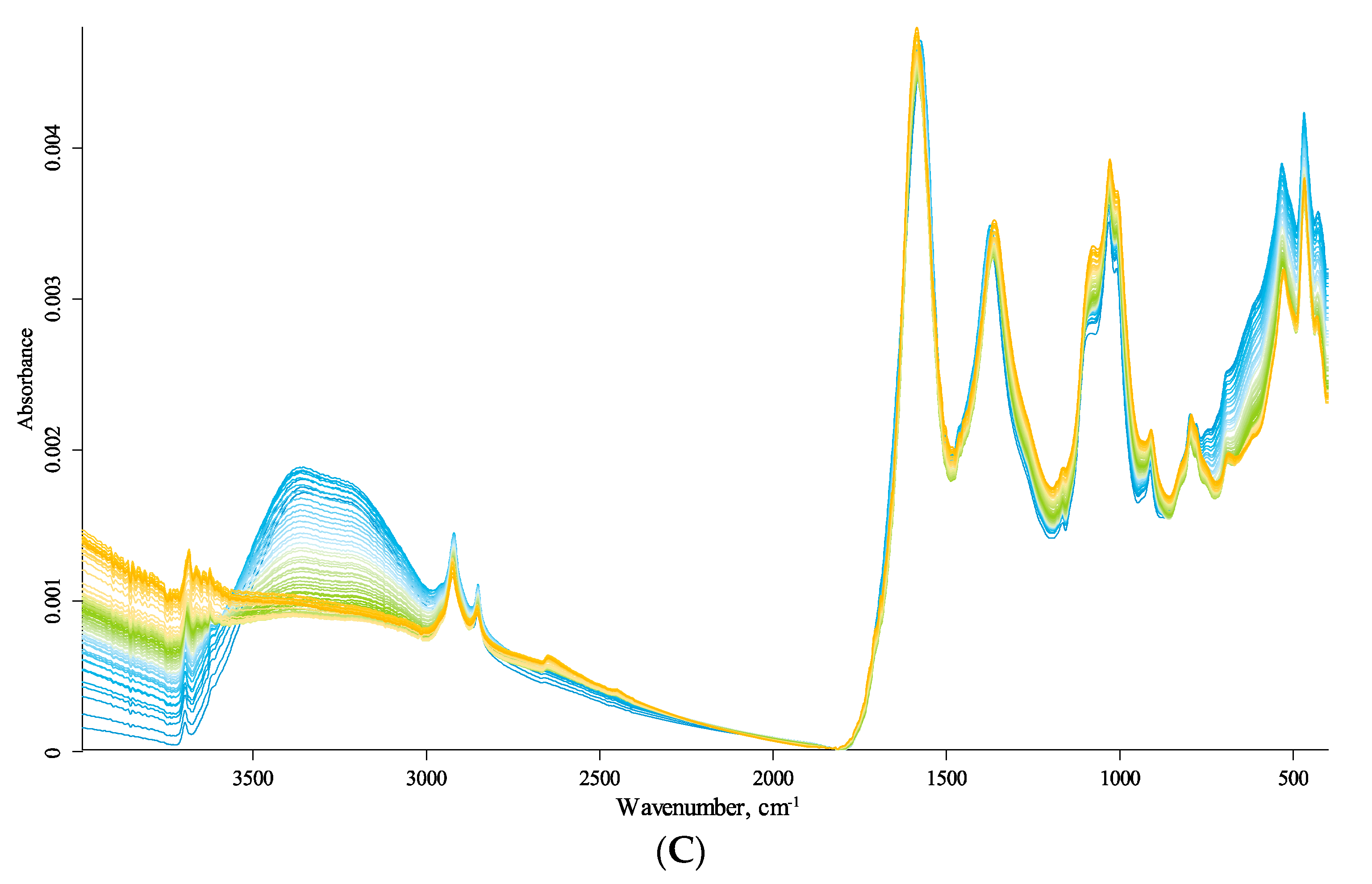
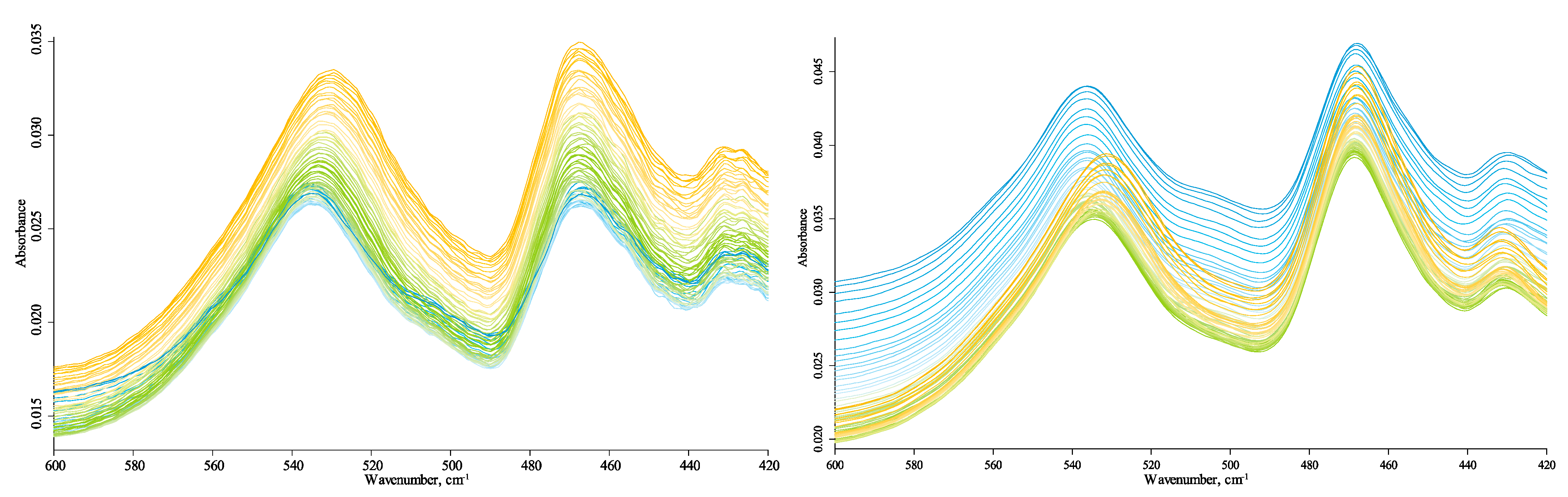
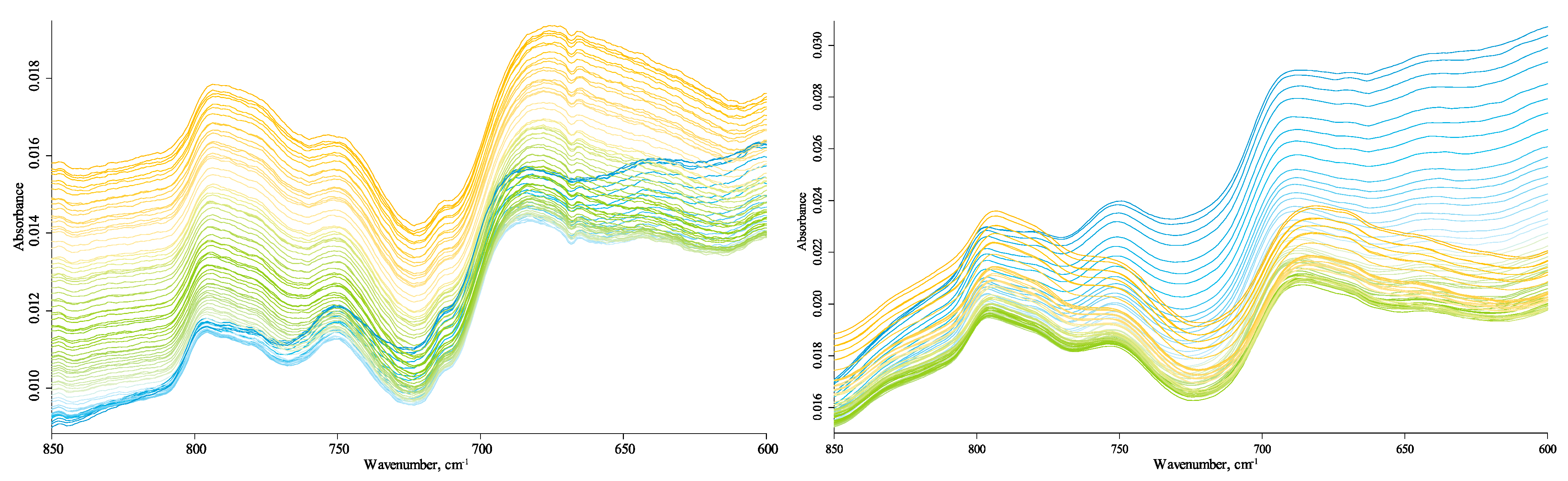
3.4. Temperature Changes
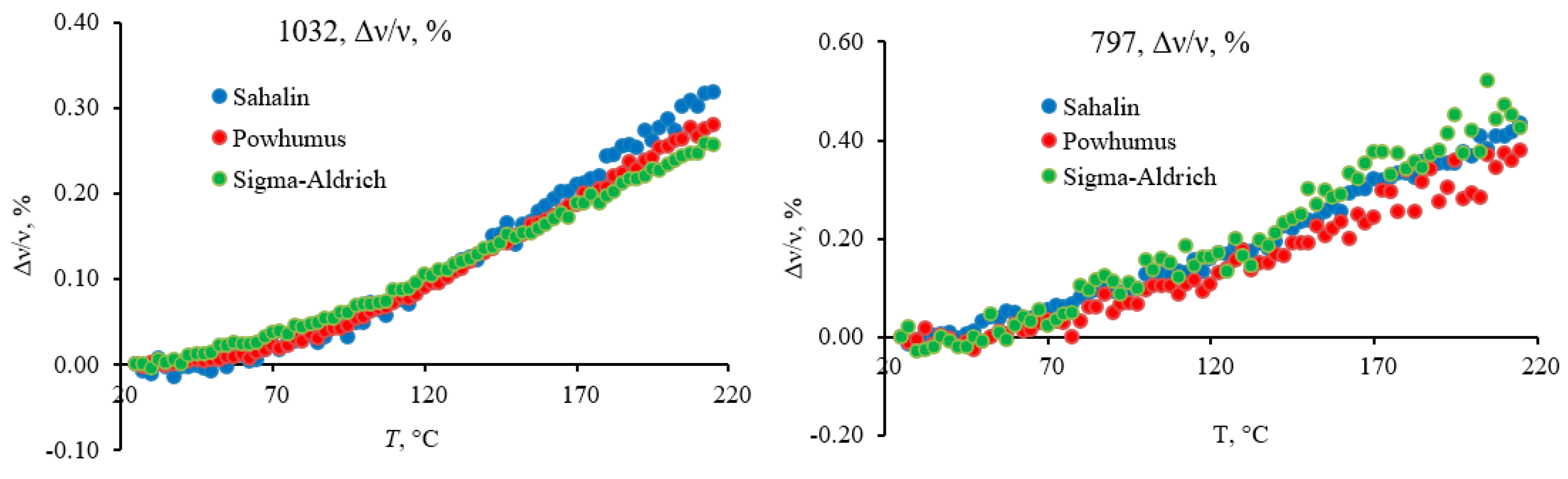
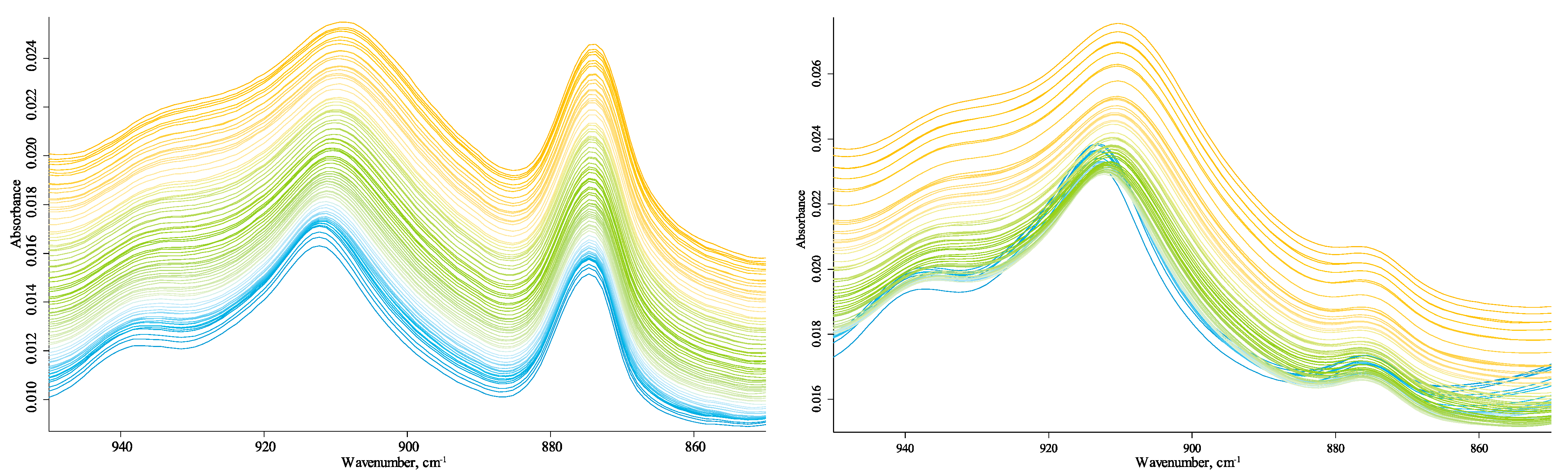
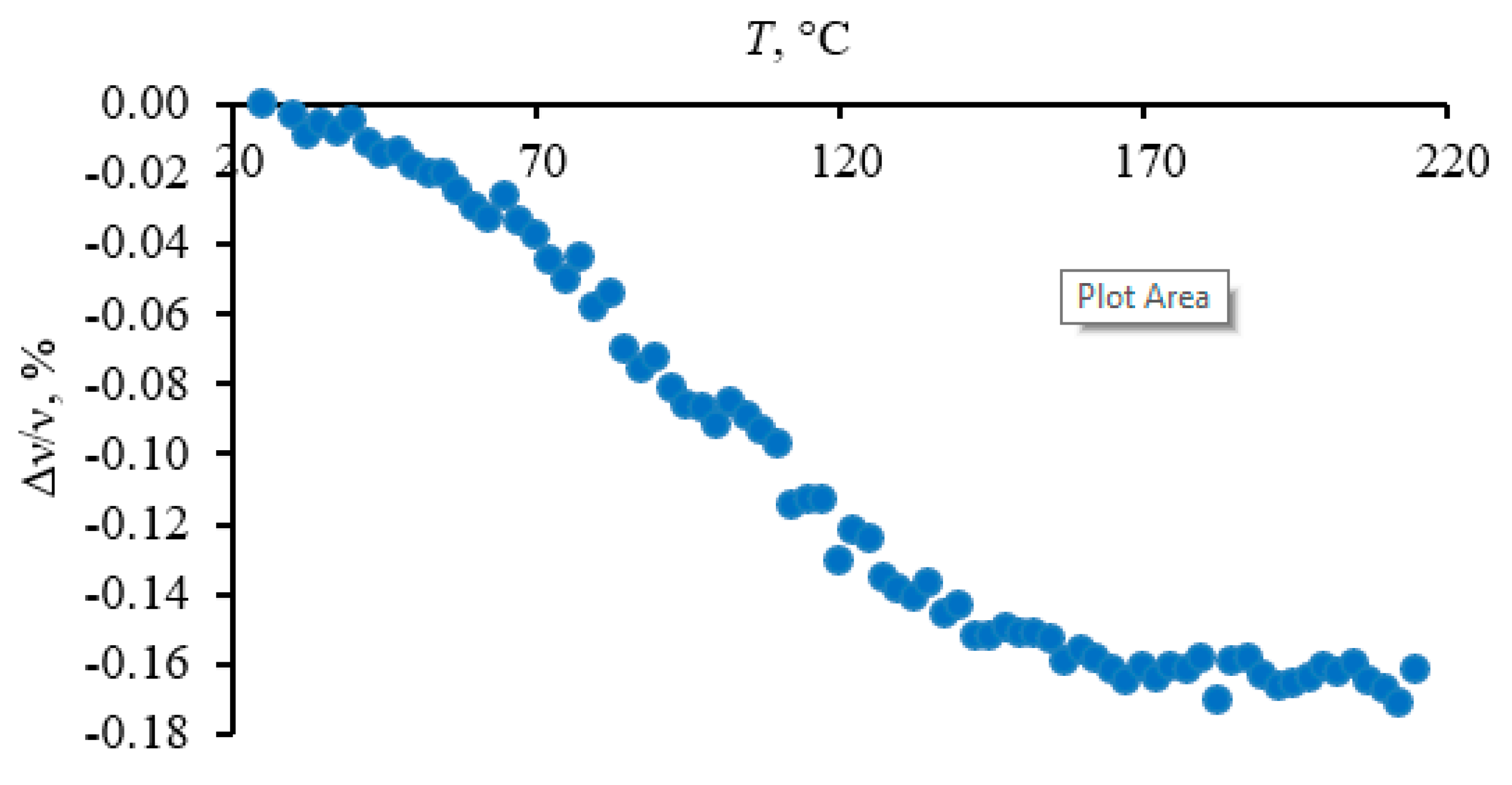
3.4.1. Quartz Lattice Region (800–260 cm−1)
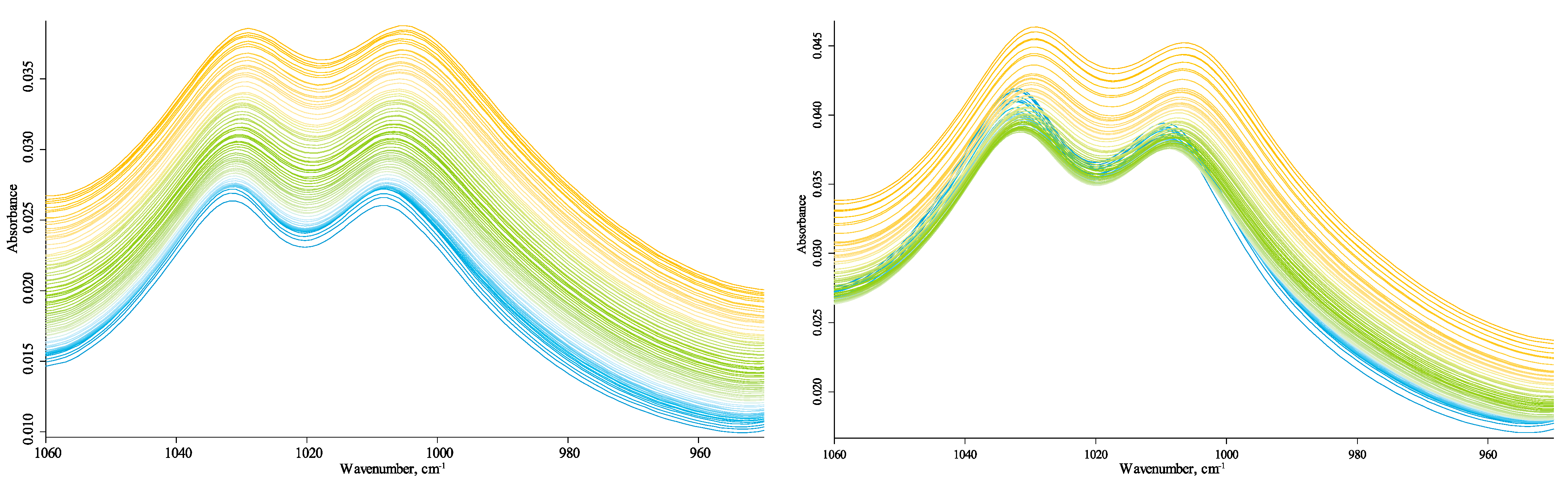
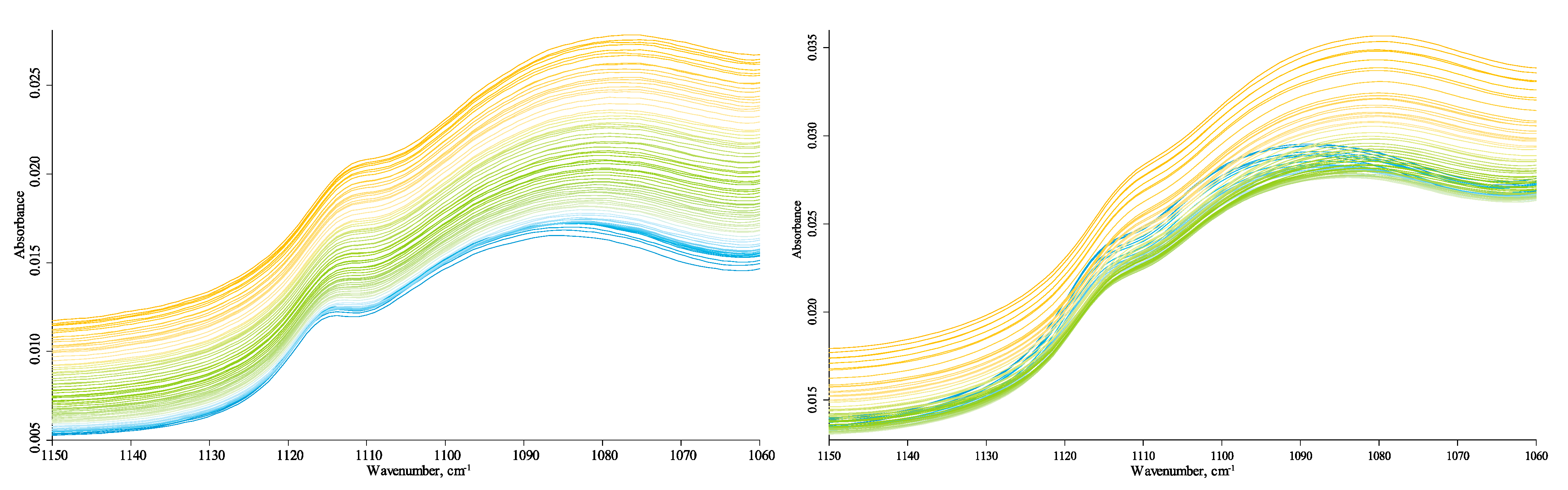
3.4.2. Quartz-Overtone-Band Region (1270–800 cm−1)
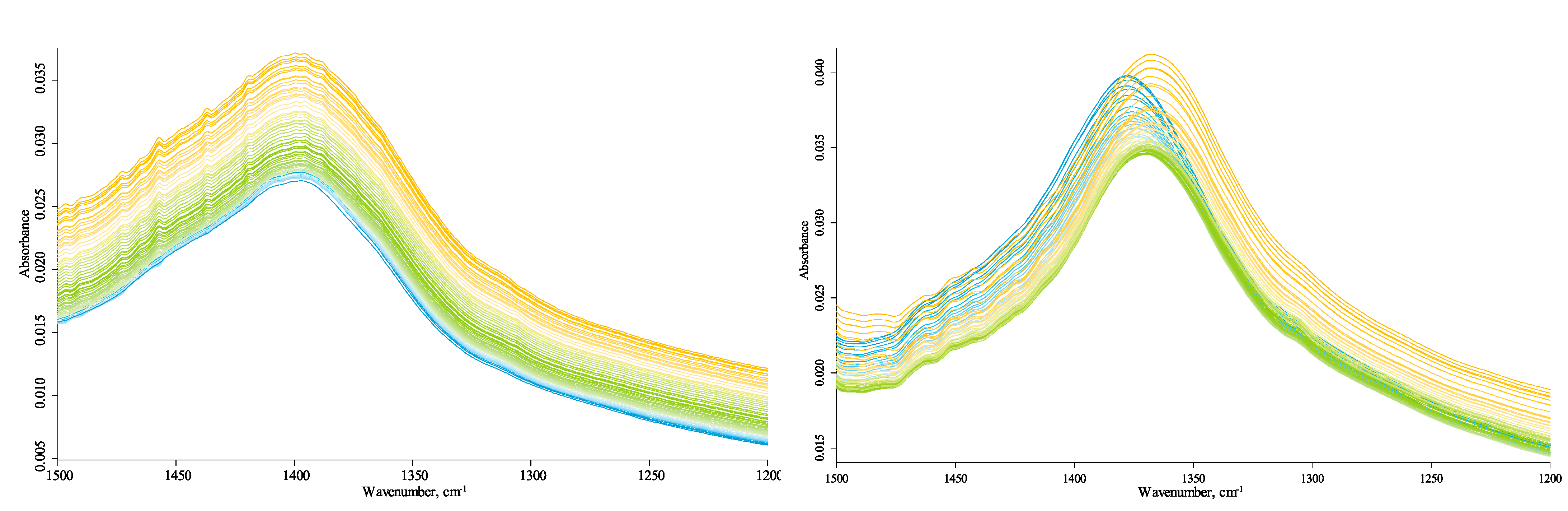
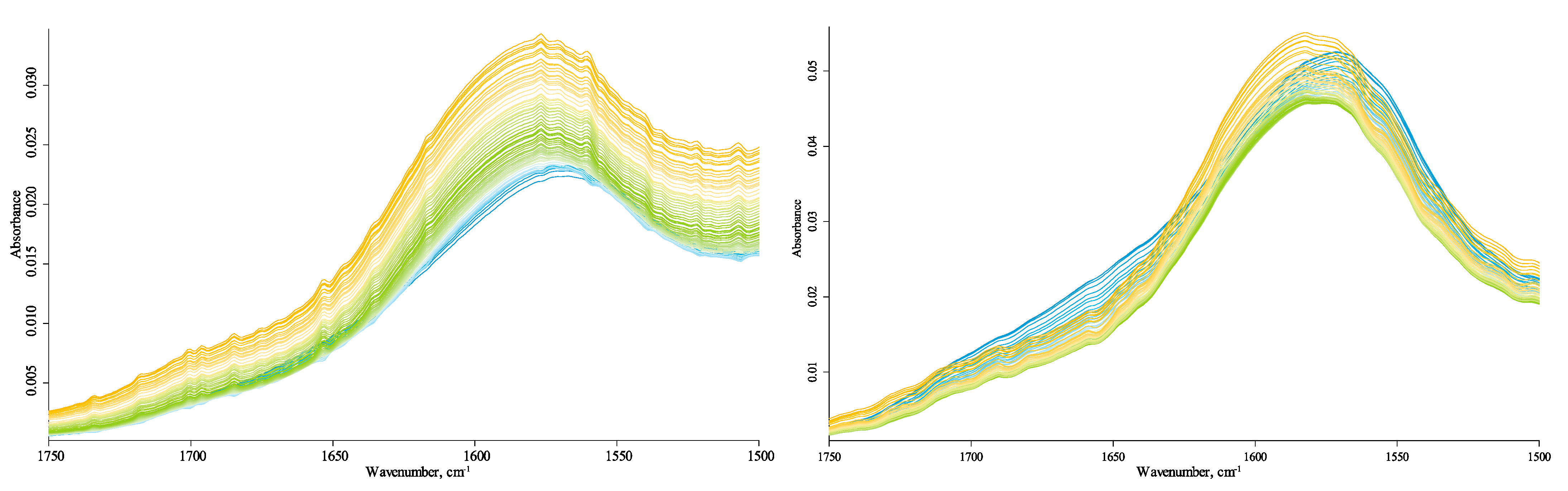
3.4.3. HSOM Region (1780–1270 cm−1)
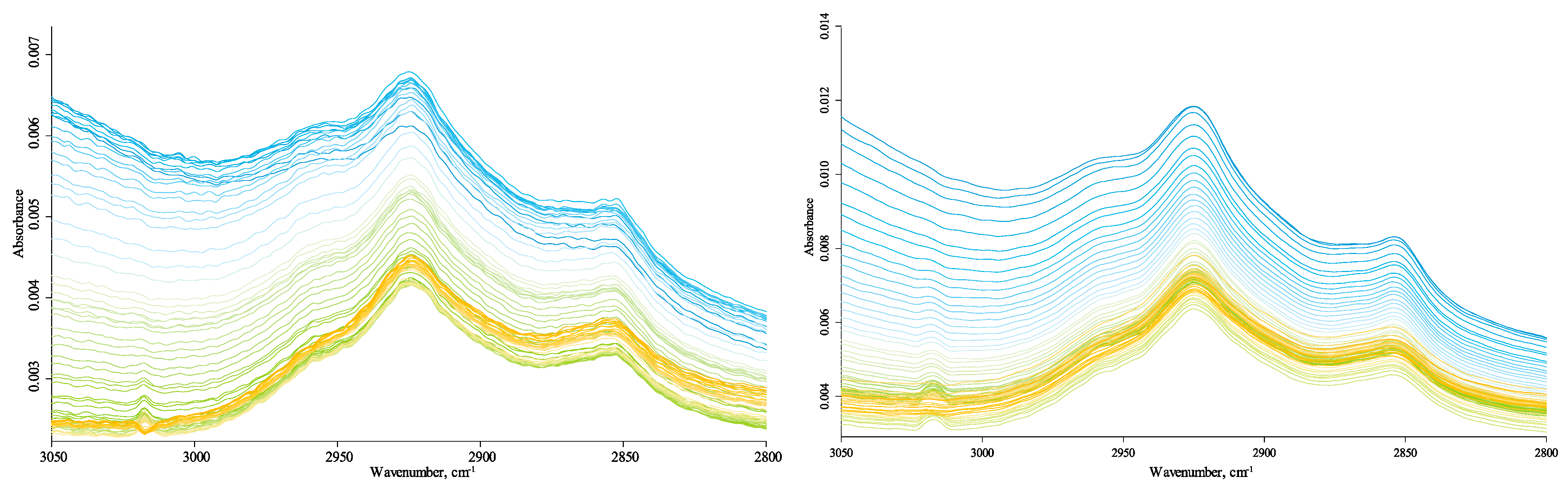
3.4.4. Quartz-Combination-Band and CH-Speciation Regions
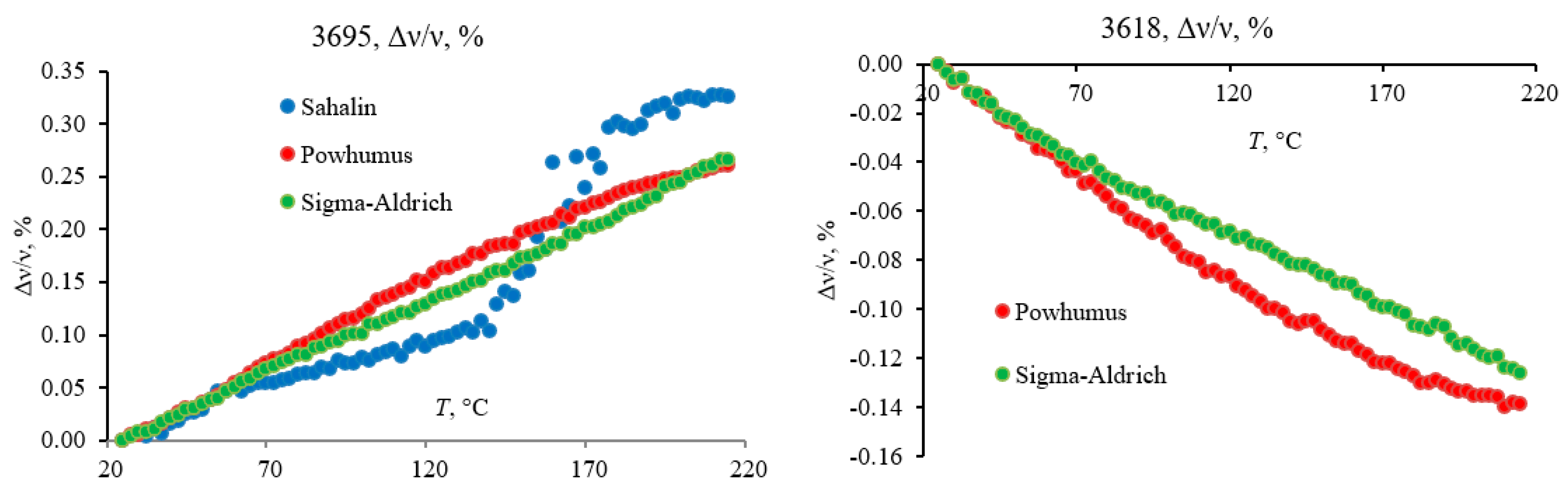
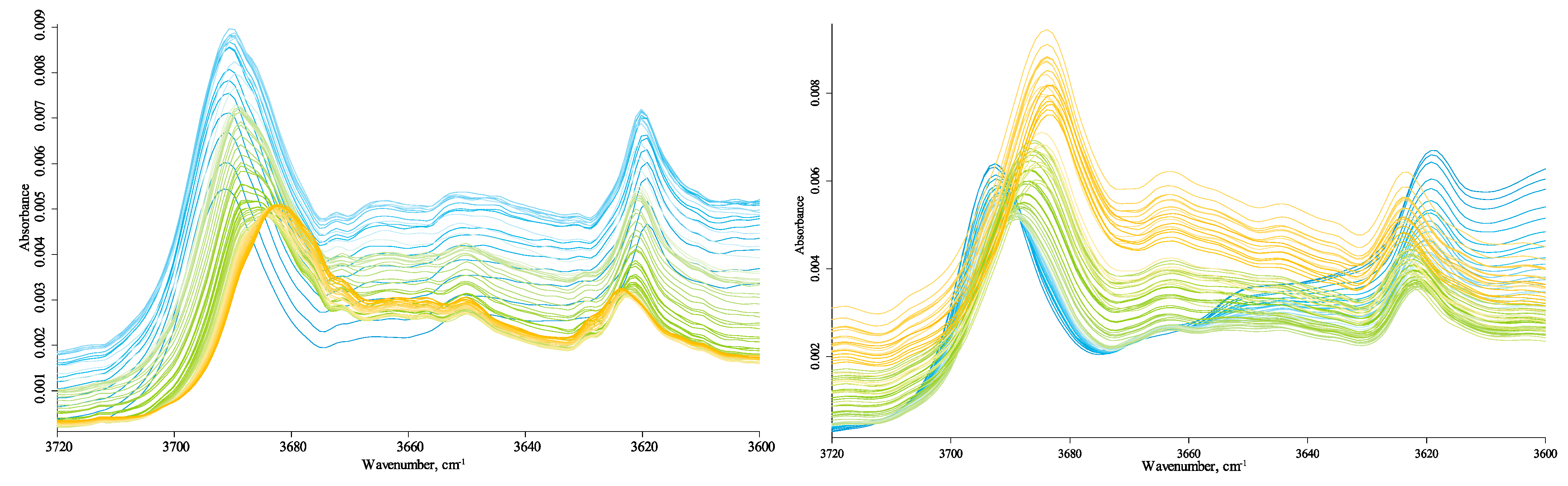
3.4.5. Hydrogen-Speciation Region (4000–3100 cm−1)
3.5. Differences between HS Samples
3.6. Uncentrifuged and Centrifuged Samples
4. Discussion
4.1. Components
4.1.1. HSOM
4.1.2. Water
4.1.3. Quartz Bulk
4.1.4. Quartz Surface (Si–OH) Groups
4.2. Comparison of HS Samples
4.3. Summary
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malcolm, R.L. The uniqueness of humic substances in each of soil, stream and marine environments. Anal. Chim. Acta 1990, 232, 19–30. [Google Scholar] [CrossRef]
- Kulikova, N.A.; Perminova, I.V. Interactions between Humic Substances and Microorganisms and Their Implications for Nature-like Bioremediation Technologies. Molecules 2021, 26, 2706. [Google Scholar] [CrossRef]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Perminova, I.V.; Garcia-Mina, J.-M.; Podgorski, D.C.; Cervantes, F.J.; Efremenko, E.N.; Domingo, J.L. Humic substances and living systems: Impact on environmental and human health. Environ. Res. 2021, 194, 110726. [Google Scholar] [CrossRef]
