
Multi-Environment Genome-Wide Association Studies of Yield Traits in Common Bean (Phaseolus vulgaris L.) × Tepary Bean (P. acutifolius A. Gray) Interspecific Advanced Lines in Humid and Dry Colombian Caribbean Subregions
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Multi-Locality Field Trials
2.3. Experimental Design and Phenotyping
2.4. Compilation of Indices for Yield Traits and Statistical Analysis
2.5. DNA Extraction and Genotyping-by-Sequencing
2.6. Sequence Processing, Alignment and SNP Calling
2.7. Analysis of Kinship and Population Structure
2.8. Identification of Loci Associated with Yield Traits
2.9. Identification of Candidate Genes and Pathways Enriched Analysis
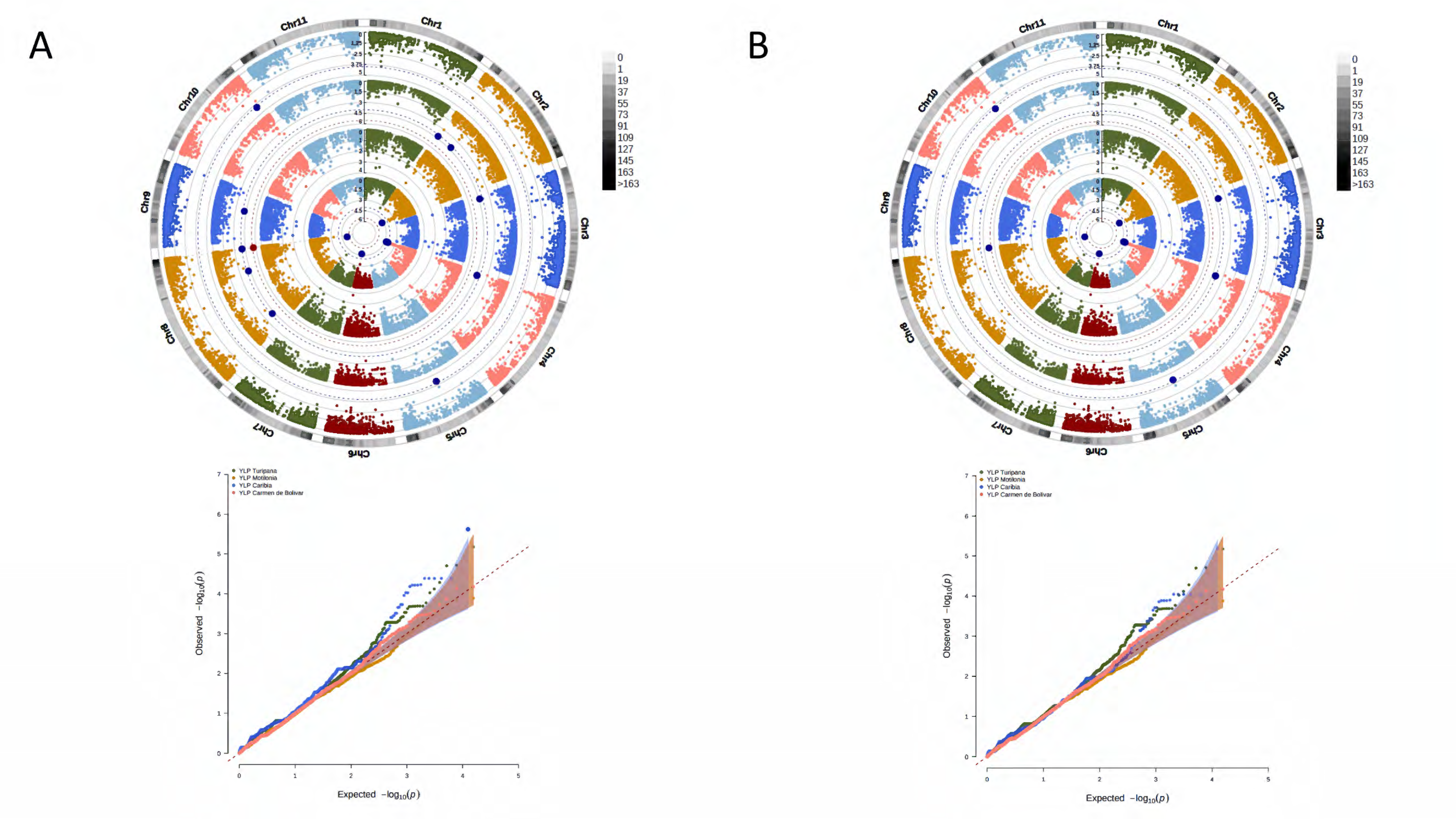
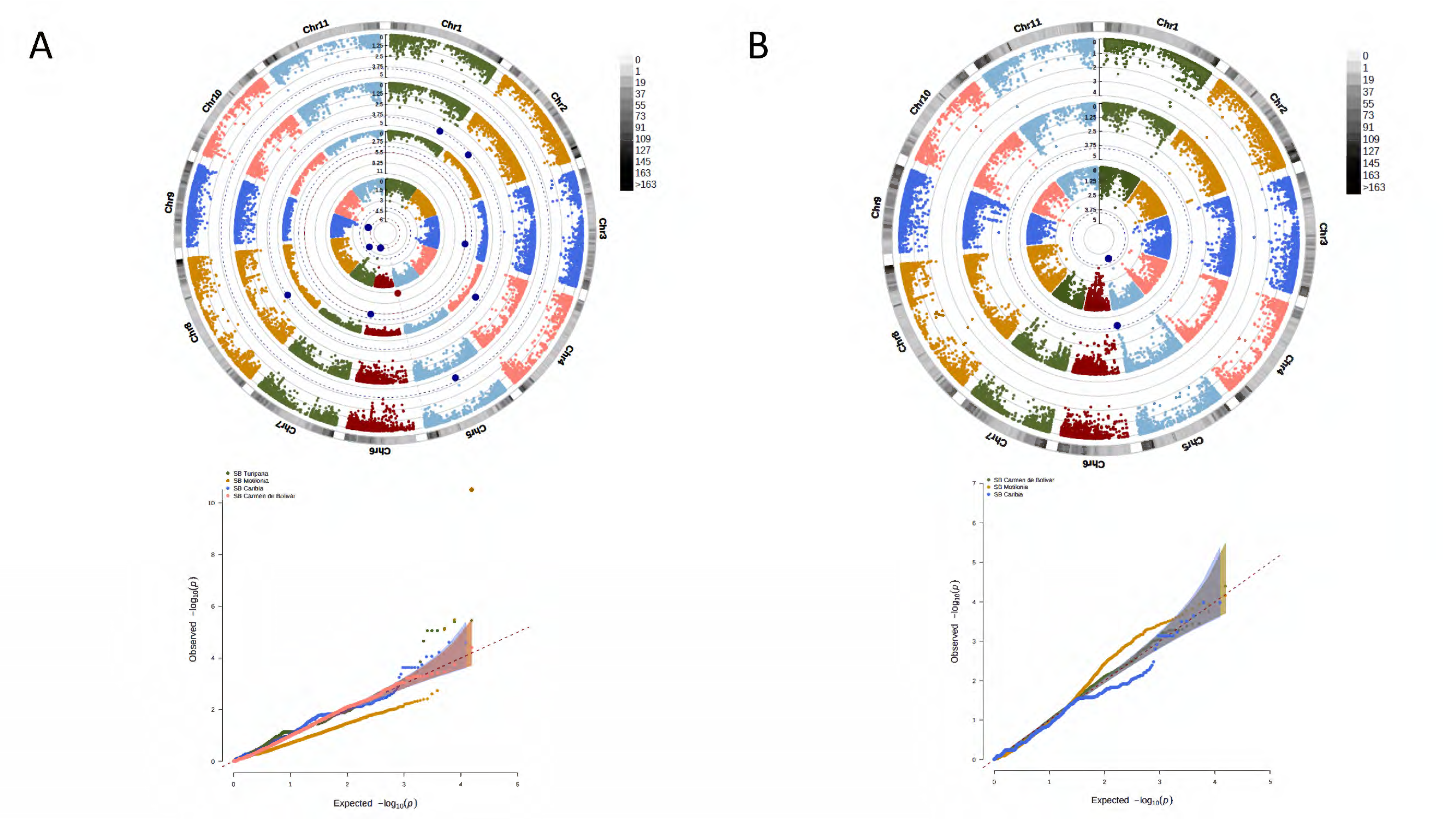
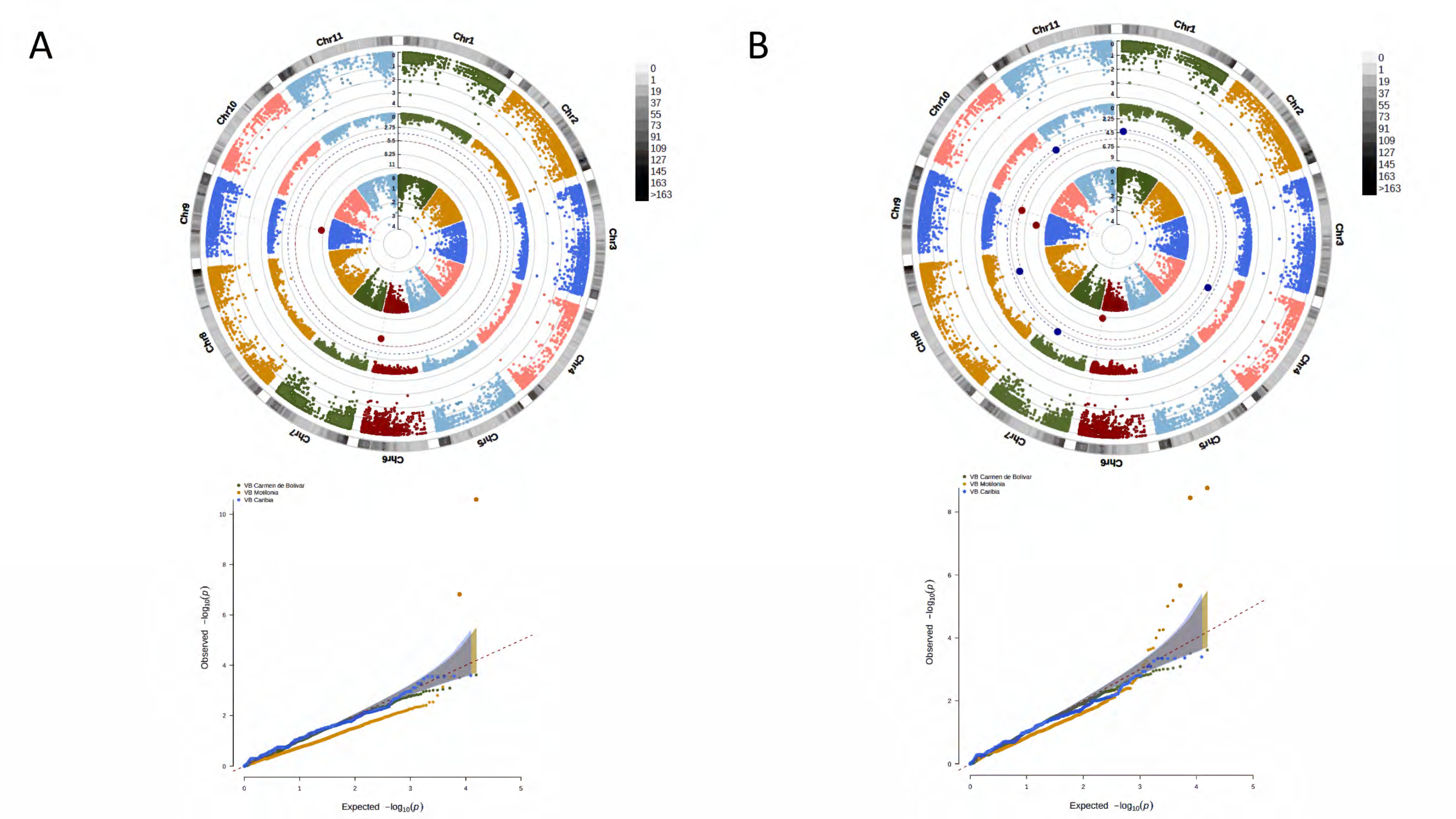
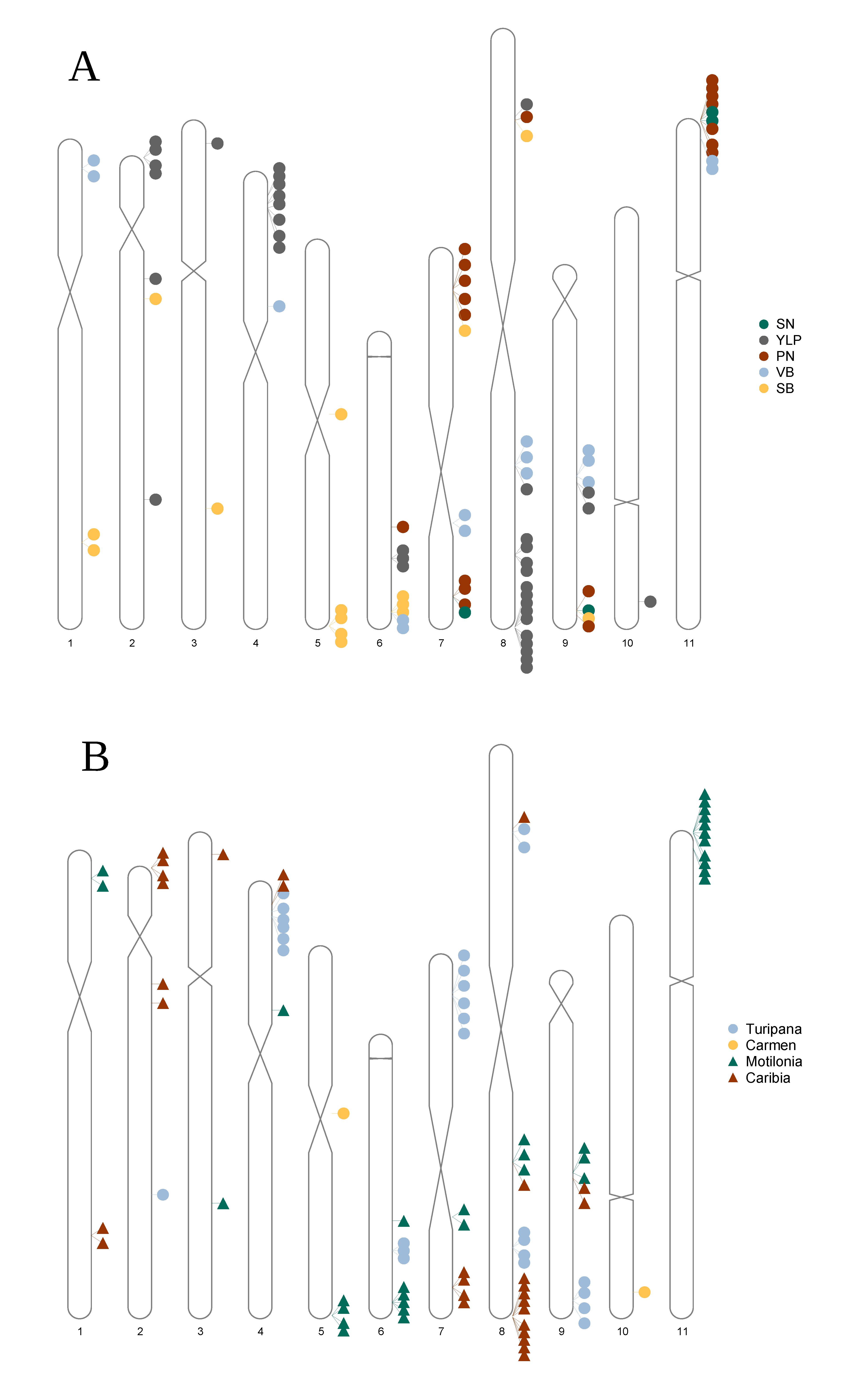
3. Results
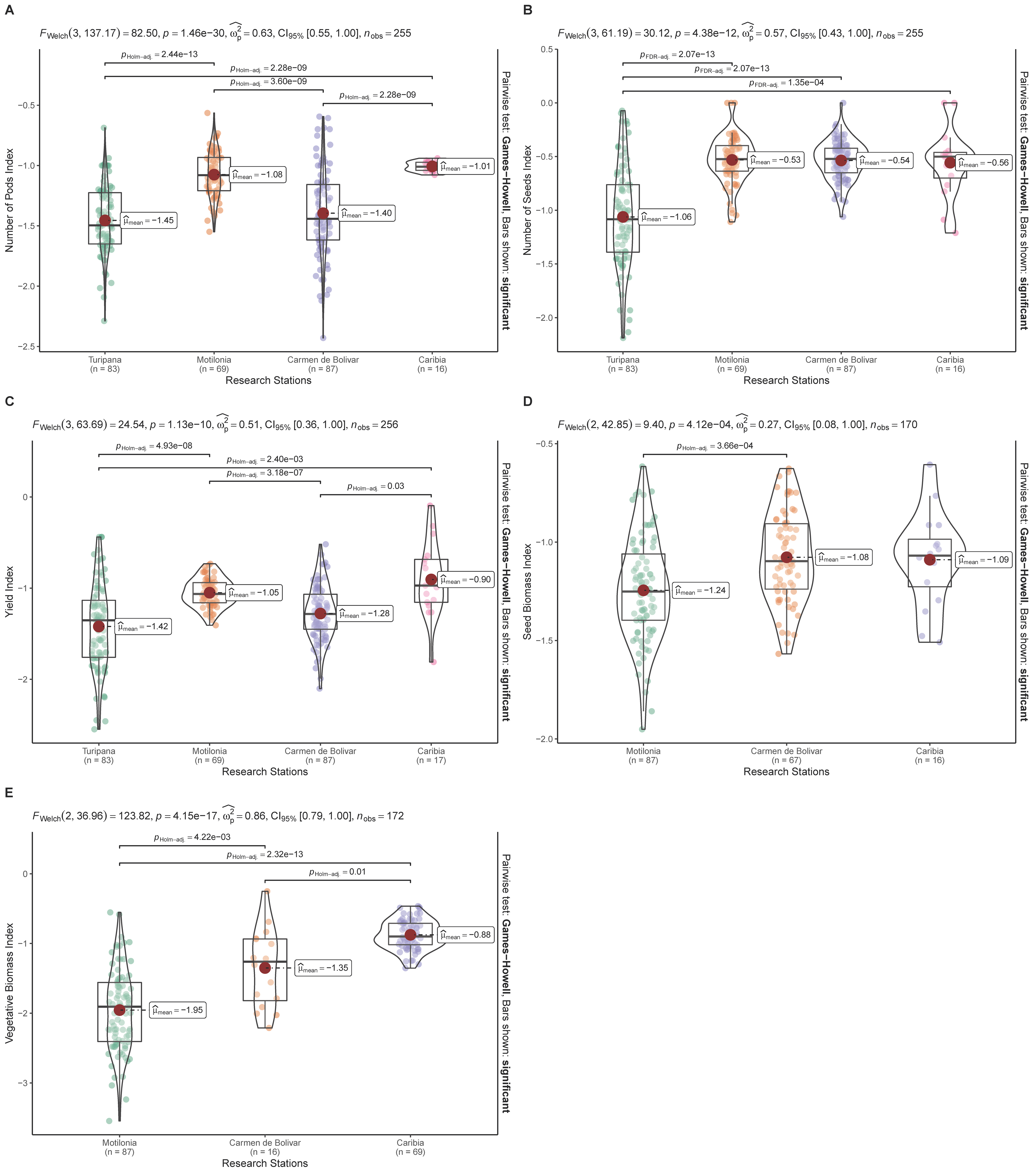
3.1. Phenotypic Segregation across Localities
3.2. A Total of 15,645 SNP Markers Were Recovered from the Interspecific Panel Using GBS

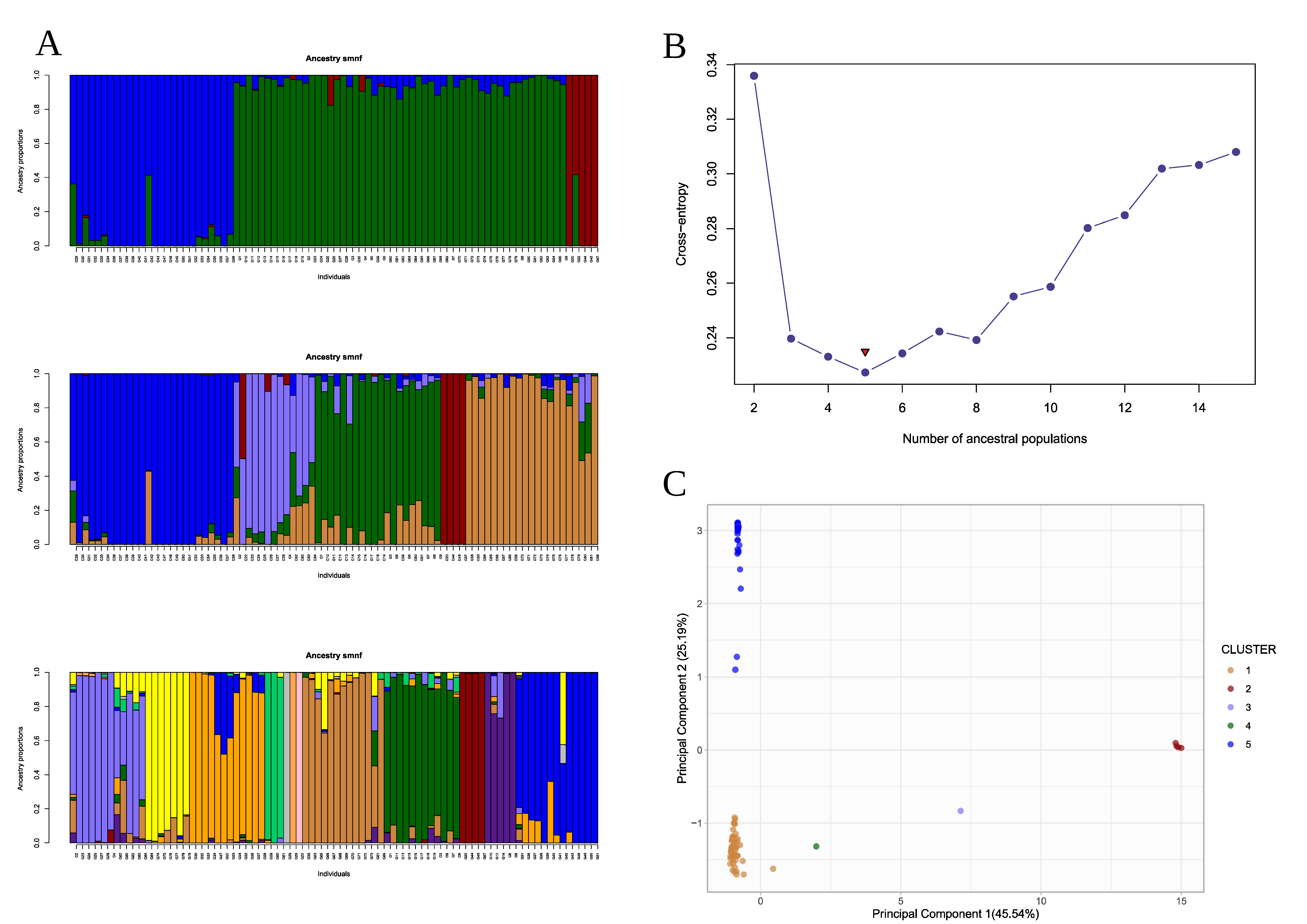
3.3. Genetic Structure and Kinship Relationships Suggested Five Demographic Clusters

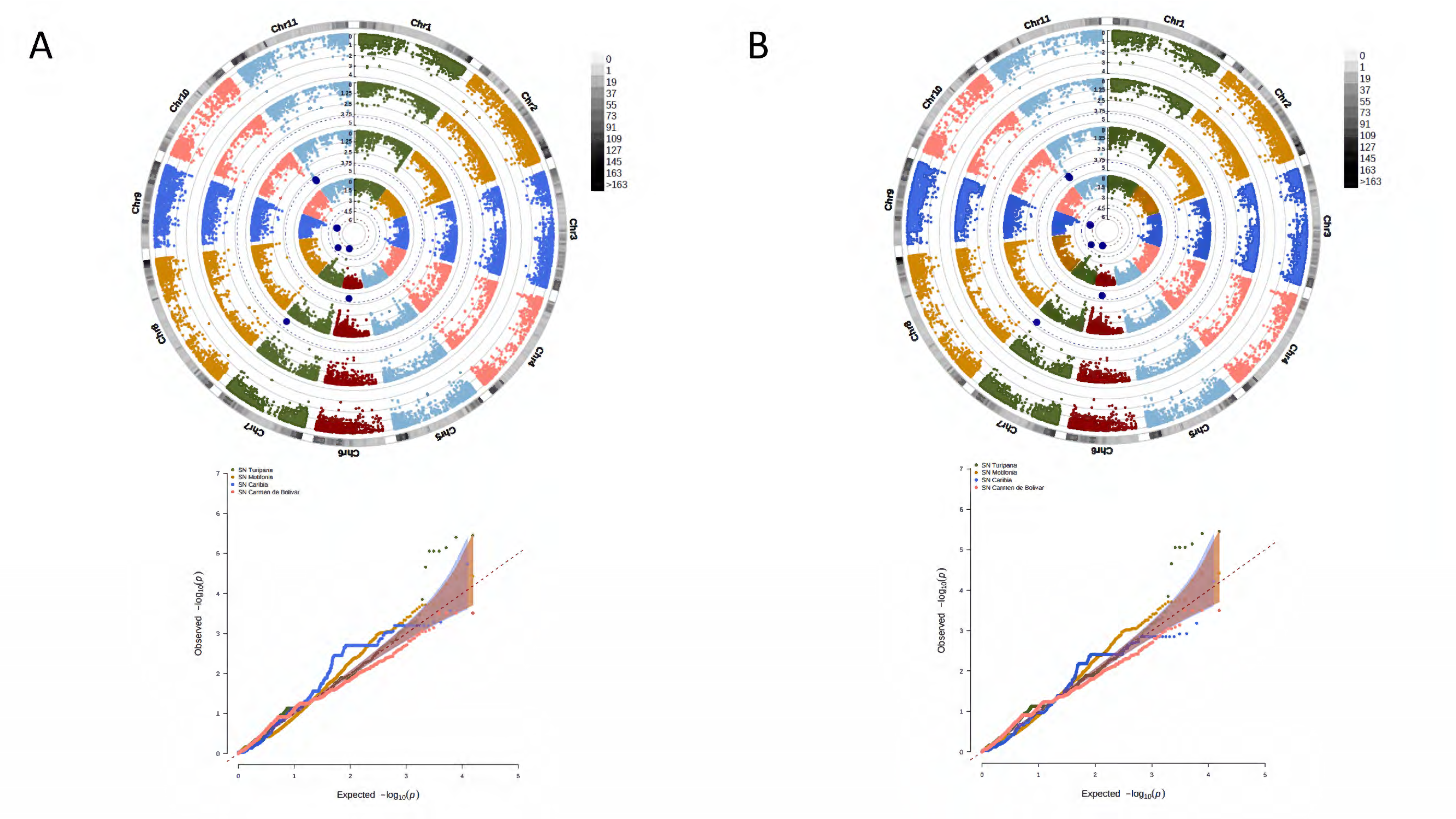
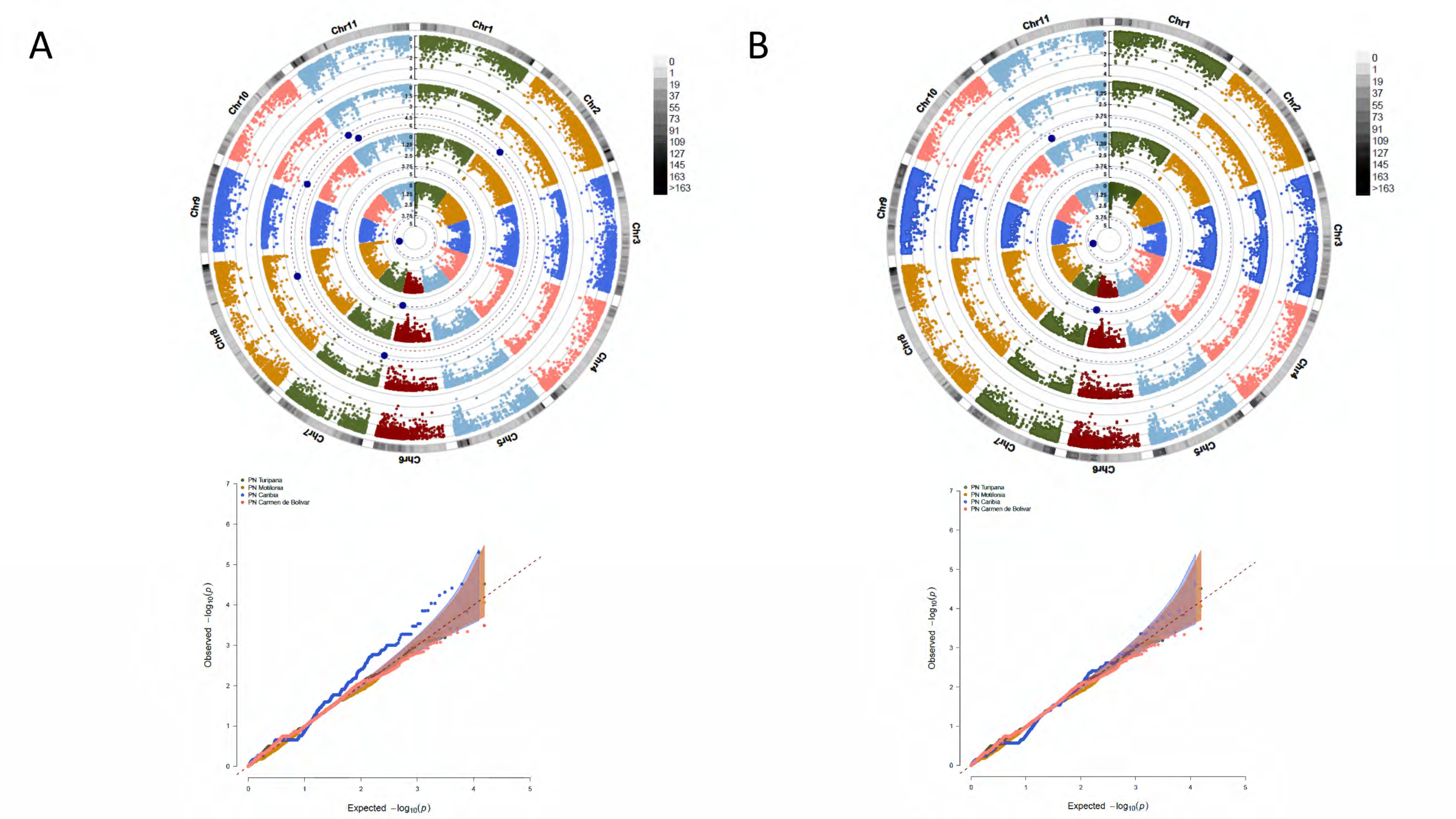
3.4. A Total of 47 Loci and 90 Genes Led to Environmentally Dependent Polygenic Adaptation
3.5. Associated Genes Enriched Drought Tolerance Response Pathways







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Gene ID | p-Value | Pathway ID | Pathway Name |
|---|---|---|---|---|
| NP | PAC:37165399 | 1.6578 × e−5 | FASYN-ELONG-PWY | fatty acid elongation—saturated |
| NP | PAC:37165399 | 2.3272 × e−4 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| NP | PAC:37165399 | 1.1329 × e−4 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| NP | PAC:37165399 | 2.7244 × e−5 | PWY-5973 | cis-vaccenate biosynthesis |
| NP | PAC:37165399 | 1.1324× e−4 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| NP | PAC:37165399 | 3.1750 × e−5 | PWY-6282 | palmitoleate biosynthesis I (from (5Z)-dodec-5-enoate) |
| NP | PAC:37165399 | 3.2671 × e−5 | PWY-7388 | octanoyl-[acyl-carrier protein] synthesis (mitochondria) |
| NP | PAC:37165399 | 1.5583 × e−5 | PWY-7663 | gondoate biosynthesis (anaerobic) |
| NP | PAC:37167444 | 1.6578 × e−5 | FASYN-ELONG-PWY | fatty acid elongation—saturated |
| NP | PAC:37167444 | 2.3272 × e−4 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| NP | PAC:37167444 | 1.1329 × e−4 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| NP | PAC:37167444 | 2.7244 × e−5 | PWY-5973 | cis-vaccenate biosynthesis |
| NP | PAC:37167444 | 1.1324 × e−4 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| NP | PAC:37167444 | 3.1750 × e−5 | PWY-6282 | palmitoleate biosynthesis I (from (5Z)-dodec-5-enoate) |
