
Molecular Characterization and Genetic Structure Evaluation of Breeding Populations of Fennel (Foeniculum vulgare Mill.)




Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Genomic DNA Isolation and SSR Marker Analysis
2.3. Genetic Diversity and Differentiation Statistics and Population Genetic Structure Analysis
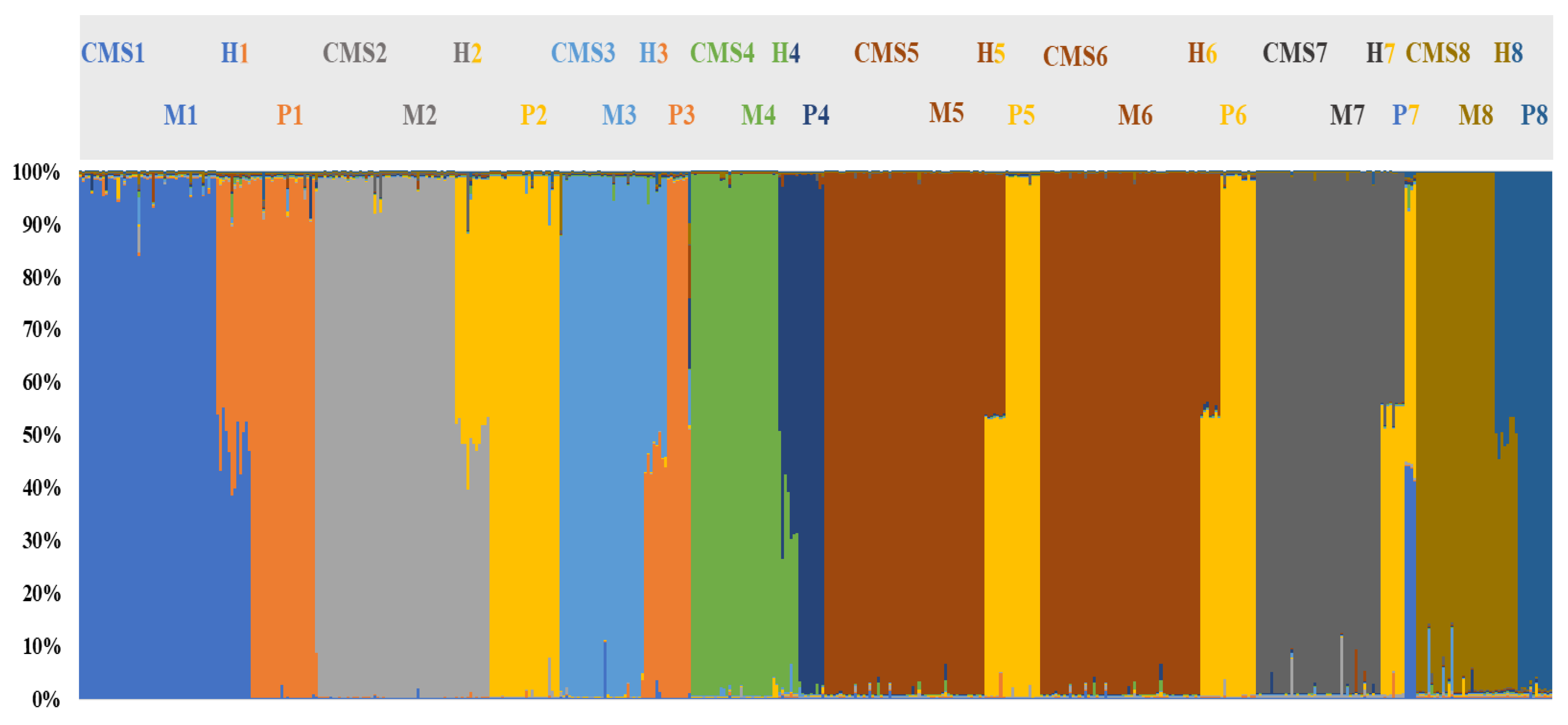
3. Results and Discussion
3.1. SSR Marker Descriptive Statistics and Genetic Variability
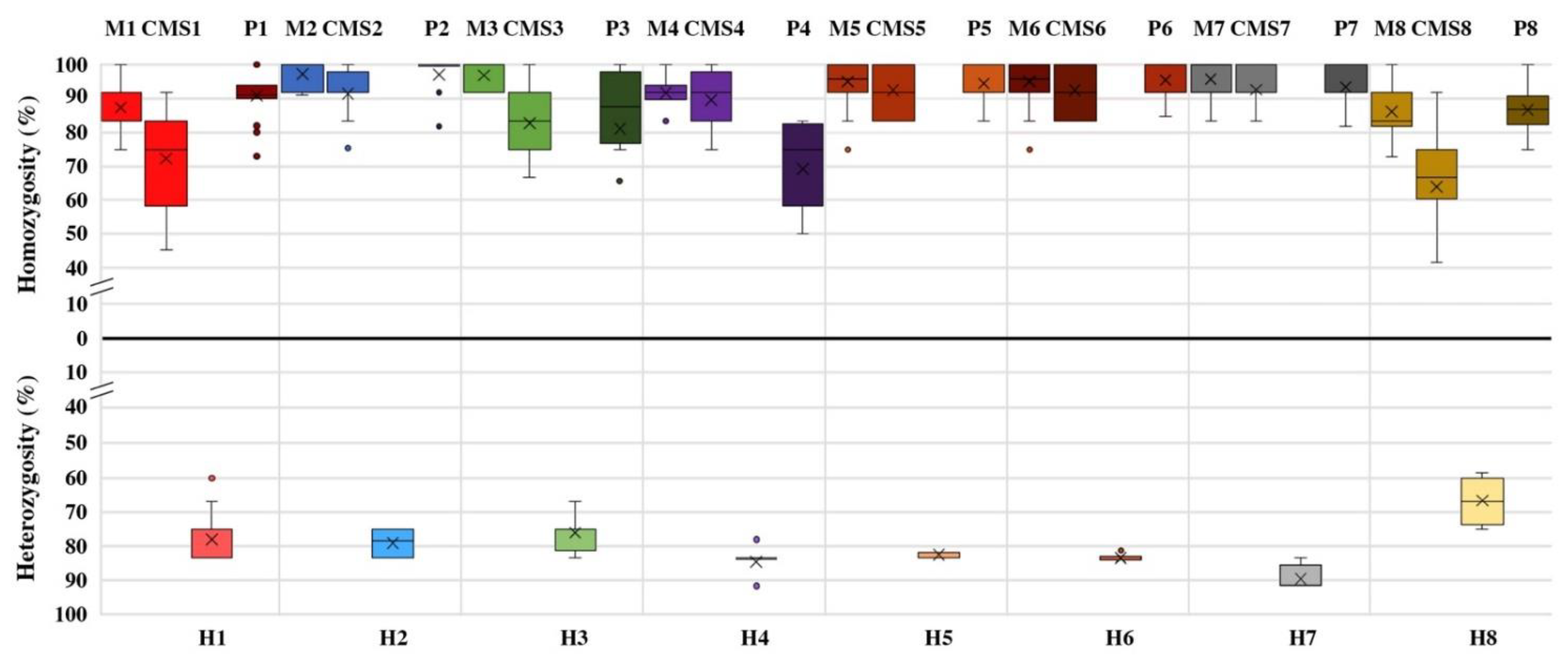
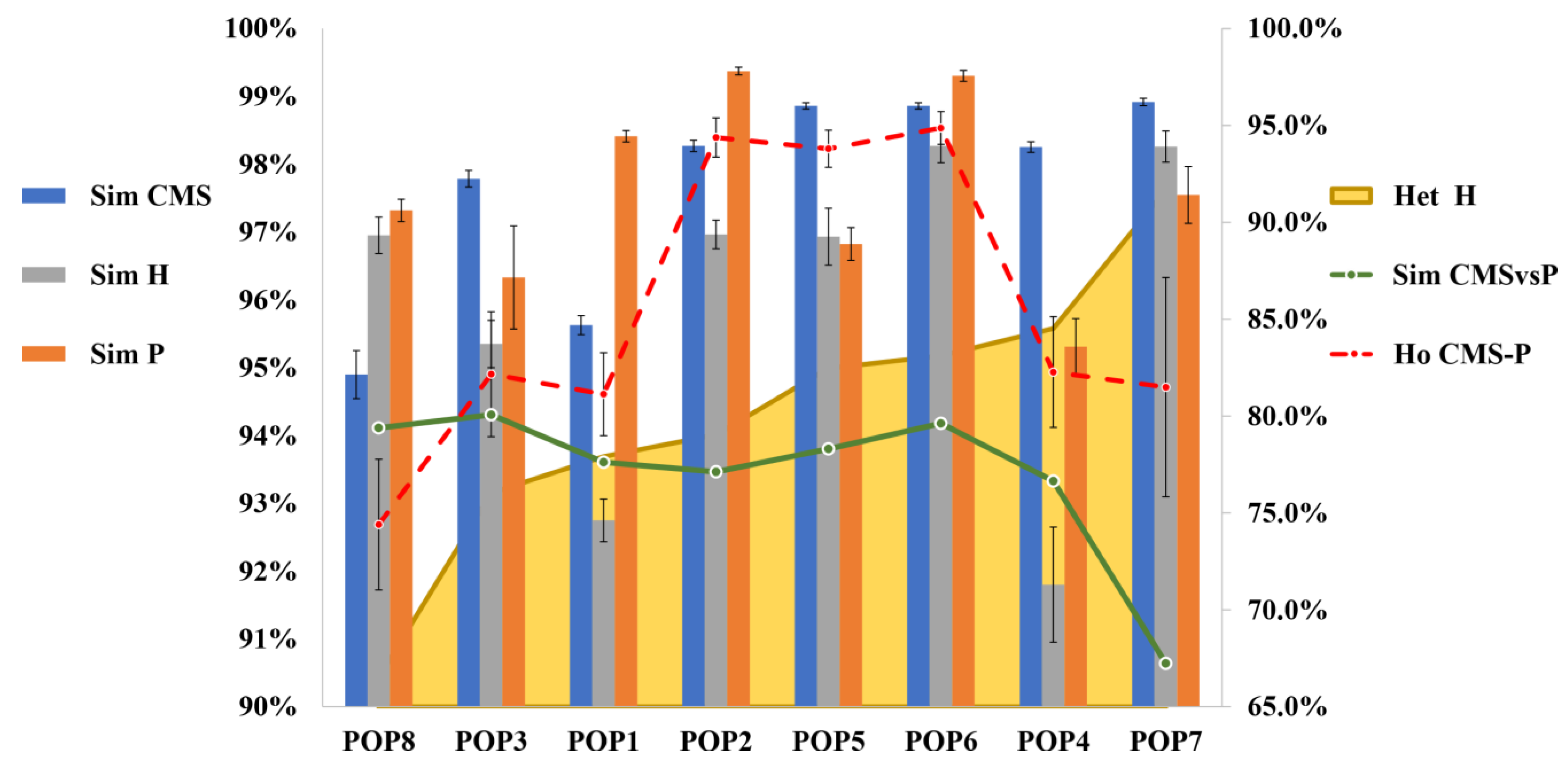
3.2. Genetic Stability of Parental Lines and Distinctiveness of F1 Hybrids
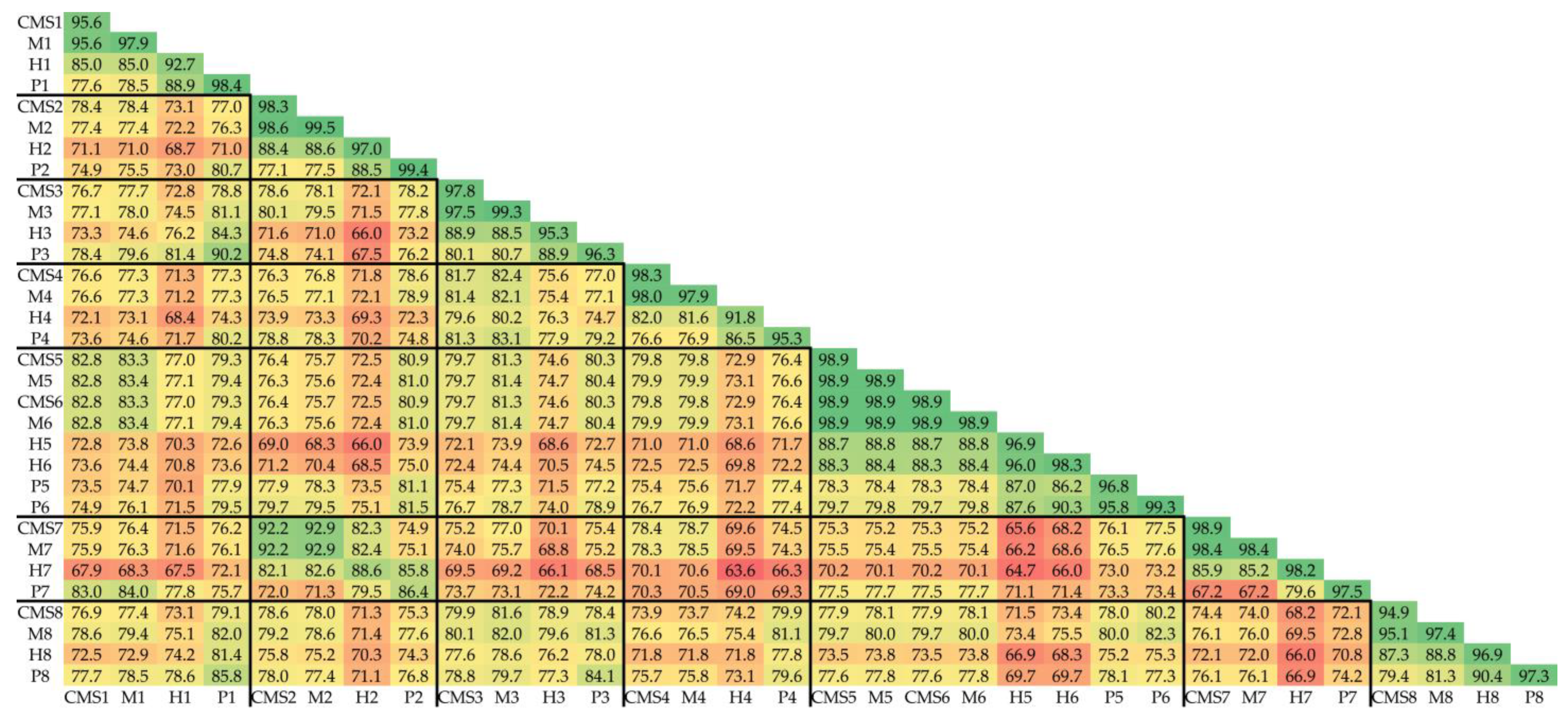
3.3. Genetic Dissimilarity among Parental Lines and Heterozygosity of F1 Hybrids
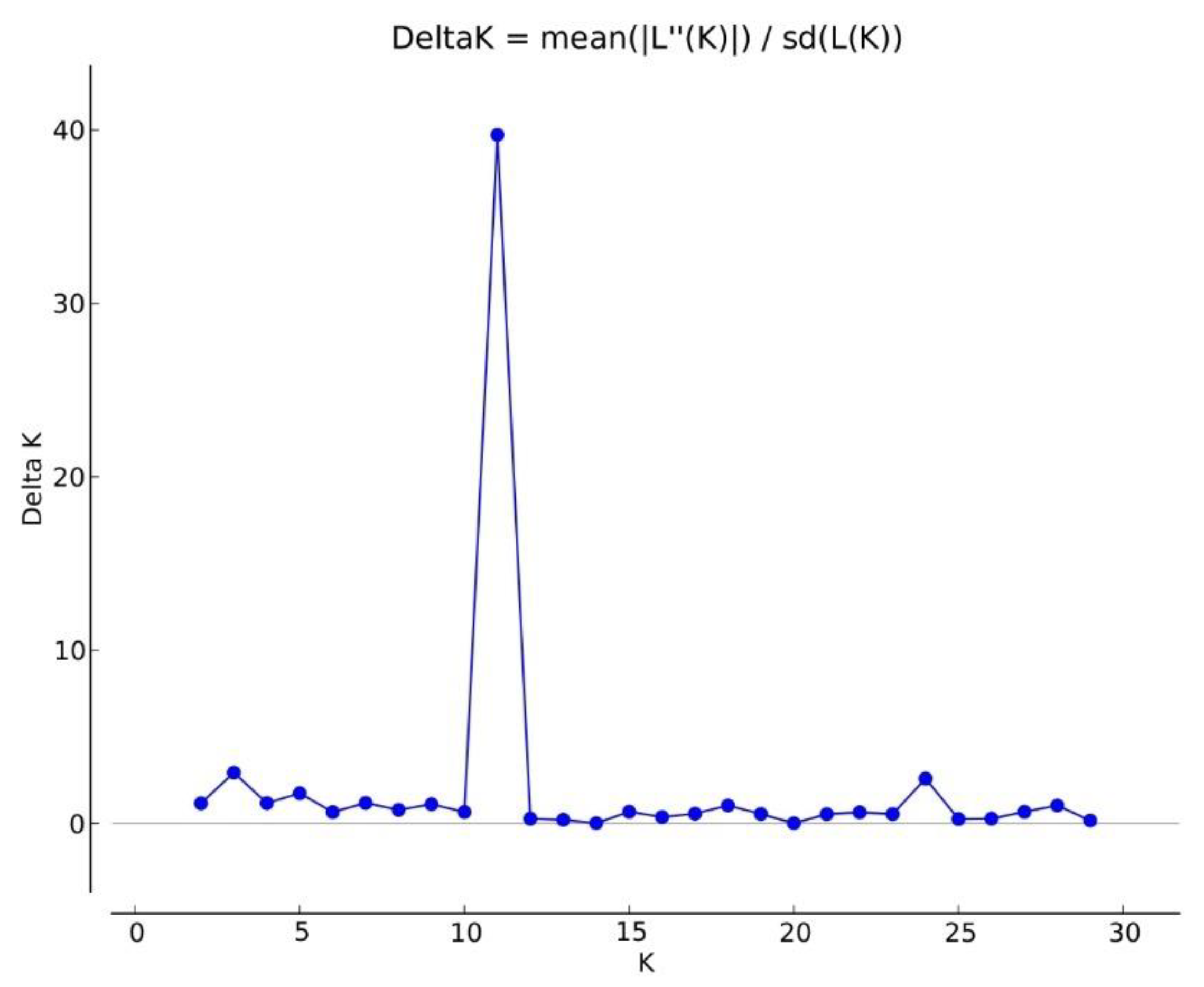
3.4. Genetic Structure of the Core Collection and Genetic Distinctiveness of Breeding Stocks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Badgujar, S.B.; Patel, V.V.; Bandivdekar, A.H. Foeniculum vulgare Mill: A review of its botany, phytochemistry, pharmacology, contemporary application, and toxicology. BioMed Res. Int. 2014, 2014, 842674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Maroto, M.C.; Perez-Coello, M.S.; Esteban, J.; Sanz, J. Comparison of the volatile composition of wild fennel samples (Foeniculum vulgare Mill.) from central Spain. J. Agric. Food Chem. 2006, 54, 6814–6818. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, F.; Vannozzi, A.; Vitulo, N.; Lucchin, M.; Barcaccia, G. The leaf transcriptome of fennel (Foeniculum vulgare Mill.) enables characterization of the t-anethole pathway and the discovery of microsatellites and single-nucleotide variants. Sci. Rep. 2018, 8, 10459. [Google Scholar] [CrossRef] [PubMed]
- Senatore, F.; Oliviero, F.; Scandolera, E.; Taglialatela-Scafati, O.; Roscigno, G.; Zaccardelli, M.; De Falco, E. Chemical composition, antimicrobial and antioxidant activities of anethole-rich oil from leaves of selected varieties of fennel [Foeniculum vulgare Mill. ssp. vulgare var. azoricum (Mill.) Thell]. Fitoterapia 2013, 90, 214–219. [Google Scholar] [CrossRef]
- Tognolini, M.; Ballabeni, V.; Bertoni, S.; Bruni, R.; Impicciatore, M.; Barocelli, E. Protective effect of Foeniculum vulgare essential oil and anethole in an experimental model of thrombosis. Pharm. Res. 2007, 56, 254–260. [Google Scholar] [CrossRef]
- FAO. FAOstat. Available online: http://www.fao.org/home/en (accessed on 22 December 2021).
- Palumbo, F.; Vannozzi, A.; Barcaccia, G. Impact of Genomic and Transcriptomic Resources on Apiaceae Crop Breeding Strategies. Int. J. Mol. Sci. 2021, 22, 9713. [Google Scholar] [CrossRef] [PubMed]
- Koul, P.; Sharma, N.; Koul, A.K. Pollination Biology of Apiaceae. Curr. Sci. 1993, 65, 219–222. [Google Scholar]
- Palumbo, F.; Vitulo, N.; Vannozzi, A.; Magon, G.; Barcaccia, G. The Mitochondrial Genome Assembly of Fennel (Foeniculum vulgare) Reveals Two Different atp6 Gene Sequences in Cytoplasmic Male Sterile Accessions. Int. J. Mol. Sci. 2020, 21, 4664. [Google Scholar] [CrossRef]