- Winkler, J.; Ghosh, S. Therapeutic Potential of Fulvic Acid in Chronic Inflammatory Diseases and Diabetes. J. Diabetes Res. 2018, 2018, 5391014. [Google Scholar] [CrossRef] [Green Version]
- Peña-Méndez, E.M.; Havel, J.; Patočka, J. Humic substances-compounds of still unknown structure: Applications in agriculture, industry, environment, and biomedicine. J. Appl. Biomed. 2005, 3, 13–24. [Google Scholar] [CrossRef] [Green Version]
- de Melo, B.A.; Motta, F.L.; Santana, M.H. Humic acids: Structural properties and multiple functionalities for novel technological developments. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 62, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Trckova, M.; Matlova, L.; Hudcova, H.; Martin, F.; Zraly, Z.; Dvorska, L.; Beran, V.; Pavlik, I. Peat as a feed supplement for animals: A review. Vet. Med.Czech. 2005, 50, 361–377. [Google Scholar] [CrossRef] [Green Version]
- Pukalchik, M.; Kydralieva, K.; Yakimenko, O.; Fedoseeva, E.; Terekhova, V. Outlining the Potential Role of Humic Products in Modifying Biological Properties of the Soil—A Review. Front. Environ. Sci. 2019, 7, 10. [Google Scholar] [CrossRef]
- Gašparovič, M.; Hrnčár, C.; Gálik, B. The effect of feed additives in pheasants fattening: A review. J. Cent. Eur. Agric. 2017, 18, 749–761. [Google Scholar] [CrossRef]