| NP | PAC:37167444 | 3.2671 × e−5 | PWY-7388 | octanoyl-[acyl-carrier protein] synthesis (mitochondria,) |
| NP | PAC:37167444 | 1.5583 × e−5 | PWY-7663 | gondoate biosynthesis (anaerobic) |
| NS | PAC:37156716 | 0.0297 | PWY-7219 | adenosine ribonucleotides de novo biosynthesis |
| NS | PAC:37156716 | 0.0297 | PWY-7229 | adenosine nucleotides de novo biosynthesis I |
| NS | PAC:37165399 | 4.9600 × e−5 | FASYN-ELONG-PWY | fatty acid elongation—saturated |
| NS | PAC:37165399 | 6.9107 × e−4 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| NS | PAC:37165399 | 3.3746 × e−4 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| NS | PAC:37165399 | 8.1448 × e−5 | PWY-5973 | cis-vaccenate biosynthesis |
| NS | PAC:37165399 | 3.3732 × e−4 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| NS | PAC:37165399 | 9.4892 × e−5 | PWY-6282 | palmitoleate biosynthesis I (from (5Z)-dodec-5-enoate) |
| NS | PAC:37165399 | 9.7640 × e−5 | PWY-7388 | octanoyl-[acyl-carrier protein] synthesis (mitochondria) |
| NS | PAC:37165399 | 4.6627 × e−5 | PWY-7663 | gondoate biosynthesis (anaerobic) |
| NS | PAC:37167444 | 4.9600 × e−5 | FASYN-ELONG-PWY | fatty acid elongation—saturated |
| NS | PAC:37167444 | 6.9107 × e−4 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| NS | PAC:37167444 | 3.3746 × e−4 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| NS | PAC:37167444 | 8.1448 × e−5 | PWY-5973 | cis-vaccenate biosynthesis |
| NS | PAC:37167444 | 3.3731 × e−4 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| NS | PAC:37167444 | 9.4892 × e−5 | PWY-6282 | palmitoleate biosynthesis I (from (5Z)-dodec-5-enoate) |
| NS | PAC:37167444 | 9.7640 × e−5 | PWY-7388 | octanoyl-[acyl-carrier protein] synthesis (mitochondria) |
| NS | PAC:37167444 | 4.6627 × e−5 | PWY-7663 | gondoate biosynthesis (anaerobic) |
| YLP | PAC:37160270 | 0.0132 | PWY1F-467 | phenylpropanoid biosynthesis, initial reactions |
| YLP | PAC:37160270 | 0.0288 | PWY-7186 | superpathway of scopolin and esculin biosynthesis |
| YLP | PAC:37160685 | 0.0434 | PWY-5690 | TCA cycle II (plants and fungi) |
| YLP | PAC:37160685 | 0.0432 | PWY-6549 | L-glutamine biosynthesis III |
| YLP | PAC:37172239 | 0.0167 | PWY-5466 | matairesinol biosynthesis |
| YLP | PAC:37172239 | 0.0045 | PWY-6824 | justicidin B biosynthesis |
| YLP | PAC:37172239 | 0.0103 | PWY-7214 | baicalein degradation (hydrogen peroxide detoxification) |
| YLP | PAC:37172239 | 0.0097 | PWY-7445 | luteolin triglucuronide degradation |
| YLP | PAC:37173372 | 0.0167 | PWY-5466 | matairesinol biosynthesis |
| YLP | PAC:37173372 | 0.0045 | PWY-6824 | justicidin B biosynthesis |
| YLP | PAC:37173372 | 0.0103 | PWY-7214 | baicalein degradation (hydrogen peroxide detoxification) |
| YLP | PAC:37173372 | 0.0097 | PWY-7445 | luteolin triglucuronide degradation |
