
Long-Term Compost Amendment Changes Interactions and Specialization in the Soil Bacterial Community, Increasing the Presence of Beneficial N-Cycling Genes in the Soil

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sampling
2.2. Soil Properties, DNA Extraction, Sequencing, Data Processing and Function Prediction
2.3. Construction and Analysis of the Microbial Network
2.4. Statistical Analysis
3. Results
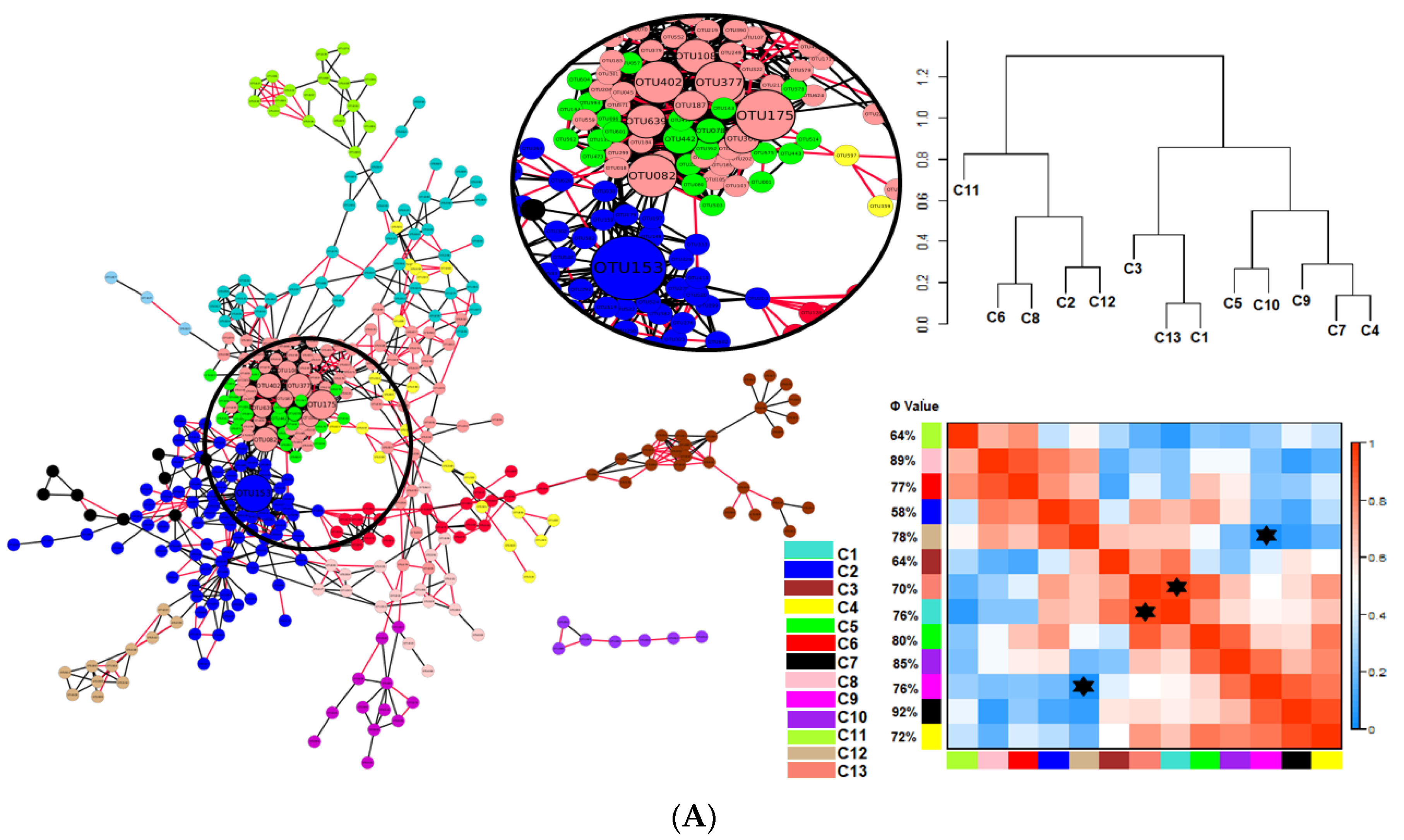
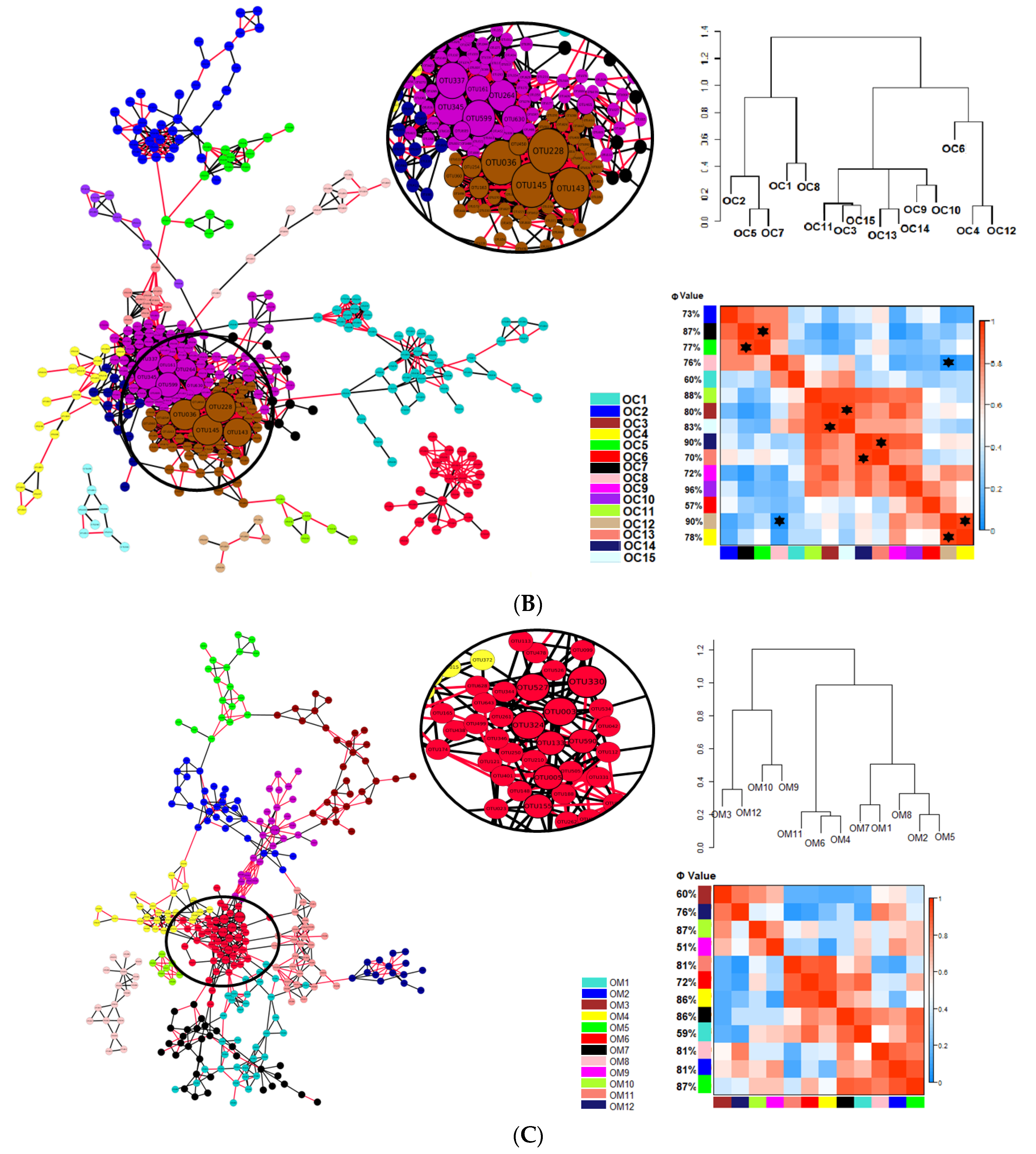
3.1. Network Analysis
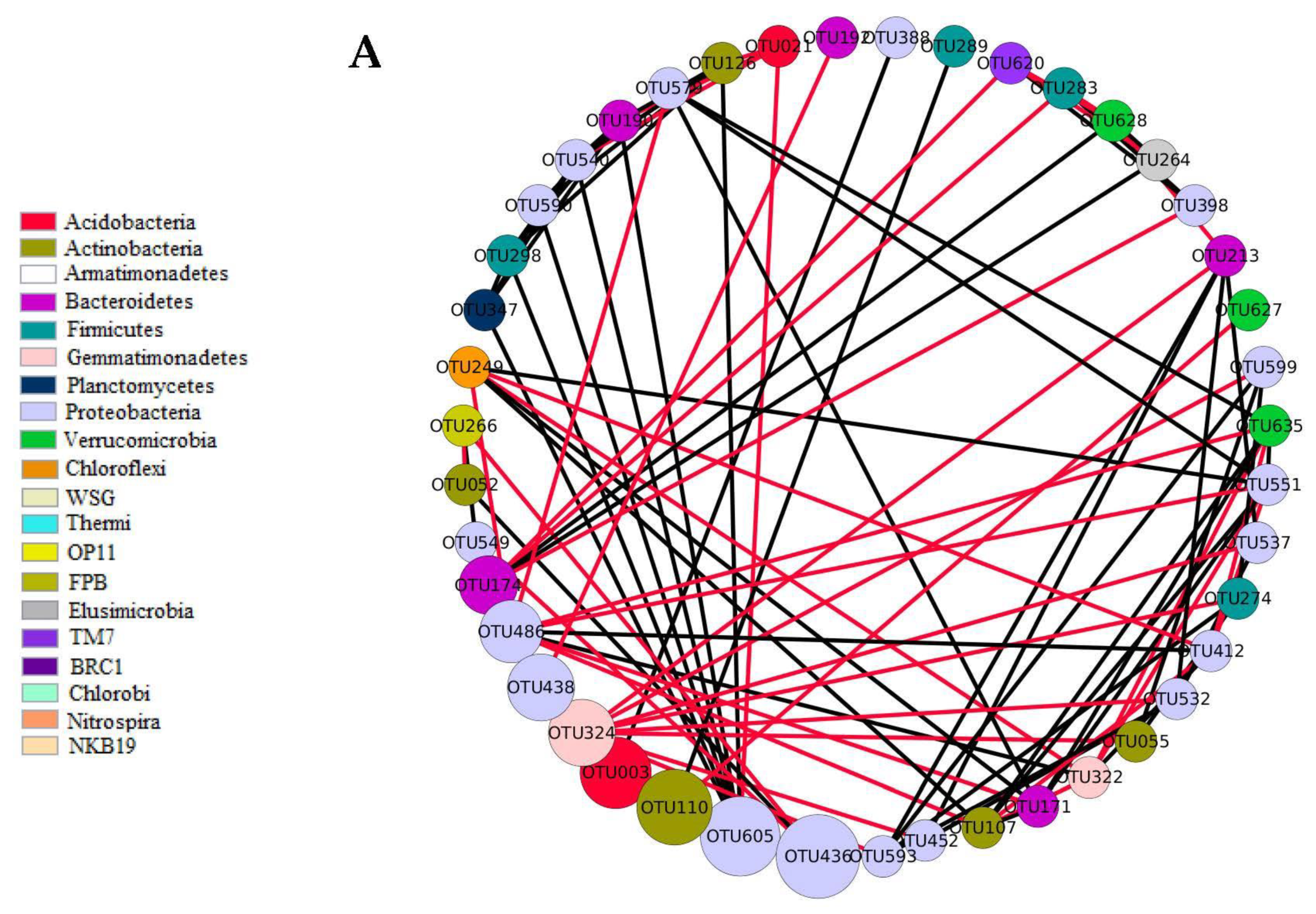
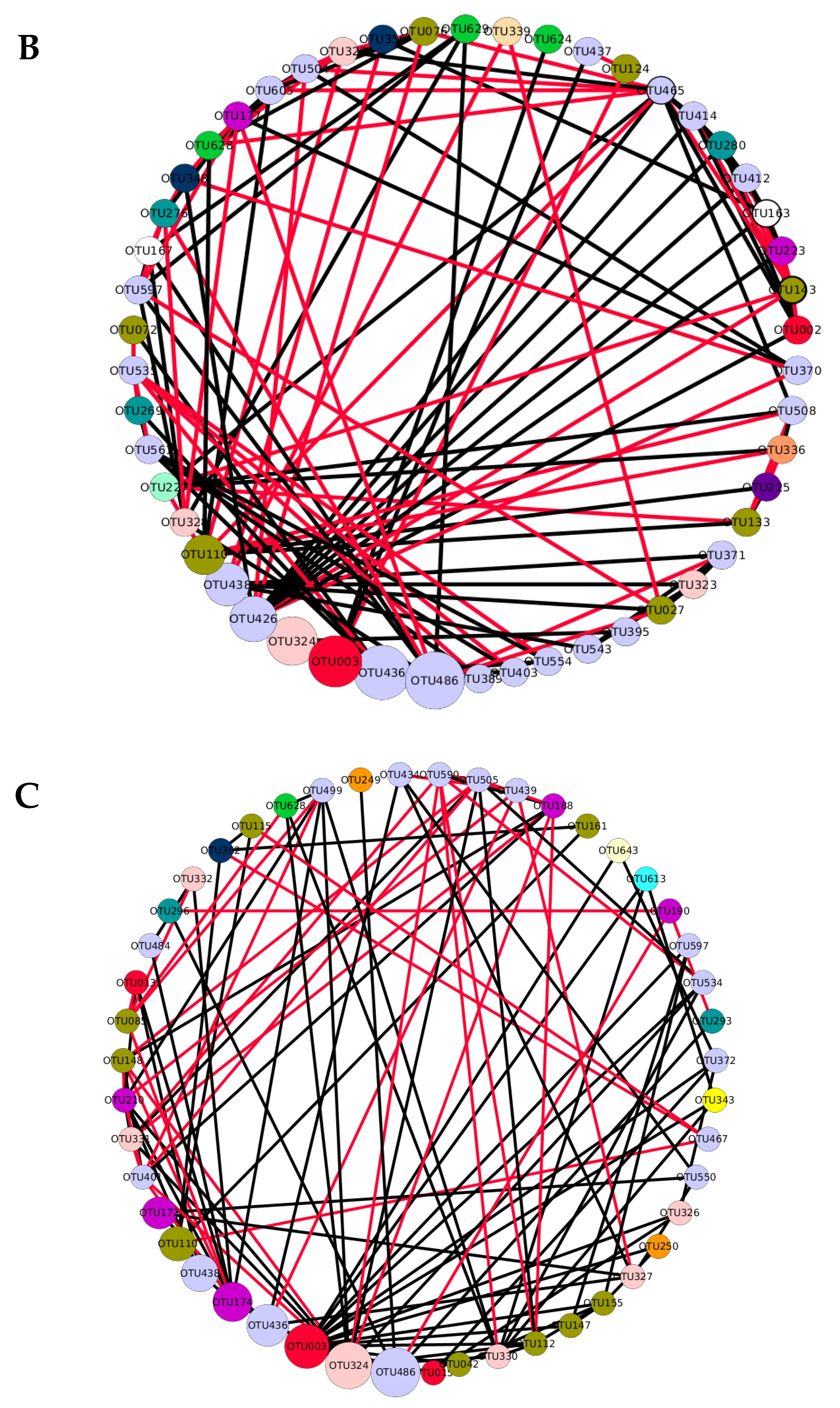
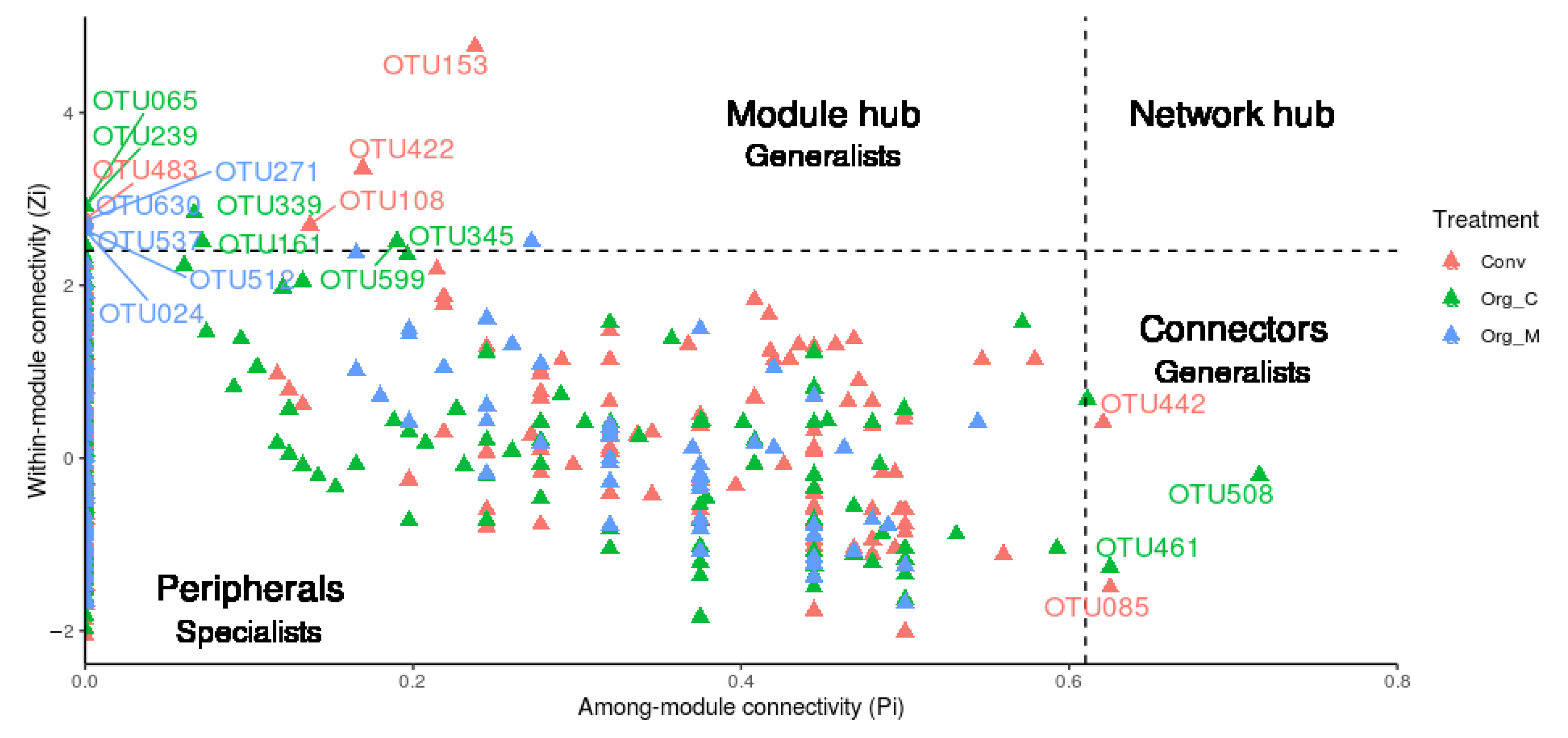
3.2. The Generalist Presence in Networks
3.3. Predictive Functional Community
3.4. Predictive Nitrogen Functional Community
3.5. Soil Properties
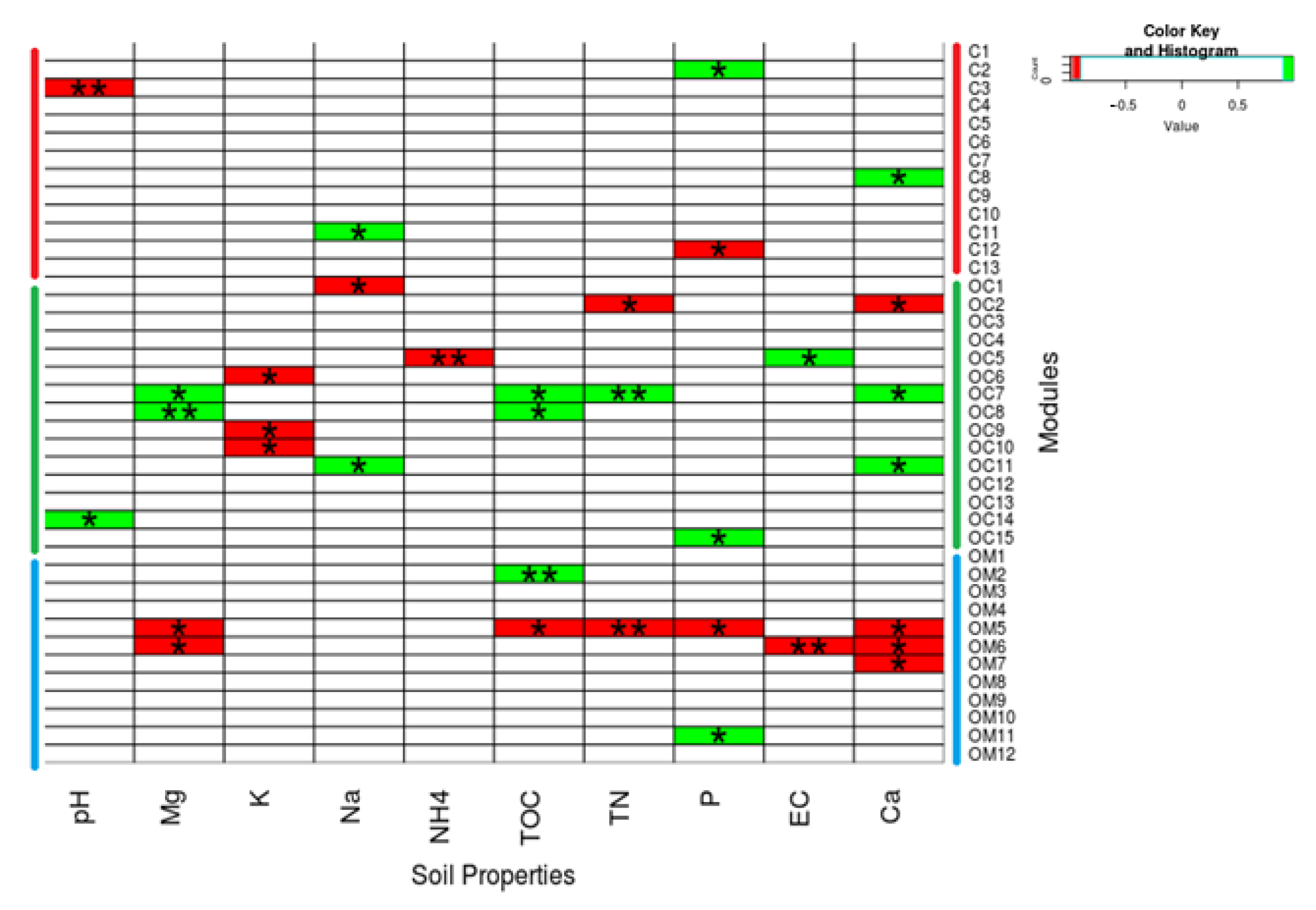
3.6. Module and Node Correlations with Soil Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pretty, J.; Benton, T.G.; Bharucha, Z.P.; Dicks, L.V.; Flora, C.B.; Godfray, H.C.J.; Goulson, D.; Hartley, S.; Lampkin, N.; Morris, C. Global assessment of agricultural system redesign for sustainable intensification. Nat. Sustain. 2018, 1, 441–446. [Google Scholar] [CrossRef]
- Gold, M.V.; Thompson, R.S.; Alternative Farming Systems Information Center. Organic Farming and Marketing: Publications from the United States Department of Agriculture, 1977–2005; Org. FARMING METHODS, Econ. Struct.; Hauppauge: New York, NY, USA, 2009; pp. 245–264. [Google Scholar]