- Pandey, A.K.; Pandey, S.D.; Misra, V.; Devi, S. Role of humic acid entrapped calcium alginate beads in removal of heavy metals. J. Hazard. Mater. 2003, 98, 177–181. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Yin, X.; Wang, N.; Chen, D. Influences of Nitrogen Application Levels on Properties of Humic Acids in Chernozem Amended with Different Types of Organic Materials. Sustainability 2019, 11, 5405. [Google Scholar] [CrossRef] [Green Version]
- Plyatsuk, L.; Chernysh, Y.; Ablieieva, I.; Bataltsev, Y.; Vaskin, R.; Roy, I.; Yakhnenko, E.; Roubík, H. Modelling and development of technological processes for low rank coal bio-utilization on the example of brown coal. Fuel 2020, 267, 117298. [Google Scholar] [CrossRef]
- Skripkina, T.S.; Bychkov, A.L.; Tikhova, V.D.; Lomovsky, O.I. Mechanochemical Solid-Phase Reactions of Humic Acids from Brown Coal with Sodium Percarbonate. Solid Fuel Chem. 2018, 52, 356–360. [Google Scholar] [CrossRef]
- Acharya, B.; Sule, I.; Dutta, A. A review on advances of torrefaction technologies for biomass processing. Biomass Convers. Biorefin. 2012, 2, 349–369. [Google Scholar] [CrossRef]
- Davis, W.M.; Erickson, C.L.; Johnston, C.T.; Delfino, J.J.; Porter, J.E. Quantitative Fourier Transform Infrared spectroscopic investigation humic substance functional group composition. Chemosphere 1999, 38, 2913–2928. [Google Scholar] [CrossRef]
- Tanaka, T.; Nagao, S.; Ogawa, H. Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) Spectroscopy of Functional Groups of Humic Acid Dissolving in Aqueous Solution. Anal. Sci. Suppl. 2002, 17icas, i1081–i1084. [Google Scholar] [CrossRef]