| SB | PAC:37165106 | 0.0151 | PWY-1121 | suberin monomers biosynthesis |
| SB | PAC:37165106 | 0.0098 | PWY-321 | cutin biosynthesis |
| SB | PAC:37165106 | 0.0095 | PWY-5136 | fatty acid and beta oxidation II (peroxisome) |
| SB | PAC:37165106 | 0.0049 | PWY-5143 | long-chain fatty acid activation |
| SB | PAC:37165106 | 0.0086 | PWY-5147 | oleate biosynthesis I (plants) |
| SB | PAC:37165106 | 0.0153 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| SB | PAC:37165106 | 0.0180 | PWY-561 | glyoxylate cycle and fatty acid degradation |
| SB | PAC:37165106 | 0.0083 | PWY-5885 | wax esters biosynthesis II |
| SB | PAC:37165106 | 0.0106 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| SB | PAC:37165106 | 0.0106 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| SB | PAC:37165106 | 0.0111 | PWY66-389 | phytol degradation |
| SB | PAC:37165106 | 0.0095 | PWY-6733 | sporopollenin precursors biosynthesis |
| SB | PAC:37165106 | 0.0126 | PWY-6803 | phosphatidylcholine acyl editing |
| VB | PAC:37165106 | 0.0447 | PWY-1121 | suberin monomers biosynthesis |
| VB | PAC:37165106 | 0.0292 | PWY-321 | cutin biosynthesis |
| VB | PAC:37165106 | 0.0281 | PWY-5136 | fatty acid and beta oxidation II (peroxisome) |
| VB | PAC:37165106 | 0.0145 | PWY-5143 | long-chain fatty acid activation |
| VB | PAC:37165106 | 0.0255 | PWY-5147 | oleate biosynthesis I (plants) |
| VB | PAC:37165106 | 0.0451 | PWY-5156 | superpathway of fatty acid biosynthesis II (plant) |
| VB | PAC:37165106 | 0.0247 | PWY-5885 | wax esters biosynthesis II |
| VB | PAC:37165106 | 0.0316 | PWY-5971 | palmitate biosynthesis II (bacteria and plants) |
| VB | PAC:37165106 | 0.0316 | PWY-5989 | stearate biosynthesis II (bacteria and plants) |
| VB | PAC:37165106 | 0.0330 | PWY66-389 | phytol degradation |
| VB | PAC:37165106 | 0.0283 | PWY-6733 | sporopollenin precursors biosynthesis |
| VB | PAC:37165106 | 0.0374 | PWY-6803 | phosphatidylcholine acyl editing |
4. Discussion
4.1. Pervasive Environmentally Dependent Polygenic Adaptation Boosted by Hybrid Breeding
4.2. Morphological, Physiological, and Metabolic Mechanisms of Adaptation to Drought & Heat
4.3. Perspectives and Recommendations for Future Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, K.F.; Gephart, J.A.; Emery, K.A.; Leach, A.M.; Galloway, J.N.; D’Odorico, P. Meeting Future Food Demand with Current Agricultural Resources. Glob. Environ. Chang. 2016, 39, 125–132. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F. Harnessing Crop Wild Diversity for Climate Change Adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef] [PubMed]
- Lascoux, M.; Glémin, S.; Savolainen, O. Local Adaptation in Plants. eLS 2016, 0025270, 1–7. [Google Scholar] [CrossRef]
- Burgarella, C.; Barnaud, A.; Kane, N.A.; Jankowski, F.; Scarcelli, N.; Billot, C.; Vigouroux, Y.; Berthouly-Salazar, C. Adaptive Introgression: An Untapped Evolutionary Mechanism for Crop Adaptation. Front. Plant Sci. 2019, 10, 4. [Google Scholar] [CrossRef]