- Sharma, D.; Yadav, K.D.; Kumar, S. Role of sawdust and cow dung on compost maturity during rotary drum composting of flower waste. Bioresour. Technol. 2018, 264, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.; Berti, M.; Lawley, Y.; Liebig, M. Integration of annual and perennial cover crops for improving soil health. In Soil Health and Intensification of Agroecosytems; Elsevier: Amsterdam, The Netherlands, 2017; pp. 127–150. [Google Scholar]
- Abdelrahman, H.M.; Zaghloul, R.A.; Abou-Aly, H.A.; Ragab, A.A.; K Elmaghraby, M.M. Application of Some Organic Farming Methods to Enhancement The Growth and Production of Green Onion. J. Agric. Chem. Biotechnol. 2021, 12, 79–89. [Google Scholar]
- Seufert, V.; Ramankutty, N.; Foley, J.A. Comparing the yields of organic and conventional agriculture. Nature 2012, 485, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Ros, M.; Klammer, S.; Knapp, B.; Aichberger, K.; Insam, H. Long-term effects of compost amendment of soil on functional and structural diversity and microbial activity. Soil Use Manag. 2006, 22, 209–218. [Google Scholar] [CrossRef]
- Van-Camp, L.; Bujarrabal, B.; Gentile, A.R.; Jones, R.J.A.; Montanarella, L.; Olazabal, C.; Selvaradjou, S. Technical working groups established under the thematic strategy for soil protection. JRC Publ. Repos. 2004, V, JRC28868. [Google Scholar]
- Kurzemann, F.R.; Plieger, U.; Probst, M.; Spiegel, H.; Sandén, T.; Ros, M.; Insam, H. Long-Term Fertilization Affects Soil Microbiota, Improves Yield and Benefits Soil. Agronomy 2020, 10, 1664. [Google Scholar] [CrossRef]