- Tatzber, M.; Stemmer, M.; Spiegel, H.; Katzlberger, C.; Haberhauer, G.; Mentler, A.; Gerzabek, M.H. FTIR-spectroscopic characterization of humic acids and humin fractions obtained by advanced NaOH, Na4P2O7, and Na2CO3 extraction procedures. J. Plant. Nutr. Soil Sci. 2007, 170, 522–529. [Google Scholar] [CrossRef]
- Yuan, Y.; Cai, X.; Tan, B.; Zhou, S.; Xing, B. Molecular insights into reversible redox sites in solid-phase humic substances as examined by electrochemical in situ FTIR and two-dimensional correlation spectroscopy. Chem. Geol. 2018, 494, 136–143. [Google Scholar] [CrossRef]
- Baes, A.U.; Bloom, P.R. Diffuse Reflectance and Transmission Fourier Transform Infrared (DRIFT) Spectroscopy of Humic and Fulvic Acids. Soil Sci. Soc. Am. J. 1989, 53, 695–700. [Google Scholar] [CrossRef]
- Vergnoux, A.; Guiliano, M.; Di Rocco, R.; Domeizel, M.; Théraulaz, F.; Doumenq, P. Quantitative and mid-infrared changes of humic substances from burned soils. Environ. Res. 2011, 111, 205–214. [Google Scholar] [CrossRef]
- Karpukhina, E.; Mikheev, I.; Perminova, I.; Volkov, D.; Proskurnin, M. Rapid quantification of humic components in concentrated humate fertilizer solutions by FTIR spectroscopy. J. Soils Sed. 2018, 19, 2729–2739. [Google Scholar] [CrossRef]
- Liu, X.Y.; Chen, W.; Qian, C.; Yu, H.Q. Interaction between Dissolved Organic Matter and Long-Chain Ionic Liquids: A Microstructural and Spectroscopic Correlation Study. Environ. Sci Technol. 2017, 51, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- MacCarthy, P.; Mark, H.B.; Griffiths, P.R. Direct measurement of the infrared spectra of humic substances in water by Fourier transform infrared spectroscopy. J. Agric. Food Chem. 1975, 23, 600–602. [Google Scholar] [CrossRef]