- Pank, F. Three approaches to the development of high performance cultivars considering the differing biological background of the starting material. In Proceedings of the International Conference on Medicinal and Aromatic Plants. Possibilities and Limitations of Medicinal and Aromatic Plant, Budapest, Hungary, 8–11 July 2001; pp. 129–137. [Google Scholar]
- Palumbo, F.; Galla, G.; Vitulo, N.; Barcaccia, G. First draft genome sequencing of fennel (Foeniculum vulgare Mill.): Identification of simple sequence repeats and their application in marker-assisted breeding. Mol. Breed. 2018, 38, 122. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, F.; Galla, G.; Martinez-Bello, L.; Barcaccia, G. Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production. Diversity 2017, 9, 15. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, F.; Galla, G.; Barcaccia, G. Developing a Molecular Identification Assay of Old Landraces for the Genetic Authentication of Typical Agro-Food Products: The Case Study of the Barley ’Agordino’. Food Technol. Biotechnol. 2017, 55, 29–39. [Google Scholar] [CrossRef]
- Yeh, F.C.; Yang, R.C.; Boyle, T.B.J.; Ye, Z.H.; Mao, J.X. POPGENE, The User-Friendly Shareware for Population Genetic Analysis; University of Alberta: Edmonton, AB, Canada, 1998; Volume 10, pp. 295–301. [Google Scholar]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; Rabaiolli, S.M.D.S.; Stefanel, C.M.; Lemos, S.C.M.; Silveira, R.L.R.; Buuron, S.K.; Dos Santos, R.S.M.; Moro, S.C. Determining the Polymorphism Information Content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef]