- Ling, N.; Zhu, C.; Xue, C.; Chen, H.; Duan, Y.; Peng, C.; Guo, S.; Shen, Q. Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis. Soil Biol. Biochem. 2016, 99, 137–149. [Google Scholar] [CrossRef]
- Wright, S. The importance of soil microorganisms in aggregate stability. In Proceedings of the North Central Extension Industry Soil Fertility Conference Proceedings, Monticello, IL, USA, 19 November 2003. [Google Scholar]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. MBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Tu, Q.; Zhi, X. Functional molecular ecological networks. MBio 2010, 1, e00169-10. [Google Scholar] [CrossRef] [Green Version]
- Kirchmann, H.; Bergström, L.; Kätterer, T.; Mattsson, L.; Gesslein, S. Comparison of Long-Term Organic and Conventional Crop–Livestock Systems on a Previously Nutrient-Depleted Soil in Sweden. Agron. J. 2007, 99, 960–972. [Google Scholar] [CrossRef]
- Ren, C.; Zhao, F.; Shi, Z.; Chen, J.; Han, X.; Yang, G.; Feng, Y.; Ren, G. Differential responses of soil microbial biomass and carbon-degrading enzyme activities to altered precipitation. Soil Biol. Biochem. 2017, 115, 1–10. [Google Scholar] [CrossRef]
- Kuypers, M.M.M.; Marchant, H.K.; Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 2018, 16, 263. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Luo, C.; Jiang, L.; Song, M.; Zhang, D.; Li, J.; Li, Y.; Ostle, N.J.; Zhang, G. Land-use changes alter soil bacterial composition and diversity in tropical forest soil in China. Sci. Total Environ. 2020, 712, 136526. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Huang, S.; Zhang, H.; Tong, Y.; Wen, D.; Xia, X.; Wang, H.; Luo, Y.; Barceló, D. Response of soil enzyme activities and bacterial communities to the accumulation of microplastics in an acid cropped soil. Sci. Total Environ. 2020, 707, 135634. [Google Scholar] [CrossRef]
- Dube, J.P.; Valverde, A.; Steyn, J.M.; Cowan, D.A.; Van der Waals, J.E. Differences in bacterial diversity, composition and function due to long-term agriculture in soils in the eastern free State of South Africa. Diversity 2019, 11, 61. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xia, Q.; Yang, T.; Shi, W. Eighteen-year farming management moderately shapes the soil microbial community structure but promotes habitat-specific taxa. Front. Microbiol. 2018, 9, 1776. [Google Scholar] [CrossRef] [Green Version]
- García Algarra, F.J. Modelos de Redes Cooperativas; ETS de Ingeniería Agronómica, Alimentaria y de Biosistemas (UPM): Madrid, Spain, 2016. [Google Scholar]
- Montoya, J.M.; Pimm, S.L.; Solé, R. V Ecological networks and their fragility. Nature 2006, 442, 259–264. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Leff, J.W.; Adams, B.J.; Nielsen, U.N.; Bates, S.T.; Lauber, C.L.; Owens, S.; Gilbert, J.A.; Wall, D.H.; Caporaso, J.G. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 2012, 109, 21390–21395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Zhou, G.; Zhang, J.; Wang, W. Microbial community responses to biochar addition when a green waste and manure mix are composted: A molecular ecological network analysis. Bioresour. Technol. 