- Stevenson, F.J.; Goh, K.M. Infrared spectra of humic acids: Elimination of interference due to hygroscopic moisture and structural changes accompanying heating with KBr. Soil Sci. 1974, 117, 34–41. [Google Scholar] [CrossRef]
- Katon, J.E.; Phillips, D.B. Infrared Spectroscopy at Subambient Temperatures. Appl. Spectrosc. Rev. 1973, 7, 1–45. [Google Scholar] [CrossRef]
- Xu, B.; Mei, Y.; Xiao, Z.; Kang, Z.; Wang, R.; Sun, D. Monitoring thermally induced structural deformation and framework decomposition of ZIF-8 through in situ temperature dependent measurements. Phys. Chem. Chem. Phys. 2017, 19, 27178–27183. [Google Scholar] [CrossRef]
- Verma, P.K.; Kundu, A.; Puretz, M.S.; Dhoonmoon, C.; Chegwidden, O.S.; Londergan, C.H.; Cho, M. The Bend+Libration Combination Band Is an Intrinsic, Collective, and Strongly Solute-Dependent Reporter on the Hydrogen Bonding Network of Liquid Water. J. Phys. Chem. B 2018, 122, 2587–2599. [Google Scholar] [CrossRef]
- Koike, C.; Mutschke, H.; Suto, H.; Naoi, T.; Chihara, H.; Henning, T.; Jäger, C.; Tsuchiyama, A.; Dorschner, J.; Okuda, H. Temperature effects on the mid-and far-infrared spectra of olivine particles. Astron. Astrophys. 2006, 449, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Tielens, A.G.G.M.; Allamandola, L.J. Composition, Structure, and Chemistry of Interstellar Dust; Springer: Dordrecht, The Netherlands; pp. 397–470.