- Kimura, M. Population Genetics, Molecular Evolution, and the Neutral Theory: Selected Papers; University of Chicago Press: Chicago, IL, USA, 1994. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Mcdermott, J.M.; Mcdonald, B.A. Gene Flow in Plant Pathosystems. Annu. Rev. Phytopathol. 1993, 31, 353–373. [Google Scholar] [CrossRef]
- Barcaccia, G.; Lucchin, M.; Parrini, P. Characterization of a flint maize (Zea mays var. indurata) Italian landrace, II. Genetic diversity and relatedness assessed by SSR and Inter-SSR molecular markers. Genet. Resour. Crop. Evol. 2003, 50, 253–271. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System Version 2.1: Owner Manual; Exeter Publishing: Setauket, NY, USA, 2000. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Mitton, J.B. Gene Flow. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 192–196. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus Name | Primer Forward | Primer Reverse | Motif | Min Size | Max Size | Anchor |
|---|---|---|---|---|---|---|
| FV_2 | CAAAGAATGGAAAACATGCTG | CAAAGAATGGAAAACATGCTG | CAA | 129 | 152 | PAN1 |
| FV_6 | TATGTTCTCAGATTCGGGTTA | TATGTTCTCAGATTCGGGTTA | TC | 214 | 226 | M13 |
| FV_253 | TTGTAGAGATACAGGGTCGAA | TTGTAGAGATACAGGGTCGAA | TC | 196 | 252 | PAN1 |