2019, 273, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xue, D.; Li, X.; Deng, Y.; Rui, J.; Feng, K.; Wang, Z. The responses and adaptations of microbial communities to salinity in farmland soils: A molecular ecological network analysis. Appl. Soil Ecol. 2017, 120, 239–246. [Google Scholar] [CrossRef]
- Anjos, L.; Gaistardo, C.; Deckers, J.; Dondeyne, S.; Eberhardt, E.; Gerasimova, M.; Harms, B.; Jones, A.; Krasilnikov, P.; Reinsch, T.; et al. World Reference Base for Soil Resources 2014 (Update 2015), International Soil Classification System for Naming Soils and Creating Legends for Soil Maps; World Soil Resour. Reports; FAO: Rome, Italy, 2015. [Google Scholar]
- Cuartero, J.; Özbolat, O.; Sánchez-Navarro, V.; Egea-Cortines, M.; Zornoza, R.; Canfora, L.; Orrù, L.; Pascual, J.A.; Vivo, J.-M.; Ros, M. Changes on Bacterial and Fungal Soil Communities in Long-Term Organic Cropping Systems. Agriculture 2021, 11, 445. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012, 40, D109–D114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimera, R.; Amaral, L.A.N. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2016. [Google Scholar]
- Oksanen, J.; Blanchet, G.; Friendly, M.; Kindt, R.; Pierre, L.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.5-7. Available online: https://cran.r-project.org/package=vegan (accessed on 10 December 2020).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; v 4.0. [Google Scholar]
- Ogle, D.; Ogle, M.D. FSA: Fisheries Stock Analysis; R Package Version 0.8.30. Available online: https://github.com/droglenc/FSA (accessed on 15 December 2020).