- Plendl, J.N. Characteristic Energy Absorption Spectra of Solids; Springer: Boston, MA, USA, 1970; pp. 387–450. [Google Scholar]
- Day, K.L. Temperature Dependence of Mid-Infrared Silicate Absorption. Astrophys. J. 1976, 203, L99. [Google Scholar] [CrossRef]
- Zaikowski, A.; Knacke, R.F.; Porco, C.C. On the presence of phyllosilicate minerals in the interstellar grains. Astrophys. Space Sci. 1975, 35, 97–115. [Google Scholar] [CrossRef]
- Mennella, V.; Brucato, J.R.; Colangeli, L.; Palumbo, P.; Rotundi, A.; Bussoletti, E. Temperature Dependence of the Absorption Coefficient of Cosmic Analog Grains in the Wavelength Range 20 Microns to 2 Millimeters. Astrophys. J. 1998, 496, 1058–1066. [Google Scholar] [CrossRef] [Green Version]
- Bowey, J.E.; Lee, C.; Tucker, C.; Hofmeister, A.M.; Ade, P.A.R.; Barlow, M.J. Temperature effects on the 15–85 μm spectra of olivines and pyroxenes. Mon. Not. R. Astron. Soc. 2001, 325, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Johnston, C.T.; Kogel, J.E.; Bish, D.L.; Kogure, T.; Murray, H.H. Low-temperature Ftir Study of Kaolin-Group Minerals. Clays Clay Miner. 2008, 56, 470–485. [Google Scholar] [CrossRef]
- Balan, E.; Delattre, S.; Roche, D.; Segalen, L.; Morin, G.; Guillaumet, M.; Blanchard, M.; Lazzeri, M.; Brouder, C.; Salje, E.K.H. Line-broadening effects in the powder infrared spectrum of apatite. Phys. Chem. Miner. 2011, 38, 111–122. [Google Scholar] [CrossRef]
- Petrov, A.K.; Sechkarev, A.V. Temperature dependence of infrared spectra. Sov. Phys. J. 1965, 8, 62–64. [Google Scholar] [CrossRef]
- Sirotiak, M.; Bartošová, A. Changes in Structure and Content Humic Substances in Soil During the Laboratory Simulated Fires. Trans. VSB 2016, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Nanda, S.; Reddy, S.N.; Hunter, H.N.; Butler, I.S.; Kozinski, J.A. Supercritical Water Gasification of Lactose as a Model Compound for Valorization of Dairy Industry Effluents. Ind. Eng. Chem. Res. 2015, 54, 9296–9306. [Google Scholar] [CrossRef]
- Zuyi, T.; Shifang, L.; Jianjun, Z.; Fenling, S. Studies of Thermal Transformations of Humic and Fulvic Acids in Soils I. Infrared Spectroscopy and Temperature-Programmed Pyrolysis Mass Spectrometry. Chem. Ecol. 1997, 13, 237–248. [Google Scholar] [CrossRef]
- Kolokassidou, C.; Pashalidis, I.; Costa, C.N.; Efstathiou, A.M.; Buckau, G. Thermal stability of solid and aqueous solutions of humic acid. Thermochim. Acta 2007, 454, 78–83. [Google Scholar] [CrossRef]
- Malcolm, R.L.; MacCarthy, P. Limitations in the use of commercial humic acids in water and soil research. Environ. Sci. Technol. 1986, 20, 904–911. [Google Scholar] [CrossRef]
- Nunn, S.; Nishikida, K. Advanced ATR Correction Algorithm. In Thermo Fisher Scientific Application Note 50581; Thermo Fisher Scientific: Madison WI, USA, 2008. [Google Scholar]
- Baigorri, R.; Fuentes, M.; González-Gaitano, G.; García-Mina, J.M.; Almendros, G.; González-Vila, F.J. Complementary Multianalytical Approach To Study the Distinctive Structural Features of the Main Humic Fractions in Solution: Gray Humic Acid, Brown Humic Acid, and Fulvic Acid. J. Agric. Food Chem. 2009, 57, 3266–3272. [Google Scholar] [CrossRef] [Green Version]
- Campanella, L.; Tomassetti, M.; Piccolo, A. Thermogravimetric and IR analysis of different extracts of humic substances. Thermochim. Acta 1990, 170, 67–80. [Google Scholar] [CrossRef]
- Provenzano, M.R.; Senesi, N. Thermal Properties of Standard and Reference Humic Substances by Differential Scanning Calorimetry. J. Therm. Anal. Calorim. 1999, 57, 517–526. [Google Scholar] [CrossRef]
- Provenzano, M.R.; Senesi, N.; Miikki, V. Characterization of Composts and Humic Acids from Pulp and Paper Mill Biosludges by DSC in Association with FT-IR Spectroscopy. J. Therm. Anal. Calorim. 1998, 52, 1037–1046. [Google Scholar] [CrossRef]
- Janoš, P.; Kozler, J. Thermal stability of humic acids and some of their derivatives. Fuel 1995, 74, 708–713. [Google Scholar] [CrossRef]
- Cihlář, Z.; Kucerik, J. Regenerated humic acids obtained by the air oxidation of south moravian lignite. part. 2. thermoanalytical characterization of products. Pet. Coal 2010, 52, 254–260. [Google Scholar]
- Giovanela, M.; Parlanti, E.; Soriano-Sierra, E.J.; Soldi, M.S.; Sierra, M.M.D. Elemental compositions, FT-IR spectra and thermal behavior of sedimentary fulvic and humic acids from aquatic and terrestrial environments. Geochem. J. 2004, 38, 255–264. [Google Scholar] [CrossRef]
- Rotaru, A.; Nicolaescu, I.; Rotaru, P.; Neaga, C. Thermal characterization of humic acids and other components of raw coal. J. Therm. Anal. Calorim. 2008, 92, 297–300. [Google Scholar] [CrossRef]
- Sirbu, C.; Cioroianu, T.; Rotaru, P. About the humic acids and thermal behaviour of some humic acids. Ann. Univ. Craiova Phys. 2010, 20, 120–126. [Google Scholar]
- Hoffmann, K.; Huculak-Maczka, M.; Grek, E. INVESTIGATION OF THE PROPERTY OF HUMIC ACIDS BY THERMAL ANALYSIS METHOD. Ecol. Chem. Eng. A 2013, 20, 261–269. [Google Scholar] [CrossRef]
- Boguta, P.; Sokołowska, Z.; Skic, K. Use of thermal analysis coupled with differential scanning calorimetry, quadrupole mass spectrometry and infrared spectroscopy (TG-DSC-QMS-FTIR) to monitor chemical properties and thermal stability of fulvic and humic acids. PLoS ONE 2017, 12, e0189653. [Google Scholar] [CrossRef] [Green Version]
- Larkin, P. Instrumentation and Sampling Methods. In Infrared and Raman Spectroscopy; Larkin, P., Ed.; Elsevier: Oxford, JD, USA, 2011; pp. 27–54. [Google Scholar] [CrossRef]
- Laskina, O.; Young, M.A.; Kleiber, P.D.; Grassian, V.H. Infrared Optical Constants of Organic Aerosols: Organic Acids and Model Humic-Like Substances (HULIS). Aerosol Sci. Technol. 2014, 48, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Dinar, E.; Abo Riziq, A.; Spindler, C.; Erlick, C.; Kiss, G.; Rudich, Y. The complex refractive index of atmospheric and model humic-like substances (HULIS) retrieved by a cavity ring down aerosol spectrometer (CRD-AS). Faraday Discuss. 2008, 137, 279–295. [Google Scholar] [CrossRef]
- Kwon, D.; Sovers, M.J.; Grassian, V.H.; Kleiber, P.D.; Young, M.A. Optical Properties of Humic Material Standards: Solution Phase and Aerosol Measurements. ACS Earth Space Chem. 2018, 2, 1102–1111. [Google Scholar] [CrossRef]
- Russell, J.D.; Fraser, A.R. Infrared methods. In Clay Mineralogy: Spectroscopic and Chemical Determinative Methods; Wilson, M.J., Ed.; Springer: Dordrecht, The Netherlands, 1994; pp. 11–67. [Google Scholar]
- Madejová, J.; Komadel, P. Baseline studies of the clay minerals society source clays: Infrared methods. Clays Clay Miner. 2001, 49, 410–432. [Google Scholar] [CrossRef]
- Senesi, N.; D’Orazio, V.; Ricca, G. Humic acids in the first generation of EUROSOILS. Geoderma 2003, 116, 325–344. [Google Scholar] [CrossRef]