- Tai, A.P.K.; Martin, M.V.; Heald, C.L. Threat to Future Global Food Security from Climate Change and Ozone Air Pollution. Nat. Clim. Chang. 2014, 4, 817–821. [Google Scholar] [CrossRef]
- FAO. Panorama de La Seguridad Alimentaria y Nutricional; FAO: Rome, Italy, 2020. [Google Scholar]
- Sgarbieri, V.C.; Whitaker, J.R. Physical, Chemical, and Nutritional Properties of Common Bean (Phaseolus) Proteins. Adv. Food Res. 1982, 28, 93–166. [Google Scholar] [CrossRef]
- Cortés, A.J.; Monserrate, F.A.; Ramírez-Villegas, J.; Madriñán, S.; Blair, M.W. Drought Tolerance in Wild Plant Populations: The Case of Common Beans (Phaseolus vulgaris L.). PLoS ONE 2013, 8, e62898. [Google Scholar] [CrossRef]
- Tofiño Rivera, A.; Ospina Cortés, D.A.; Rozo Leguizamón, Y. Compatibility of Ancestral and Innovative Agricultural Practices in the Kankuamo People of Colombia. Ambient. Soc. 2021, 24, 1–24. [Google Scholar] [CrossRef]
- Shukla, P.R.; Skea, J.; Buendia, E.C.; Masson-Delmotte, V.; Pörtner, H.-O.; Roberts, D.C.; Zhai, P.; Slade, R.; Connors, S.; van Diemen, R.; et al. Climate Change and Land: An IPCC Special Report; IPPC: Geneva, Switzerland, 2019; pp. 1–864. [Google Scholar]
- Teichmann, C.; Eggert, B.; Elizalde, A.; Haensler, A.; Jacob, D.; Kumar, P.; Moseley, C.; Pfeifer, S.; Rechid, D.; Remedio, A.R.; et al. How Does a Regional Climate Model Modify the Projected Climate Change Signal of the Driving GCM: A Study over Different CORDEX Regions Using REMO. Atmosphere 2013, 4, 214–236. [Google Scholar] [CrossRef]
- Molina, O.D.; Bernhofer, C. Projected Climate Changes in Four Different Regions in Colombia. Environ. Syst. Res. 2019, 8, 33. [Google Scholar] [CrossRef]
- Beebe, S.E.; Rao, I.M.; Blair, M.W.; Acosta-Gallegos, J.A. Phenotyping Common Beans for Adaptation to Drought. Front. Physiol. 2013, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Buitrago-Bitar, M.A.; Cortés, A.J.; López-Hernández, F.; Londoño-Caicedo, J.M.; Muñoz-Florez, J.E.; Carmenza Muñoz, L.; Blair, M.W. Allelic Diversity at Abiotic Stress Responsive Genes in Relationship to Ecological Drought Indices for Cultivated Tepary Bean, Phaseolus Acutifolius a. Gray, and Its Wild Relatives. Genes 2021, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Mhlaba, Z.B.; Mashilo, J.; Shimelis, H.; Assefa, A.B.; Modi, A.T. Progress in Genetic Analysis and Breeding of Tepary Bean (Phaseolus acutifolius A. Gray): A Review. Sci. Hortic. 2018, 237, 112–119. [Google Scholar] [CrossRef]
- Jiri, O.; Mafongoya, P.L.; Chivenge, P. Climate Smart Crops for Food and Nutritional Security for Semi-Arid Zones of Zimbabwe. Afr. J. Food Agric. Nutr. Dev. 2017, 17, 12280–12294. [Google Scholar] [CrossRef]