- Morrissey, E.M.; Mau, R.L.; Schwartz, E.; Caporaso, J.G.; Dijkstra, P.; Van Gestel, N.; Koch, B.J.; Liu, C.M.; Hayer, M.; McHugh, T.A. Phylogenetic organization of bacterial activity. ISME J. 2016, 10, 2336–2340. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.-E.T.; Kim, D.Y.; Sachdeva, R.; Caron, D.A.; Fuhrman, J.A. Top-down controls on bacterial community structure: Microbial network analysis of bacteria, T4-like viruses and protists. ISME J. 2014, 8, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, S.; Lyu, T.; Ding, Y.; Li, Z.; Wang, D.; You, S.; Xie, Q. Campus sewage treatment in multilayer horizontal subsurface flow constructed wetlands: Nitrogen removal and microbial community distribution. CLEAN–Soil Air Water 2017, 45, 1700254. [Google Scholar] [CrossRef]
- Wood, S.A.; Gilbert, J.A.; Leff, J.W.; Fierer, N.; D’Angelo, H.; Bateman, C.; Gedallovich, S.M.; Gillikin, C.M.; Gradoville, M.R.; Mansor, P. Consequences of tropical forest conversion to oil palm on soil bacterial community and network structure. Soil Biol. Biochem. 2017, 112, 258–268. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, B.; Li, H.; Liu, M.; Chen, L.; Zhou, S. Aggregate-related changes in network patterns of nematodes and ammonia oxidizers in an acidic soil. Soil Biol. Biochem. 2015, 88, 101–109. [Google Scholar] [CrossRef]
- Shi, L.; Huang, Y.; Zhang, M.; Yu, Y.; Lu, Y.; Kong, F. Bacterial community dynamics and functional variation during the long-term decomposition of cyanobacterial blooms in-vitro. Sci. Total Environ. 2017, 598, 77–86. [Google Scholar] [CrossRef]
- Yang, H.; Ma, J.; Rong, Z.; Zeng, D.; Wang, Y.; Hu, S.; Ye, W.; Zheng, X. Wheat straw return influences nitrogen-cycling and pathogen associated soil microbiota in a wheat–soybean rotation system. Front. Microbiol. 2019, 10, 1811. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.; Zhang, Z.; Cai, W.; Liu, W.; Xu, M.; Yin, H.; Wang, A.; He, Z.; Deng, Y. Biodiversity and species competition regulate the resilience of microbial biofilm community. Mol. Ecol. 2017, 26, 6170–6182. [Google Scholar] [CrossRef]
- Ye, Z.; Li, J.; Wang, J.; Zhang, C.; Liu, G. Diversity and co-occurrence network modularization of bacterial communities determine soil fertility and crop yields in arid fertigation agroecosystems. Biol. Fertil. Soils 2021, 57, 809–824. [Google Scholar] [CrossRef]
- Lo, C.-C. Effect of pesticides on soil microbial community. J. Environ. Sci. Heal. Part B 2010, 45, 348–359. [Google Scholar] [CrossRef]
- Meron, D.; Atias, E.; Kruh, L.I.; Elifantz, H.; Minz, D.; Fine, M.; Banin, E. The impact of reduced pH on the microbial community of the coral Acropora eurystoma. ISME J. 2011, 5, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röttjers, L.; Faust, K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 2018, 42, 761–780. [Google Scholar] [CrossRef] [Green Version]
- Mundra, S.; Kjønaas, O.J.; Morgado, L.N.; Krabberød, A.K.; Ransedokken, Y.; Kauserud, H. Soil depth matters: Shift in composition and inter-kingdom co-occurrence patterns of microorganisms in forest soils. FEMS Microbiol. Ecol. 2021, 97, fiab022. [Google Scholar] [CrossRef] [PubMed]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yin, S.; Liu, X.; Zhang, W.; Gu, T.; Shen, Q.; Qiu, H. Fungal networks in yield-invigorating and-debilitating soils induced by prolonged potato monoculture. Soil Biol. Biochem. 2013, 65, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Siles, J.A.; García-Sánchez, M.; Gómez-Brandón, M. Studying Microbial Communities through Co-Occurrence Network Analyses during Processes of Waste Treatment and in Organically Amended Soils: A Review. Microorganisms 2021, 9, 1165. [Google Scholar] [CrossRef]
- Zhou, Z.; Gao, T.; Zhu, Q.; Yan, T.; Li, D.; Xue, J.; Wu, Y. Increases in bacterial community network complexity induced by biochar-based fertilizer amendments to karst calcareous soil. Geoderma 2019, 337, 691–700. [Google Scholar] [CrossRef]
- Mehrani, M.-J.; Sobotka, D.; Kowal, P.; Ciesielski, S.; Makinia, J. The occurrence and role of Nitrospira in nitrogen removal systems. Bioresour. Technol. 2020, 303, 122936. [Google Scholar] [CrossRef]
- Pinto, A.J.; Marcus, D.N.; Ijaz, U.Z.; Bautista-de Lose Santos, Q.M.; Dick, G.J.; Raskin, L. Metagenomic evidence for the presence of comammox Nitrospira-like bacteria in a drinking water system. Msphere 2016, 1, e00054-15. [Google Scholar] [CrossRef] [Green Version]
- Dahal, B.; NandaKafle, G.; Perkins, L.; Brözel, V.S. Diversity of free-Living nitrogen fixing Streptomyces in soils of the badlands of South Dakota. Microbiol. Res. 2017, 195, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ide, H.; Ishii, K.; Fujitani, H.; Tsuneda, S. Draft genome sequence of Acidovorax sp. strain NB1, isolated from a nitrite-oxidizing enrichment culture. Microbiol. Resour. Announc. 2019, 8, e00547-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, L.C.; Lajtha, K.; Rousk, J. Nutrient limitation may induce microbial mining for resources from persistent soil organic matter. Ecology 2021, 102, e03328. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ye, G.; Kuzyakov, Y.; Liu, D.; Fan, J.; Ding, W. Long-term manure application increases soil organic matter and aggregation, and alters microbial community structure and keystone taxa. Soil Biol. Biochem. 2019, 134, 187–196. [Google Scholar] [CrossRef]
- Quilty, J.R.; Cattle, S.R. Use and understanding of organic amendments in Australian agriculture: A review. Soil Res. 2011, 49, 1–26. [Google Scholar] [CrossRef]
- Zhao, J.; Ni, T.; Xun, W.; Huang, X.; Huang, Q.; Ran, W.; Shen, B.; Zhang, R.; Shen, Q. Influence of straw incorporation with and without straw decomposer on soil bacterial community structure and function in a rice-wheat cropping system. Appl. Microbiol. Biotechnol. 2017, 101, 4761–4773. [Google Scholar] [CrossRef]