- Dhillon, G.S.; Gillespie, A.; Peak, D.; Van Rees, K.C.J. Spectroscopic investigation of soil organic matter composition for shelterbelt agroforestry systems. Geoderma 2017, 298, 1–13. [Google Scholar] [CrossRef]
- Lucas, S.; Tognonvi, M.T.; Gelet, J.L.; Soro, J.; Rossignol, S. Interactions between silica sand and sodium silicate solution during consolidation process. J. Non-Cryst. Solids 2011, 357, 1310–1318. [Google Scholar] [CrossRef]
- Lønstad Bleken, B.-T.; Mino, L.; Giordanino, F.; Beato, P.; Svelle, S.; Lillerud, K.P.; Bordiga, S. Probing the surface of nanosheet H-ZSM-5 with FTIR spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 13363–13370. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, G.E.; Laviano, R.; Sabbatini, L.; Zambonin, P.G. Infrared spectroscopy in the mineralogical characterization of ancient pottery. J. Cult. Herit. 2002, 3, 177–186. [Google Scholar] [CrossRef]
- Roeges, N.P.G. A Guide to the Complete Interpretation of Infrared Spectral of Organic Structures; Wiley: Hoboken, NJ, USA, 1994. [Google Scholar]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistry; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar]
- Workman, J. The Handbook of Organic Compounds, Three-Volume Set: NIR, IR, R, and UV-Vis Spectra Featuring Polymers and Surfactants; Elsevier Science: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Frost, R.L.; Vassallo, A.M. The Dehydroxylation of the Kaolinite Clay Minerals using Infrared Emission Spectroscopy. Clays Clay Miner. 1996, 44, 635–651. [Google Scholar] [CrossRef]
- Ma, F.; Zeng, Y.; Du, C.; Shen, Y.; Ma, H.; Xu, S.; Zhou, J. Soil variability description using Fourier transform mid-infrared photoacoustic spectroscopy coupling with RGB method. Catena 2017, 152, 190–197. [Google Scholar] [CrossRef]
- Krivoshein, P.K.; Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. FTIR photoacoustic spectroscopy for identification and assessment of soil components: Chernozems and their size fractions. Photoacoustics 2020, 18, 100162. [Google Scholar] [CrossRef]
- Maréchal, Y. The molecular structure of liquid water delivered by absorption spectroscopy in the whole IR region completed with thermodynamics data. J. Mol. Struct. 2011, 1004, 146–155. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., McKelvy, M.L., Eds.; Wiley: Hoboken, NJ, USA, 2006; p. a5606. [Google Scholar] [CrossRef]
- Bock, J.; Su, G.-J. Interpretation of the Infrared Spectra of Fused Silica. J. Am. Ceram. Soc. 1970, 53, 69–73. [Google Scholar] [CrossRef]
- Vaculíková, L.; Plevová, E.; Vallová, S.; Koutník, I. Characterization and differentiation of kaolinites from selected czech deposits using infrared spectroscopy and differential thermal analysis. Acta Geodyn. Et Geomater. 2011, 8, 59–67. [Google Scholar]
- Inoue, A.; Watanabe, T. Infrared Spectra of Interstratified Illite/Smectite from Hydrothermally Altered Tuffs (Shinzan, Japan) and Diagenetic Bentonites (Kinnekulle, Sweden). Clay Sci. 1989, 7, 263–275. [Google Scholar] [CrossRef]
- Wilson, M.J. Dissolution and formation of quartz in soil environments: A review. Soil Sci. Annu. 2020, 71, 99–110. [Google Scholar] [CrossRef]
- Vettegren, V.I.; Sobolev, G.A.; Kireenkova, S.M.; Morozov, Y.A.; Smul’skaya, A.I.; Mamalimov, R.I.; Kulik, V.B. Effect of water on the α-β phase transition in a surface quartz layer. Phys. Sol. State 2014, 56, 1228–1233. [Google Scholar] [CrossRef]
- Spitzer, W.G.; Kleinman, D.A. Infrared Lattice Bands of Quartz. Phys. Rev. 1961, 121, 1324–1335. [Google Scholar] [CrossRef]
- Max, J.J.; Chapados, C. Isotope effects in liquid water by infrared spectroscopy. III. H2O and D2O spectra from 6000 to 0 cm−1. J. Chem. Phys. 2009, 131, 184505. [Google Scholar] [CrossRef]
- Heaney, P.J.; Kronenberg, A.K.; Prewitt, C.T.; Gibbs, G.V. Chapter 4. Hydrogen Speciation and Chemical Weakening of Quartz. In Silica; De Gruyter: Berlin, Germany, 1994; pp. 123–176. [Google Scholar] [CrossRef]
- Cabaniss, S.E.; McVey, I.F. Aqueous infrared carboxylate absorbances: Aliphatic monocarboxylates. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 1995, 51, 2385–2395. [Google Scholar] [CrossRef]
- Cabaniss, S.E.; Leenheer, J.A.; McVey, I.F. Aqueous infrared carboxylate absorbances: Aliphatic di-acids. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 1998, 54, 449–458. [Google Scholar] [CrossRef]
- Dunn, G.E.; McDonald, R.S. Infrared spectra of aqueous sodium benzoates and salicylates in the carboxyl-stretching region: Chelation in aqueous sodium salicylates. Can. J. Chem. 1969, 47, 4577–4588. [Google Scholar] [CrossRef]
- Calderón, F.J.; Mikha, M.M.; Vigil, M.F.; Nielsen, D.C.; Benjamin, J.G.; Reeves, J.B. Diffuse-Reflectance Mid-infrared Spectral Properties of Soils under Alternative Crop Rotations in a Semi-arid Climate. Commun. Soil Sci. Plant. Anal. 2011, 42, 2143–2159. [Google Scholar] [CrossRef]
- Nardi, S.; Ertani, A.; Francioso, O. Soil-root cross-talking: The role of humic substances. J. Plant. Nutr. Soil Sci. 2017, 180, 5–13. [Google Scholar] [CrossRef]
- Bronnikov, S.V.; Vettegren, V.I.; Frenkel, S.Y. Description of thermal and mechanical properties of drawn polymers over a wide temperature range. Polym. Eng. Sci. 1992, 32, 1204–1208. [Google Scholar] [CrossRef]
- Poulet, H.; Mathieu, J.P. Vibration Spectra and Symmetry of Crystals; Gordon and Breach: New York, NY, USA; London, UK; Paris, France, 1976. [Google Scholar]
- Scott, J.F.; Porto, S.P.S. Longitudinal and Transverse Optical Lattice Vibrations in Quartz. Phys. Rev. 1967, 161, 903–910. [Google Scholar] [CrossRef]
- Tan, C.Z. Optical interference in overtones and combination bands in α-quartz. J. Phys. Chem. Solids 2003, 64, 121–125. [Google Scholar] [CrossRef]
- Zhuang, J.; Li, M.; Pu, Y.; Ragauskas, A.; Yoo, C. Observation of Potential Contaminants in Processed Biomass Using Fourier Transform Infrared Spectroscopy. Appl. Sci. 2020, 10, 4345. [Google Scholar] [CrossRef]
- Palacios, E.G.; Juárez-López, G.; Monhemius, A.J. Infrared spectroscopy of metal carboxylates: II. Analysis of Fe(III), Ni and Zn carboxylate solutions. Hydrometallurgy 2004, 72, 139–148. [Google Scholar] [CrossRef]
- Palacios, E.G.; Monhemius, A.J. Infrared spectroscopy of metal carboxylates: I. Determination of free acid in solution. Hydrometallurgy 2001, 62, 135–143. [Google Scholar] [CrossRef]
- Herrera-Gómez, A.; Velázquez-Cruz, G.; Martín-Polo, M.O. Analysis of the water bound to a polymer matrix by infrared spectroscopy. J. Appl. Phys. 2001, 89, 5431–5437. [Google Scholar] [CrossRef]
- Honghong, H.; Meifang, G.; Juan, L.; Ortín, A.; Yau, W.W. Analysis of Propylene-1-Butene Copolymer Composition by GPC with Online Detectors. Macromol. Symp. 2015, 356, 110–121. [Google Scholar] [CrossRef]