- Moghaddam, S.M.; Oladzad, A.; Koh, C.; Ramsay, L.; Hart, J.P.; Mamidi, S.; Hoopes, G.; Sreedasyam, A.; Wiersma, A.; Zhao, D.; et al. The Tepary Bean Genome Provides Insight into Evolution and Domestication under Heat Stress. Nat. Commun. 2021, 12, 2638. [Google Scholar] [CrossRef]
- Mwale, S.E.; Shimelis, H.; Mafongoya, P.; Mashilo, J. Breeding Tepary Bean (Phaseolus acutifolius) for Drought Adaptation: A Review. Plant Breed. 2020, 139, 821–833. [Google Scholar] [CrossRef]
- Muñoz, L.C.; Duque, M.C.; Debouck, D.G.; Blair, M.W. Taxonomy of Tepary Bean and Wild Relatives as Determined by Amplified Fragment Length Polymorphism (AFLP) Markers. Crop Sci. 2006, 46, 1744–1754. [Google Scholar] [CrossRef]
- Migicovsky, Z.; Myles, S. Exploiting Wild Relatives for Genomics-Assisted Breeding of Perennial Crops. Front. Plant Sci. 2017, 8, 460. [Google Scholar] [CrossRef]
- Burbano-Erazo, E.; León-Pacheco, R.I.; Cordero-Cordero, C.C.; López-Hernández, F.; Cortés, A.J.; Tofiño-Rivera, A.P. Multi-Environment Yield Components in Advanced Common Bean (Phaseolus vulgaris L.) × Tepary Bean (p. Acutifolius A. Gray) Interspecific Lines for Heat and Drought Tolerance. Agronomy 2021, 11, 1978. [Google Scholar] [CrossRef]
- Souter, J.R.; Gurusamy, V.; Porch, T.G.; Bett, K.E. Successful Introgression of Abiotic Stress Tolerance from Wild Tepary Bean to Common Bean. Crop Sci. 2017, 57, 1160–1171. [Google Scholar] [CrossRef]
- Belivanis, T.; Doré, C. Doré Lnterspecific Hybridization of Phaseolus vulgaris L. and Phaseolus angustissimus A. Gray Using in Vitro Embryo Culture. Plant Cell Rep. 1986, 5, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Blair, M.W. Genotyping by Sequencing and Genome–Environment Associations in Wild Common Bean Predict Widespread Divergent Adaptation to Drought. Front. Plant Sci. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Oddou-Muratorio, S.; Lalagüe, H.; Pluess, A.R.; Heiri, C.; Vendramin, G.G. Genome-Environment Association Study Suggests Local Adaptation to Climate at the Regional Scale in Fagus Sylvatica. New Phytol. 2016, 210, 589–601. [Google Scholar] [CrossRef]
- Cortés, A.J.; López-Hernández, F.; Blair, M.W. Genome–Environment Associations, an Innovative Tool for Studying Heritable Evolutionary Adaptation in Orphan Crops and Wild Relatives. Front. Genet. 2022, 13, 1562. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, F.; Cortés, A.J. Last-Generation Genome–Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.). Front. Genet. 2019, 10, 954. [Google Scholar] [CrossRef]
- Ambachew, D.; Blair, M.W. Genome Wide Association Mapping of Root Traits in the Andean Genepool of Common Bean (Phaseolus vulgaris L.) Grown With and Without Aluminum Toxicity. Front. Plant Sci. 2021, 12, 628687. [Google Scholar] [CrossRef]