- Pandit, S.N.; Kolasa, J.; Cottenie, K. Contrasts between habitat generalists and specialists: An empirical extension to the basic metacommunity framework. Ecology 2009, 90, 2253–2262. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [Green Version]
- Tahir, H.A.S.; Gu, Q.; Wu, H.; Niu, Y.; Huo, R.; Gao, X. Bacillus volatiles adversely affect the physiology and ultra-structure of Ralstonia solanacearum and induce systemic resistance in tobacco against bacterial wilt. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Tan, H.; Zhou, S.; Deng, Z.; He, M.; Cao, L. Ribosomal-sequence-directed selection for endophytic streptomycete strains antagonistic to Ralstonia solanacearum to control tomato bacterial wilt. Biol. Control 2011, 59, 245–254. [Google Scholar] [CrossRef]
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Suominen, S.; van Vliet, D.M.; Sánchez-Andrea, I.; van der Meer, M.T.J.; Sinninghe Damste, J.S.; Villanueva, L. Organic matter type defines the composition of active microbial communities originating from anoxic Baltic Sea sediments. Front. Microbiol. 2021, 12, 978. [Google Scholar] [CrossRef] [PubMed]
- Sagova-Mareckova, M.; Zadorova, T.; Penizek, V.; Omelka, M.; Tejnecky, V.; Pruchova, P.; Chuman, T.; Drabek, O.; Buresova, A.; Vanek, A. The structure of bacterial communities along two vertical profiles of a deep colluvial soil. Soil Biol. Biochem. 2016, 101, 65–73. [Google Scholar] [CrossRef]
- Mo, Y.; Zhang, W.; Wilkinson, D.M.; Yu, Z.; Xiao, P.; Yang, J. Biogeography and co-occurrence patterns of bacterial generalists and specialists in three subtropical marine bays. Limnol. Oceanogr. 2021, 66, 793–806. [Google Scholar] [CrossRef]
- Lin, Z.; Zhen, Z.; Wu, Z.; Yang, J.; Zhong, L.; Hu, H.; Luo, C.; Bai, J.; Li, Y.; Zhang, D. The impact on the soil microbial community and enzyme activity of two earthworm species during the bioremediation of pentachlorophenol-contaminated soils. J. Hazard. Mater. 2016, 301, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Ventura, M.; Canchaya, C.; Fitzgerald, G.F.; Gupta, R.S.; van Sinderen, D. Genomics as a means to understand bacterial phylogeny and ecological adaptation: The case of bifidobacteria. Antonie Van Leeuwenhoek 2007, 91, 351–372. [Google Scholar] [CrossRef]
- Kersters, K.; De Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the Proteobacteria. In The Prokaryotes: A Handbook on the Biology of Bacteria; Springer: Berlin, Germany, 2006; Volume 5, pp. 3–37. ISBN 0387307451. [Google Scholar]
- Wiedenbeck, J.; Cohan, F.M. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 2011, 35, 957–976. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ryals, R.; Silver, W.L. Effects of organic matter amendments on net primary productivity and greenhouse gas emissions in annual grasslands. Ecol. Appl. 2013, 23, 46–59. [Google Scholar] [CrossRef]
- Fierer, N.; Ladau, J.; Clemente, J.C.; Leff, J.W.; Owens, S.M.; Pollard, K.S.; Knight, R.; Gilbert, J.A.; McCulley, R.L. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 2013, 342, 621–624. [Google Scholar] [CrossRef] [Green Version]
- Bossolani, J.W.; Crusciol, C.A.C.; Merloti, L.F.; Moretti, L.G.; Costa, N.R.; Tsai, S.M.; Kuramae, E.E. Long-term lime and gypsum amendment increase nitrogen fixation and decrease nitrification and denitrification gene abundances in the rhizosphere and soil in a tropical no-till intercropping system. Geoderma 2020, 375, 114476. [Google Scholar] [CrossRef]
- Zhaoxiang, W.; Huihu, L.; Qiaoli, L.; Changyan, Y.; Faxin, Y. Application of bio-organic fertilizer, not biochar, in degraded red soil improves soil nutrients and plant growth. Rhizosphere 2020, 16, 100264. [Google Scholar] [CrossRef]
- Fan, F.; Yu, B.; Wang, B.; George, T.S.; Yin, H.; Xu, D.; Li, D.; Song, A. Microbial mechanisms of the contrast residue decomposition and priming effect in soils with different organic and chemical fertilization histories. Soil Biol. Biochem. 2019, 135, 213–221. [Google Scholar] [CrossRef]
- Igiehon, N.O.; Babalola, O.O. Rhizosphere microbiome modulators: Contributions of nitrogen fixing bacteria towards sustainable agriculture. Int. J. Environ. Res. Public Health 2018, 15, 574. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Cheng, H.; Yang, L.; Su, S.; Wang, H.; Wang, S.; Wang, A. Coupled sulfur and iron (II) carbonate-driven autotrophic denitrification for significantly enhanced nitrate removal. Environ. Sci. Technol. 2018, 53, 1545–1554. [Google Scholar] [CrossRef]
- Van Zwieten, L.; Singh, B.P.; Kimber, S.W.L.; Murphy, D.V.; Macdonald, L.M.; Rust, J.; Morris, S. An incubation study investigating the mechanisms that impact N2O flux from soil following biochar application. Agric. Ecosyst. Environ. 2014, 191, 53–62. [Google Scholar] [CrossRef]
- Putz, M.; Schleusner, P.; Rütting, T.; Hallin, S. Relative abundance of denitrifying and DNRA bacteria and their activity determine nitrogen retention or loss in agricultural soil. Soil Biol. Biochem. 2018, 123, 97–104. [Google Scholar] [CrossRef]
 ) indicates a significant (p < 0.05) correlation among modules: Conv, conventional cropping system; Org_C, organic cropping system with compost and compost tea; Org_M, organic cropping system with manure.
) indicates a significant (p < 0.05) correlation among modules: Conv, conventional cropping system; Org_C, organic cropping system with compost and compost tea; Org_M, organic cropping system with manure.
) indicates a significant (p < 0.05) correlation among modules: Conv, conventional cropping system; Org_C, organic cropping system with compost and compost tea; Org_M, organic cropping system with manure.