| FV_9919 | AGTAAAGGCATAATCTGTTGGTGG | AGTAAAGGCATAATCTGTTGGTGG | GT | 231 | 248 | PAN3 |
| FV_11537 | TTCATGTATCAACTACGCACAC | TTCATGTATCAACTACGCACAC | AG | 152 | 166 | M13 |
| FV_15981 | CTAGCGTTTCCATCTCGTCTC | CTAGCGTTTCCATCTCGTCTC | TC | 235 | 245 | PAN1 |
| FV_18902 | GTTTGAACTCGAATGACCACCT | GTTTGAACTCGAATGACCACCT | TC | 410 | 424 | PAN2 |
| FV_179837 | ATTCACCATGACATCACCTC | ATTCACCATGACATCACCTC | TC | 320 | 336 | M13 |
| FV_217218 | ACAAACGTACCTCTGTACGAA | ACAAACGTACCTCTGTACGAA | AG | 345 | 360 | M13 |
| FV_217225 | AAAGAATGGAGAGAAGAATGG | AAAGAATGGAGAGAAGAATGG | AG | 309 | 344 | PAN1 |
| FV_290063 | TGATTTCTCAAAGGCATTCTA | TGATTTCTCAAAGGCATTCTA | GA | 294 | 324 | PAN3 |
| FV_290202 | AGGGCTGAGATTAGTTTCTAGTT | AGGGCTGAGATTAGTTTCTAGTT | TA | 139 | 210 | PAN2 |
| Locus Name | PIC | N. Alleles | Highest Allele Frequency |
|---|---|---|---|
| FV_2 | 0.73 | 7 | 0.366 |
| FV_6 | 0.65 | 7 | 0.514 |
| FV_253 | 0.86 | 13 | 0.234 |
| FV_9919 | 0.69 | 5 | 0.343 |
| FV_11537 | 0.80 | 6 | 0.290 |
| FV_15981 | 0.63 | 5 | 0.472 |
| FV_18902 | 0.80 | 8 | 0.279 |
| FV_179837 | 0.79 | 9 | 0.346 |
| FV_217218 | 0.75 | 8 | 0.405 |
| FV_217225 | 0.85 | 11 | 0.194 |
| FV_290063 | 0.77 | 8 | 0.288 |
| FV_290202 | 0.89 | 15 | 0.186 |
| Mean | 0.77 | 8.5 | 0.326 |
| Population ID | N | npl | %pl | na | ne | Ho | He | H | Nm |
|---|---|---|---|---|---|---|---|---|---|
| CMS1 | 24 | 10 | 83.3% | 2.33 | 1.39 | 0.72 | 0.77 | 0.23 | 0.32 |