- Diaz, S.; Ariza-Suarez, D.; Izquierdo, P.; Lobaton, J.D.; de la Hoz, J.F.; Acevedo, F.; Duitama, J.; Guerrero, A.F.; Cajiao, C.; Mayor, V.; et al. Genetic Mapping for Agronomic Traits in a MAGIC Population of Common Bean (Phaseolus vulgaris L.). BMC Genom. 2020, 21, 799. [Google Scholar] [CrossRef]
- Wu, X.; Islam, A.S.M.F.; Limpot, N.; Mackasmiel, L.; Mierzwa, J.; Cortés, A.J.; Blair, M.W. Genome-Wide SNP Identification and Association Mapping for Seed Mineral Concentration in Mung Bean (Vigna radiata L.). Front. Genet. 2020, 11, 656. [Google Scholar] [CrossRef]
- Scott, M.F.; Ladejobi, O.; Amer, S.; Bentley, A.R.; Biernaskie, J.; Boden, S.A.; Clark, M.; Dell’Acqua, M.; Dixon, L.E.; Filippi, C.V.; et al. Multi-Parent Populations in Crops: A Toolbox Integrating Genomics and Genetic Mapping with Breeding. Heredity 2020, 125, 396–416. [Google Scholar] [CrossRef]
- McCouch, S. Diversifying Selection in Plant Breeding. PLoS Biol. 2004, 2, e347. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the Missing Heritability of Complex Diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. The Case of the Missing Heritability. Nature 2022, 456, 18. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Hermisson, J.; Schlötterer, C. Polygenic Adaptation: A Unifying Framework to Understand Positive Selection. Nat. Rev. Genet. 2020, 21, 769–781. [Google Scholar] [CrossRef]
- Cruz, S.; Lobatón, J.; Milan, U.O.; Ariza-Suárez, D.; Raatz, B.; Aparicio, J.; Gloria, M.; Beebe, S. Interspecific Common Bean Population Derived from Phaseolus acutifolius Using a Bridging Genotype Demonstrate Useful Adaptation to Heat Tolerance. Front. Plant Sci. 2023, 14, 1145858. [Google Scholar] [CrossRef]
- Karger, D.N.; Wilson, A.M.; Mahony, C.; Zimmermann, N.E.; Jetz, W. Global Daily 1 Km Land Surface Precipitation Based on Cloud Cover-Informed Downscaling. Sci. Data 2021, 8, 307. [Google Scholar] [CrossRef]
- Blair, M.W.; Soler, A.; Cortés, A.J. Diversification and Population Structure in Common Beans (Phaseolus vulgaris L.). PLoS ONE 2012, 7, e49488. [Google Scholar] [CrossRef] [PubMed]
- Tukey, J.W. Exploratory Data Analysis; Mosteller, F., Ed.; Addison-Wesley Publishing Company: Boston, MA, USA, 1977; Volume 33, ISBN 0-201-07616-0. [Google Scholar]
- Mangiafico, S.S.; Package ‘Rcompanion’: Functions to Support Extension Education Program Evaluation. Rutgers Cooperative Extension, New Brunswick, New Jersey. Version 2.4.30. Available online: https://CRAN.R-project.org/package=rcompanion/ (accessed on 15 May 2023).
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef]
- Goh, L.; Yap, V.B. Effects of Normalization on Quantitative Traits in Association Test. BMC Bioinform. 2009, 10, 415. [Google Scholar] [CrossRef]
- Gross, J.; Ligges, U. Package ‘Nortest’. Available online: https://cran.r-project.org/web/packages/nortest/index.html (accessed on 15 May 2023).