) indicates a significant (p < 0.05) correlation among modules: Conv, conventional cropping system; Org_C, organic cropping system with compost and compost tea; Org_M, organic cropping system with manure.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Empirical Networks | Random Networks | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of Original OTUs | Similarity Threshold St | Network Size | R Square of Power-Law | Avg Connectivity | Node | Edge | Average Path Distance (GD) | Avg Clustering Coefficient | Modularity (No. of Modules) | Avg Path Distance ±SD | Avg Clustering Coefficient ± SD | Avg Modularity ± SD | |
| Conv | 539 | 0.97 | 453 | 0.83 | 6.04 | 404 | 1220 | 6.821 b | 0.368 b | 0.646 (32) c | 3.430 ± 0.031 | 0.037 ± 0.004 | 0.369 ± 0.005 |
| Org_C | 539 | 0.97 | 452 | 0.80 | 6.80 | 400 | 1360 | 6.896 b | 0.395 a | 0.698 (36) b | 3.300 ± 0.031 | 0.044 ± 0.005 | 0.337 ± 0.005 |
| Org_M | 439 | 0.97 | 396 | 0.75 | 4.08 | 357 | 729 | 8.262 a | 0.355 c | 0.824 (29) a | 4.176 ± 0.038 | 0.014 ± 0.04 | 0.497 ± 0.008 |
| Gene | Conv | Org_C | Org_M | ANOVA | Kruskal—Wallis | |
|---|---|---|---|---|---|---|
| N-fixation | nifH | 0.42 ± 0.06 ab | 0.47 ± 0.07 a | 0.32 ± 0.10 b | * | - |
| nifD | 0.43 ± 0.07 ab | 0.52 ± 0.06 a | 0.36 ± 0.06 b | ** | - | |
| nifK | 0.43 ± 0.03 b | 0.59 ± 0.13 a * | 0.28 ± 0.05 c * | *** | - | |
| Denitrification | narG | 0.38 ± 0.06 | 0.49 ± 0.11 | 0.36 ± 0.14 | ns | - |
| narH | 0.38 ± 0.05 b | 0.49 ± 0.05 a | 0.36 ± 0.04 b | - | ** | |
| nrfA | 0.09 ± 0.00 ab | 0.15 ± 0.07 a | 0.07 ± 0.02 b | * | - | |
| nirK | 0.35 ± 0.08 a | 0.32 ± 0.09 a | 0.19 ± 0.05 b * | * | - | |
| norB | 0.48 ± 0.06 a | 0.43 ± 0.05 a | 0.29 ± 0.04 b *** | *** | - | |
| nosZ | 0.20 ± 0.03 | 0.25 ± 0.07 | 0.16 ± 0.04 | ns | - | |
| Nitrification | amoC | 0.01 ± 0.00 a | 0.01 ± 0.00 a | 0.00 ± 0.00 b | - | *** |
| amoB | 0.01 ± 0.00 a | 0.01 ± 0.00 a | 0.00 ± 0.00 b | - | *** | |
| amoA | 0.01 ± 0.00 a | 0.01 ± 0.00 a | 0.00 ± 0.00 b | - | *** | |
| hao | 0.08 ± 0.01 b | 0.13 ± 0.01 a | 0.08 ± 0.01 b | - | ** |
| Soil Properties | Cropping System | ||||
|---|---|---|---|---|---|
| Conv | Org_C | Org_M | ANOVA | Kruskal—Wallis | |
| pH | 8.39 ± 0.17 b | 8.47 ± 0.14 ab | 8.70 ± 0.10 a | * | - |
| EC (dS m−1) | 0.54 ± 0.15 | 0.52 ± 0.13 | 0.38 ± 0.04 | - | ns |
| TOC (g kg−1) | 11.49 ± 0.28 ab | 15.64 ± 3.37 a | 9.01 ± 3.49 b | ** | - |
| TN (g kg−1) | 1.13 ± 0.19 b | 1.59 ± 0.34 a | 0.93 ± 0.24 b | ** | - |
| p (mg kg−1) | 20.15 ± 5.24 | 14.65 ± 7.71 | 14.33 ± 7.48 | ns | - |
| NH4+ (mg kg−1) | 0.10 ± 0.23 b | 1.33 ± 0.15 b | 0.00 ± 0.00 a | - | ** |
| Mg (cmol kg−1) | 3.54 ± 0.11 ab | 4.39 ± 1.09 a | 3.13 ± 0.54 b | * | - |
| K (cmol kg−1) | 0.62 ± 0.15 | 0.85 ± 0.17 | 0.78 ± 0.06 | * | - |
| Na (cmol kg−1) | 2.12 ± 0.32 | 2.19 ± 0.86 | 1.64 ± 0.23 | ns | - |
| Ca (cmol kg−1) | 8.44 ± 0.83 | 10.03 ± 2.40 | 7.19 ± 1.49 | - | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuartero, J.; Özbolat, O.; Sánchez-Navarro, V.; Weiss, J.; Zornoza, R.; Pascual, J.A.; Vivo, J.-M.; Ros, M. Long-Term Compost Amendment Changes Interactions and Specialization in the Soil Bacterial Community, Increasing the Presence of Beneficial N-Cycling Genes in the Soil. Agronomy 2022, 12, 316. https://doi.org/10.3390/agronomy12020316
Cuartero J, Özbolat O, Sánchez-Navarro V, Weiss J, Zornoza R, Pascual JA, Vivo J-M, Ros M. Long-Term Compost Amendment Changes Interactions and Specialization in the Soil Bacterial Community, Increasing the Presence of Beneficial N-Cycling Genes in the Soil. Agronomy. 2022; 12(2):316. https://doi.org/10.3390/agronomy12020316
Chicago/Turabian StyleCuartero, Jessica, Onurcan Özbolat, Virginia Sánchez-Navarro, Julia Weiss, Raúl Zornoza, José Antonio Pascual, Juana-María Vivo, and Margarita Ros. 2022. "Long-Term Compost Amendment Changes Interactions and Specialization in the Soil Bacterial Community, Increasing the Presence of Beneficial N-Cycling Genes in the Soil" Agronomy 12, no. 2: 316. https://doi.org/10.3390/agronomy12020316
APA StyleCuartero, J., Özbolat, O., Sánchez-Navarro, V., Weiss, J., Zornoza, R., Pascual, J. A., Vivo, J.-M., & Ros, M. (2022). Long-Term Compost Amendment Changes Interactions and Specialization in the Soil Bacterial Community, Increasing the Presence of Beneficial N-Cycling Genes in the Soil. Agronomy, 12(2), 316. https://doi.org/10.3390/agronomy12020316