| CMS2 | 24 | 4 | 33.3% | 1.67 | 1.11 | 0.91 | 0.92 | 0.08 | 0.33 |
| CMS3 | 16 | 7 | 58.3% | 1.58 | 1.21 | 0.83 | 0.86 | 0.13 | 0.44 |
| CMS4 | 16 | 5 | 41.7% | 1.42 | 1.15 | 0.90 | 0.91 | 0.09 | 0.33 |
| CMS5 | 27 | 4 | 33.3% | 1.33 | 1.10 | 0.94 | 0.94 | 0.06 | 0.24 |
| CMS6 | 27 | 4 | 33.3% | 1.33 | 1.10 | 0.94 | 0.94 | 0.06 | 0.24 |
| CMS7 | 24 | 5 | 41.7% | 1.50 | 1.09 | 0.93 | 0.94 | 0.06 | 0.34 |
| CMS8 | 14 | 11 | 91.7% | 2.25 | 1.47 | 0.64 | 0.72 | 0.28 | 0.43 |
| Average CMS | 20.7 | 6.6 | 54.8% | 1.73 | 1.22 | 0.84 | 0.87 | 0.13 | 0.35 |
| M1 | 23 | 7 | 58.3% | 1.75 | 1.16 | 0.87 | 0.89 | 0.11 | 0.32 |
| M2 | 24 | 4 | 33.3% | 1.33 | 1.03 | 0.97 | 0.97 | 0.03 | 0.14 |
| M3 | 13 | 4 | 33.3% | 1.33 | 1.03 | 0.97 | 0.97 | 0.03 | 0.28 |
| M4 | 14 | 3 | 25.0% | 1.42 | 1.19 | 0.92 | 0.91 | 0.09 | 0.21 |
| M5 | 28 | 5 | 41.7% | 1.42 | 1.10 | 0.95 | 0.95 | 0.05 | 0.23 |
| M6 | 28 | 5 | 41.7% | 1.42 | 1.10 | 0.95 | 0.95 | 0.05 | 0.23 |
| M7 | 19 | 5 | 41.7% | 1.75 | 1.09 | 0.93 | 0.93 | 0.07 | 0.32 |
| M8 | 13 | 5 | 41.7% | 1.50 | 1.21 | 0.86 | 0.87 | 0.13 | 0.30 |
| Average M | 19.1 | 4.7 | 39.3% | 1.50 | 1.12 | 0.92 | 0.93 | 0.07 | 0.26 |
| P1 | 22 | 8 | 66.7% | 1.83 | 1.18 | 0.92 | 0.88 | 0.11 | 0.34 |
| P2 | 24 | 5 | 41.7% | 1.50 | 1.11 | 0.98 | 0.92 | 0.07 | 0.05 |
| P3 | 8 | 9 | 75.0% | 2.00 | 1.23 | 0.81 | 0.84 | 0.15 | 0.39 |
| P4 | 9 | 8 | 66.7% | 1.90 | 1.49 | 0.69 | 0.75 | 0.24 | 0.42 |
| P5 | 12 | 3 | 25.0% | 1.50 | 1.27 | 0.92 | 0.87 | 0.13 | 0.10 |