- Patil, I. Visualizations with Statistical Details: The “ggstatsplot” Approach. J. Open Source Softw. 2021, 6, 3167. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 May 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; Van Den Beek, M.; Bouvier, D.; Ech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for Association Mapping of Complex Traits in Diverse Samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Sekula, M.; Datta, S.; Datta, S. OptCluster: An R Package for Determining the Optimal Clustering Algorithm. Bioinformation 2017, 13, 101–103. [Google Scholar] [CrossRef]
- Frichot, E.; François, O. LEA: An R Package for Landscape and Ecological Association Studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Zhou, Y.; Summers, R.M. BLINK: A Package for the next Level of Genome-Wide Association Studies with Both Individuals and Markers Meng Huang. Gigascience 2018, 8, giy154. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Joo, J.W.J.; Hormozdiari, F.; Han, B.; Eskin, E. Multiple Testing Correction in Linear Mixed Models. Genome Biol. 2016, 17, 29–33. [Google Scholar] [CrossRef]
- Pasam, R.K.; Sharma, R.; Malosetti, M.; van Eeuwijk, F.A.; Haseneyer, G.; Kilian, B.; Graner, A. Genome-Wide Association Studies for Agronomical Traits in a World Wide Spring Barley Collection. BMC Plant Biol. 2012, 12, 16. [Google Scholar] [CrossRef]
- Oladzad, A.; Porch, T.; Rosas, J.C.; Ma, S.; Beaver, J.; Beebe, S.E.; Burridge, J.; Jochua, C.N.; Miguel, M.A.; Miklas, P.N.; et al. Single and Multi-Trait GWAS Identify Genetic Factors Associated with Production Traits in Common Bean Under Abiotic Stress Environments. G3 Genes Genomes Genet. 2019, 9, 1881–1892. [Google Scholar]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG Graphics to Visualize and Map Genome-Wide Data on the Idiograms. PeerJ. Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Slate, J.; Gratten, J.; Beraldi, D.; Stapley, J.; Hale, M.; Pemberton, J.M. Gene Mapping in the Wild with SNPs: Guidelines and Future Directions. Genetica 2009, 136, 97–107. [Google Scholar] [CrossRef]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A Reference Genome for Common Bean and Genome-Wide Analysis of Dual Domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef]
- Reimand, J.; Isser, R.; Voisin, V.; Kucera, M.; Tannus-lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape and EnrichmentMap-ZLO FAJN ČLANEK, TUDI RAZLAGE POSAMEZNIH TERMINOV. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- Asakura, H.; Yamakawa, T.; Tamura, T.; Ueda, R.; Taira, S.; Saito, Y.; Abe, K.; Asakura, T. Transcriptomic and Metabolomic Analyses Provide Insights into the Upregulation of Fatty Acid and Phospholipid Metabolism in Tomato Fruit under Drought Stress. J. Agric. Food Chem. 2021, 69, 2894–2905. [Google Scholar] [CrossRef]
- Blair, M.W.; Cortés, A.J.; Farmer, A.D.; Huang, W.; Ambachew, D.; Varma Penmetsa, R.; Carrasquilla-Garcia, N.; Assefa, T.; Cannon, S.B. Uneven Recombination Rate and Linkage Disequilibrium across a Reference SNP Map for Common Bean (Phaseolus vulgaris L.). PLoS ONE 2018, 13, e0189597. [Google Scholar] [CrossRef] [PubMed]
- Galeano, C.H.; Cortés, A.J.; Fernández, A.C.; Soler, Á.; Franco-Herrera, N.; Makunde, G.; Vanderleyden, J.; Blair, M.W. Gene-Based Single Nucleotide Polymorphism Markers for Genetic and Association Mapping in Common Bean. BMC Genet. 2012, 13, 48. [Google Scholar] [CrossRef]
- Seehausen, O. Hybridization and Adaptive Radiation. Trends Ecol. Evol. 2004, 19, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Jiménez, A.; Muñoz, C.; Jacobsen, H.J.; Roca, W.M.; Singh, S.P. Interspecific Hybridization between Common and Tepary Beans: Increased Hybrid Embryo Growth, Fertility, and Efficiency of Hybridization through Recurrent and Congruity Backcrossing. Theor. Appl. Genet. 1994, 88, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.C.; Blair; Duque, W.; Tohme, M.C.; Roca, J. Introgression in Common Bean—Tepary Bean Interspecific Congruity-Backcross Lines as Measured by AFLP Markers. Crop Sci. 2004, 44, 637–645. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Bresadola, L.; Caseys, C.; Castiglione, S.; Buerkle, C.A.; Wegmann, D.; Lexer, C. Admixture Mapping in Interspecific Populus Hybrids Identifies Classes of Genomic Architectures for Phytochemical, Morphological and Growth Traits. New Phytol. 2019, 223, 2076–2089. [Google Scholar] [CrossRef]
- Vasylyk, I.; Gorislavets, S.; Matveikina, E.; Lushchay, E.; Lytkin, K.; Grigoreva, E.; Karzhaev, D.; Volkov, V.; Volodin, V.; Spotar, G.; et al. SNPs Associated with Foliar Phylloxera Tolerance in Hybrid Grape Populations Carrying Introgression from Muscadinia. Horticulturae 2022, 8, 16. [Google Scholar] [CrossRef]
- Osorio-Guarín, J.A.; Garzón-Martínez, G.A.; Delgadillo-Duran, P.; Bastidas, S.; Moreno, L.P.; Enciso-Rodríguez, F.E.; Cornejo, O.E.; Barrero, L.S. Genome-Wide Association Study (GWAS) for Morphological and Yield-Related Traits in an Oil Palm Hybrid (Elaeis oleifera x Elaeis guineensis) Population. BMC Plant Biol. 2019, 19, 533. [Google Scholar] [CrossRef]
- Sarfraz, Z.; Iqbal, M.S.; Geng, X.; Iqbal, M.S.; Nazir, M.F.; Ahmed, H.; He, S.; Jia, Y.; Pan, Z.; Sun, G.; et al. GWAS Mediated Elucidation of Heterosis for Metric Traits in Cotton (Gossypium hirsutum L.) Across Multiple Environments. Front. Plant Sci. 2021, 12, 565552. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.P.; Wu, T.H.; Chan, Y.K.; van Zonneveld, M.; Schafleitner, R. De Novo SNP Calling Reveals the Genetic Differentiation and Morphological Divergence in Genus Amaranthus. Plant Genome 2022, 15, e20206. [Google Scholar] [CrossRef]
- Nagasaka, K.; Nishiyama, S.; Fujikawa, M.; Yamane, H.; Shirasawa, K.; Babiker, E.; Tao, R. Genome-Wide Identification of Loci Associated With Phenology-Related Traits and Their Adaptive Variations in a Highbush Blueberry Collection. Front. Plant Sci. 2022, 12, 3147. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Baenziger, P.S. Genome-Wide Association Study Reveals Novel Genomic Regions for Grain Yield and Yield-Related Traits in Drought-Stressed Synthetic Hexaploid Wheat. Int. J. Mol. Sci. 2018, 19, 3011. [Google Scholar] [CrossRef]
- Anderson, J.T.; Lee, C.R.; Mitchell-Olds, T. Strong Selection Genome-Wide Enhances Fitness Trade-Offs across Environments and Episodes of Selection. Evolution 2014, 68, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, O.; Lascoux, M.; Merilä, J. Ecological Genomics of Local Adaptation. Nat. Rev. Genet. 2013, 14, 807–820. [Google Scholar] [CrossRef]
- Anderson, J.T.; Willis, J.H.; Mitchell-Olds, T. Evolutionary Genetics of Plant Adaptation. Trends Genet. 2011, 27, 258–266. [Google Scholar] [CrossRef]
- Wadgymar, S.M.; Lowry, D.B.; Gould, B.A.; Byron, C.N.; Mactavish, R.M.; Anderson, J.T. Identifying Targets and Agents of Selection: Innovative Methods to Evaluate the Processes That Contribute to Local Adaptation. Methods Ecol. Evol. 2017, 8, 738–749. [Google Scholar] [CrossRef]
- Mee, J.A.; Yeaman, S. Unpacking Conditional Neutrality: Genomic Signatures of Selection on Conditionally Beneficial and Conditionally Deleterious Mutations. Am. Nat. 2019, 194, 529–540. [Google Scholar] [CrossRef]
- Orr, H.A. The Genetic Theory of Adaptation: A Brief History. Nat. Rev. Genet. 2005, 6, 119–127. [Google Scholar] [CrossRef]
- Payseur, B.A.; Rieseberg, L.H. A Genomic Perspective on Hybridization and Speciation. Mol. Ecol. 2016, 25, 2337–2360. [Google Scholar] [CrossRef] [PubMed]
- Todesco, M.; Owens, G.L.; Bercovich, N.; Légaré, J.S.; Soudi, S.; Burge, D.O.; Huang, K.; Ostevik, K.L.; Drummond, E.B.M.; Imerovski, I.; et al. Massive Haplotypes Underlie Ecotypic Differentiation in Sunflowers. Nature 2020, 584, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.; Frisch, M. Selection Strategies for Marker-Assisted Backcrossing with High-Throughput Marker Systems. Theor. Appl. Genet. 2011, 123, 251–260. [Google Scholar] [CrossRef]