- Yamagishi, H.; Nakashima, S.; Ito, Y. High temperature infrared spectra of hydrous microcrystalline quartz. Phys. Chem. Miner. 1997, 24, 66–74. [Google Scholar] [CrossRef]
- Mikhaylova, Y.; Adam, G.; Häussler, L.; Eichhorn, K.J.; Voit, B. Temperature-dependent FTIR spectroscopic and thermoanalytic studies of hydrogen bonding of hydroxyl (phenolic group) terminated hyperbranched aromatic polyesters. J. Mol. Struct. 2006, 788, 80–88. [Google Scholar] [CrossRef]
- Du, C.; Goyne, K.W.; Miles, R.J.; Zhou, J. A 1915–2011 microscale record of soil organic matter under wheat cultivation using FTIR-PAS depth-profiling. Agron. Sustain. Dev. 2013, 34, 803–811. [Google Scholar] [CrossRef]
- Kar, S.; Maity, J.P.; Jean, J.-S.; Liu, C.-C.; Nath, B.; Lee, Y.-C.; Bundschuh, J.; Chen, C.-Y.; Li, Z. Role of organic matter and humic substances in the binding and mobility of arsenic in a Gangetic aquifer. J. Environ. Sci. Health Part. A 2011, 46, 1231–1238. [Google Scholar] [CrossRef]
- Volkov, D.S.; Krivoshein, P.K.; Mikheev, I.V.; Proskurnin, M.A. Pristine detonation nanodiamonds as regenerable adsorbents for metal cations. Diam. Relat. Mater. 2020, 110, 108121. [Google Scholar] [CrossRef]
- Nguyen, T.; Janik, L.; Raupach, M. Diffuse reflectance infrared fourier transform (DRIFT) spectroscopy in soil studies. Soil Res. 1991, 29, 49–67. [Google Scholar] [CrossRef]
- Fukuda, J.-i.; Shimizu, I. Water distribution in quartz schists of the Sanbagawa Metamorphic Belt, Japan: Infrared spectroscopic mapping and comparison of the calibrations proposed for determining water contents. Earth Planets Space 2019, 71, 136. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, B.-X.; Fu, C.-S.; Sui, G.-R.; Iso, M. gamma-irradiation damage of quartz fiber and its effects on near-infrared transmission characteristics. Acta Phys. Sin. 2010, 59, 7782–7787. [Google Scholar]
- Hofmeister, A.M.; Bowey, J.E. Quantitative Infrared Spectra of Hydrosilicates and Related Minerals. Mon. Not. R. Astron. Soc. 2006, 367, 577–591. [Google Scholar] [CrossRef]
- Shinoda, K.; Aikawa, N. Absorption coefficients of overtone and combination modes of quartz. Mineral. Mag. 1994, 58, 601–606. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spectral range, cm−1 | 4000–400 |
| Resolution, cm−1 | 2 |
| Background scan | 128 |
| Sample scan | 128 |
| Acquisition mode | Double-sided, forward–backward |
| High-frequency limit | 15000 |
| Aperture setting | 8 mm |
| Phase resolution | 4 |
| Phase-correction mode | Mertz |
| Zero filling factor | 2 |
| Apodization function | Blackman–Harris 3-term |
| Sample and background pre-amplification gain | ‘Ref’ (without amplification) |
| Background signal gain | Auto |
| Sample signal gain | Auto |
| Scanner velocity | 10 kHz |
| Detector | Room temperature DLaTGS |
| Source | MIR, globar |
| Beam splitter | KBr |
| Background | Diamond crystal with a lowered pressure screw with a flat end |
| Sigma-Aldrich | Powhumus | Sakhalin | Assignment | Ref. | |||
|---|---|---|---|---|---|---|---|
| ν (25 °C) | ν (215 °C)–ν (25 °C) | ν (25 °C) | ν (215 °C)–ν (25 °C) | ν (25 °C) | ν (215 °C)–ν (25 °C) | ||
| 3691.4 | −9.84 | 3692.8 | −9.61 | 3695.9 | −12.04 | OH stretching of inner-surface hydroxyl groups, in-phase vibration with a transition moment nearly perpendicular to the (001) plane of kaolinite | [60,61] |
| 3666.7 | * | 3666.8 | * | - | - | OH stretching of inner-surface hydroxyl groups, anti-phase vibration with transition moment lying in the (001) plane of kaolinite | |
| 3650.0 | * | 3651.1 | * | - | - | ||
| 3618.7 | 4.57 | 3618.9 | 5.02 | 3613.3 | 10.63 | OH stretching of inner hydroxyl groups | |
| 2960 *** | * | 2960 *** | * | 2960 *** | * | Asymmetric and symmetric C–H stretching of aliphatic groups CHx | [18,62] |
| 2925.7 | −1.59 | 2925.2 | −0.13 | 2919.5 | 4.71 | ||
| 2854.9 | 0.47 | 2854.1 | 1.38 | 2850.7 | 2.60 | ||
| 1569.1 | 8.48 | 1571.2 | 11.15 | 1574.9 | 10.54 | Aromatic C=C stretching of the phenyl ring, CO carboxylate stretching; aromatic C=C skeletal stretching vibrations, C=O of quinone and/or H-bonded conjugated ketones; –COO– symmetric stretching | [20] |
| 1398.0 | 0.13 | - | - | - | - | 1450 –CH deformation of –CH3 and –CH bending of –CH; 1350 symmetric –COO~ stretch and/or –CH bending of aliphatics | [20] |
| - | - | 1378.1 | −10.39 | 1375.3 | −12.99 | A combination band, and is primarily attributed to CH absorption in aliphatics, as well as to CO–CH3 vibrations in lignin-derived phenols | [63] |
| 1164.5 | * | 1164.5 | * | 1164.5 | * | C–OH stretching of aliphatic O–H | [62] |
| 1114 ** | 1114 ** | - | - | - | Si–O stretching (longitudinal mode) of kaolinite | [61] | |
| 1085.2 | −8.42 | 1089.5 | −9.76 | ** | Alcoholic and polysaccharide CO stretch and OH deformation; Si–OH bend in silicate impurities | [62] | |
| 1031.5 | −2.64 | 1032.1 | −2.89 | 1032.2 | −3.27 | In-plane Si–O stretching (of kaolinite) | [61] |
| 1008.4 | −3.17 | 1009.5 | −3.06 | 1010.1 | −1.48 | In-plane Si–O stretching (of kaolinite) | [61] |
| 937.2 | * | 937.7 | * | 935 *** | * | OH deformation of the inner-surface hydroxyl group of kaolinite | [61] |
| 912.4 | −3.29 | 913.4 | −3.16 | 912.6 | −2.44 | OH deformation of inner hydroxyl groups of kaolinite | [61] |
| 875.0 | −0.80 | 875.9 | 1.03 | Si–O− or Si–O–Si bridge or carbonate, calcite, or polyaromatic bend vibrations | [60,64,65,66,67] | ||
| 795.7 | −3.38 | 796.4 | −3.02 | 797.5 | −3.46 | Si–O in kaolinite | [61] |
| 778 *** | * | 778 *** | * | 778.5 | * | Aromatic CH out of plane bending | [20] |
| 749.9 | 2.74 | 749.9 | * | 750 *** | * | Si–O, perpendicular | [61] |
| 713 | * | - | - | - | - | Al–O vibrations or C4+ alkanes | [68,69] |
| 690 ** | - | 690 ** | - | 690 ** | OH translation in kaolinite | [70] | |
| 640 *** | - | 640 *** | - | - | - | Si–O | [61] |
| 534.9 | −4.52 | 536.2 | −4.94 | 532.6 | −5.06 | Si–O–Al deformation in kaolinite | [61,70] |
| 466.6 | 0.84 | 468.3 | −0.44 | 468.9 | −0.95 | Si–O bending in kaolinite | [70] |
| 428.6 | 1.28 | 430.0 | 1.85 | Si–O deformation of kaolinite | [61] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volkov, D.S.; Rogova, O.B.; Proskurnin, M.A. Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy 2021, 11, 1822. https://doi.org/10.3390/agronomy11091822
Volkov DS, Rogova OB, Proskurnin MA. Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy. 2021; 11(9):1822. https://doi.org/10.3390/agronomy11091822
Chicago/Turabian StyleVolkov, Dmitry S., Olga B. Rogova, and Mikhail A. Proskurnin. 2021. "Temperature Dependences of IR Spectra of Humic Substances of Brown Coal" Agronomy 11, no. 9: 1822. https://doi.org/10.3390/agronomy11091822
APA StyleVolkov, D. S., Rogova, O. B., & Proskurnin, M. A. (2021). Temperature Dependences of IR Spectra of Humic Substances of Brown Coal. Agronomy, 11(9), 1822. https://doi.org/10.3390/agronomy11091822