| P6 | 12 | 2 | 16.7% | 1.17 | 1.05 | 0.96 | 0.96 | 0.03 | 0.39 |
| P7 | 4 | 11 | 91.7% | 2.08 | 1.98 | 0.10 | 0.46 | 0.47 | 4.78 |
| P8 | 12 | 6 | 50.0% | 1.50 | 1.23 | 0.86 | 0.86 | 0.13 | 0.18 |
| Average P | 11.6 | 6.3 | 52.4% | 1.67 | 1.34 | 0.76 | 0.81 | 0.18 | 0.90 |
| H1 | 12 | 10 | 83.3% | 2.75 | 2.16 | 0.22 | 0.50 | 0.48 | 0.98 |
| H2 | 12 | 11 | 91.7% | 2.25 | 1.91 | 0.21 | 0.55 | 0.43 | 2.35 |
| H3 | 8 | 11 | 91.7% | 2.42 | 1.94 | 0.24 | 0.54 | 0.44 | 1.70 |
| H4 | 7 | 11 | 91.7% | 2.75 | 2.31 | 0.16 | 0.44 | 0.52 | 1.08 |
| H5 | 7 | 10 | 83.3% | 2.17 | 2.01 | 0.17 | 0.51 | 0.45 | 1.56 |
| H6 | 7 | 10 | 83.3% | 2.00 | 1.93 | 0.17 | 0.53 | 0.43 | 3.51 |
| H7 | 8 | 12 | 100.0% | 2.17 | 1.99 | 0.06 | 0.48 | 0.49 | 5.00 |
| H8 | 8 | 9 | 75.0% | 1.92 | 1.79 | 0.32 | 0.59 | 0.38 | 0.73 |
| Average H | 8.1 | 10.6 | 88.1% | 2.24 | 1.98 | 0.19 | 0.52 | 0.45 | 2.28 |
| Overall Mean | 7.2 | 59.7% | 1.82 | 1.43 | |||||
| Among overall | 451 | 0.78 | 0.23 | 0.77 | 0.09 |
| Population ID | N | npl | %pl | na | ne | HT | HS | Ho | He | GS | Nm |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CMS1_M1 | 47 | 10 | 83.3% | 2.58 | 1.28 | 0.19 | 0.80 | 0.81 | 0.96 | 2.38 | |
| CMS2_M2 | 48 | 6 | 50.0% | 1.83 | 1.07 | 0.06 | 0.94 | 0.94 | 0.99 | 3.53 | |
| CMS3_M3 | 29 | 10 | 83.3% | 1.92 | 1.13 | 0.10 | 0.89 | 0.90 | 0.98 | 1.78 | |
| CMS4_M4 | 30 | 6 | 50.0% | 1.67 | 1.17 | 0.10 | 0.91 | 0.90 | 0.98 | 8.34 | |
| CMS5_M5 | 55 | 7 | 58.3% | 1.58 | 1.10 | 0.06 | 0.95 | 0.95 | 0.99 | 69.19 | |