- Oliveira, L.K.; Melo, L.C.; Brondani, C.; Peloso, M.J.D.; Brondani, R.P.V. Backcross Assisted by Microsatellite Markers in Common Bean. Genet. Mol. Res. 2008, 7, 1000–1010. [Google Scholar] [CrossRef]
- Blair, M.W.; Iriarte, G.; Beebe, S. QTL Analysis of Yield Traits in an Advanced Backcross Population Derived from a Cultivated Andean × Wild Common Bean (Phaseolus vulgaris L.) Cross. Theor. Appl. Genet. 2006, 112, 1149–1163. [Google Scholar] [CrossRef]
- Yang, X.; Lu, M.; Wang, Y.; Wang, Y.; Liu, Z.; Chen, S. Response Mechanism of Plants to Drought Stress. Horticulturae 2021, 7, 50. [Google Scholar] [CrossRef]
- Pollard, M.; Beisson, F.; Li, Y.; Ohlrogge, J.B. Building Lipid Barriers: Biosynthesis of Cutin and Suberin. Trends Plant Sci. 2008, 13, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhu, X.; Zhang, Y.; Du, Z.; Chen, X.; Kong, X.; Sun, W.; Chen, C. Drought Stress Modify Cuticle of Tender Tea Leaf and Mature Leaf for Transpiration Barrier Enhancement through Common and Distinct Modes. Sci. Rep. 2020, 10, 6696. [Google Scholar] [CrossRef]
- Patwari, P.; Salewski, V.; Gutbrod, K.; Kreszies, T.; Dresen-Scholz, B.; Peisker, H.; Steiner, U.; Meyer, A.J.; Schreiber, L.; Dörmann, P. Surface Wax Esters Contribute to Drought Tolerance in Arabidopsis. Plant J. 2019, 98, 727–744. [Google Scholar] [CrossRef]
- Kannangara, R.; Branigan, C.; Liu, Y.; Penfield, T.; Rao, V.; Mouille, G.; Höfte, H.; Pauly, M.; Riechmann, J.L.; Broun, P. The Transcription Factor WIN1/SHN1 Regulates Cutin Biosynthesis in Arabidopsis Thaliana. Plant Cell 2007, 19, 1278–1294. [Google Scholar] [CrossRef]
- Liu, N.; Chen, J.; Wang, T.; Li, Q.; Cui, P.; Jia, C.; Hong, Y. Overexpression of WAX INDUCER1/SHINE1 Gene Enhances Wax Accumulation under Osmotic Stress and Oil Synthesis in Brassica Napus. Int. J. Mol. Sci. 2019, 20, 4435. [Google Scholar] [CrossRef]
- Cortés, A.J.; This, D.; Chavarro, C.; Madriñán, S.; Blair, M.W. Nucleotide Diversity Patterns at the Drought-Related DREB2 Encoding Genes in Wild and Cultivated Common Bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2012, 125, 1069–1085. [Google Scholar] [CrossRef]
- Quirino, B.F.; Noh, Y.; Himelblau, E.; Amasino, R.M. Molecular Aspects of Leaf Senescence. Trends Plant Sci. 2000, 5, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, N.; Wang, X.; Meng, X.; Cui, X.; Chen, Z.; Ren, H.; Ma, J.; Liu, H. Late Embryogenesis Abundant (LEA) Gene Family in Salvia miltiorrhiza: Identification, Expression Analysis, and Response to Drought Stress. Plant Signal. Behav. 2021, 16, 1891769. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.M.; Kumar, M.; Pratap, A.; Tripathi, A. Genome-Wide Analysis of Late Embryogenesis Abundant Protein Gene Family in Vigna Species and Expression of VrLEA Encoding Genes in Vigna Glabrescens Reveal Its Role in Heat Tolerance. Front. Plant Sci. 2022, 13, 843107. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, S. Biosynthesis and Regulation of Phenylpropanoids in Plants. CRC Crit. Rev. Plant Sci. 2017, 36, 257–290. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive Oxygen Species and Antioxidant Machinery in Abiotic Stress Tolerance in Crop Plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- Cheng, M.C.; Liao, P.M.; Kuo, W.W.; Lin, T.P. The Arabidopsis Ethylene Response FACTOR1 Regulates Abiotic Stress-Responsive Gene Expression by Binding to Different Cis-Acting Elements in Response to Different Stress Signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef]
- Yang, J.; Duan, G.; Li, C.; Liu, L.; Han, G.; Zhang, Y.; Wang, C. The Crosstalks Between Jasmonic Acid and Other Plant Hormone Signaling Highlight the Involvement of Jasmonic Acid as a Core Component in Plant Response to Biotic and Abiotic Stresses. Front. Plant Sci. 2019, 10, 1349. [Google Scholar] [CrossRef]
- Dornbos, D.L.; Mullen, R.E. Soybean Seed Protein and Oil Contents and Fatty Acid Composition Adjustments by Drought and Temperature. J. Am. Oil Chem. Soc. 1992, 69, 228–231. [Google Scholar] [CrossRef]
- Kazan, K. Diverse Roles of Jasmonates and Ethylene in Abiotic Stress Tolerance. Trends Plant Sci. 2015, 20, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; Skeen, P.; Blair, M.W.; Chacón-Sánchez, M.I. Does the Genomic Landscape of Species Divergence in Phaseolus Beans Coerce Parallel Signatures of Adaptation and Domestication? Front. Plant Sci. 2018, 871, 1816. [Google Scholar] [CrossRef] [PubMed]
- Barbulescu, D.M.; Fikere, M.; Malmberg, M.M. Imputation to Whole-Genome Sequence Increases the Power of Genome Wide Association Studies for Blackleg Resistance in Canola. In AusCanola 2018 20th Australian Research Assembly on Brassicas Perth; GIWA: Perth, Australia, 2018; p. 29. [Google Scholar]
- Lang-Mladek, C.; Popova, O.; Kiok, K.; Berlinger, M.; Rakic, B.; Aufsatz, W.; Jonak, C.; Hauser, M.-T.; Luschnig, C. Transgenerational Inheritance and Resetting of Stress-Induced Loss of Epigenetic Gene Silencing in Arabidopsis. Mol. Plant 2010, 3, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Pecinka, A.; Dinh, H.Q.; Baubec, T.; Rosa, M.; Lettner, N.; Mittelsten Scheid, O. Epigenetic Regulation of Repetitive Elements Is Attenuated by Prolonged Heat Stress in Arabidopsis. Plant Cell 2010, 22, 3118–3129. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, C.; Zhang, F.; Mendez, J.; Lozano, Y.; Chatpar, K.; Irish, V.F.; Jacob, Y. Increased Efficiency of Targeted Mutagenesis by CRISPR/Cas9 in Plants Using Heat Stress. Plant J. 2018, 93, 377–386. [Google Scholar] [CrossRef]
- Assefa, T.; Assibi Mahama, A.; Brown, A.V.; Cannon, E.K.S.; Rubyogo, J.C.; Rao, I.M.; Blair, M.W.; Cannon, S.B. A Review of Breeding Objectives, Genomic Resources, and Marker-Assisted Methods in Common Bean (Phaseolus vulgaris L.). Mol. Breed. 2019, 39, 20. [Google Scholar] [CrossRef]
- Pelaáz, D.; Aguilar, P.A.; Mercado, M.; Loápez-Hernaández, F.; Guzmaán, M.; Burbano-Erazo, E.; Denning-James, K.; Medina, C.I.; Blair, M.W.; De Vega, J.J.; et al. Genotype Selection, and Seed Uniformity and Multiplication to Ensure Common Bean (Phaseolus vulgaris L.) var. Liborino. Agronomy 2022, 12, 2285. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Hernández, F.; Burbano-Erazo, E.; León-Pacheco, R.I.; Cordero-Cordero, C.C.; Villanueva-Mejía, D.F.; Tofiño-Rivera, A.P.; Cortés, A.J. Multi-Environment Genome-Wide Association Studies of Yield Traits in Common Bean (Phaseolus vulgaris L.) × Tepary Bean (P. acutifolius A. Gray) Interspecific Advanced Lines in Humid and Dry Colombian Caribbean Subregions. Agronomy 2023, 13, 1396. https://doi.org/10.3390/agronomy13051396
López-Hernández F, Burbano-Erazo E, León-Pacheco RI, Cordero-Cordero CC, Villanueva-Mejía DF, Tofiño-Rivera AP, Cortés AJ. Multi-Environment Genome-Wide Association Studies of Yield Traits in Common Bean (Phaseolus vulgaris L.) × Tepary Bean (P. acutifolius A. Gray) Interspecific Advanced Lines in Humid and Dry Colombian Caribbean Subregions. Agronomy. 2023; 13(5):1396. https://doi.org/10.3390/agronomy13051396
Chicago/Turabian StyleLópez-Hernández, Felipe, Esteban Burbano-Erazo, Rommel Igor León-Pacheco, Carina Cecilia Cordero-Cordero, Diego F. Villanueva-Mejía, Adriana Patricia Tofiño-Rivera, and Andrés J. Cortés. 2023. "Multi-Environment Genome-Wide Association Studies of Yield Traits in Common Bean (Phaseolus vulgaris L.) × Tepary Bean (P. acutifolius A. Gray) Interspecific Advanced Lines in Humid and Dry Colombian Caribbean Subregions" Agronomy 13, no. 5: 1396. https://doi.org/10.3390/agronomy13051396
APA StyleLópez-Hernández, F., Burbano-Erazo, E., León-Pacheco, R. I., Cordero-Cordero, C. C., Villanueva-Mejía, D. F., Tofiño-Rivera, A. P., & Cortés, A. J. (2023). Multi-Environment Genome-Wide Association Studies of Yield Traits in Common Bean (Phaseolus vulgaris L.) × Tepary Bean (P. acutifolius A. Gray) Interspecific Advanced Lines in Humid and Dry Colombian Caribbean Subregions. Agronomy, 13(5), 1396. https://doi.org/10.3390/agronomy13051396