| CMS6_M6 | 55 | 7 | 58.3% | 1.58 | 1.10 | 0.06 | 0.95 | 0.95 | 0.99 | 69.19 | |
| CMS7_M7 | 43 | 7 | 58.3% | 1.92 | 1.09 | 0.07 | 0.93 | 0.94 | 0.98 | 4.39 | |
| CMS8_M8 | 27 | 11 | 91.7% | 2.42 | 1.39 | 0.23 | 0.74 | 0.77 | 0.95 | 1.98 | |
| Mean CMS-M | 279 | 8.1 | 67.9% | 1.99 | 1.18 | 0.71 | |||||
| St. Dev. | 2.1 | 17.6% | 2.68 | 1.15 | |||||||
| Among CMS-M | 0.89 | 0.29 | 0.79 | 0.05 | |||||||
| St. Dev. | 0.07 | 0.07 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scariolo, F.; Palumbo, F.; Barcaccia, G. Molecular Characterization and Genetic Structure Evaluation of Breeding Populations of Fennel (Foeniculum vulgare Mill.). Agronomy 2022, 12, 542. https://doi.org/10.3390/agronomy12030542
Scariolo F, Palumbo F, Barcaccia G. Molecular Characterization and Genetic Structure Evaluation of Breeding Populations of Fennel (Foeniculum vulgare Mill.). Agronomy. 2022; 12(3):542. https://doi.org/10.3390/agronomy12030542
Chicago/Turabian StyleScariolo, Francesco, Fabio Palumbo, and Gianni Barcaccia. 2022. "Molecular Characterization and Genetic Structure Evaluation of Breeding Populations of Fennel (Foeniculum vulgare Mill.)" Agronomy 12, no. 3: 542. https://doi.org/10.3390/agronomy12030542
APA StyleScariolo, F., Palumbo, F., & Barcaccia, G. (2022). Molecular Characterization and Genetic Structure Evaluation of Breeding Populations of Fennel (Foeniculum vulgare Mill.). Agronomy, 12(3), 542. https://doi.org/10.3390/agronomy12030542