1. Introduction
NUE in maize is defined as an incremental increase in grain yield with incremental increases in N fertilizer [
1,
2]. Grain yield responds to N fertility in a linear-plateau fashion [
3]. NUE is greater at lower inputs of N [
1,
4,
5] where grain yield responses approach linearity, and NUE is very low past the inflexion point where little grain yield increases result from increased N fertility. NUE in maize ranges from 50–10% [
6,
7,
8] but field sources of N are not limited to fertilizer input. Field environments may provide up to 185 kg ha
−1 of non-fertilizer N [
9] with N mineralization and N carryover as sources of non-fertilizer N input. These additional sources of N vary from season to season based on soil temperature and moisture, organic matter, and performance of the previous crop. Precise control of N input in a field environment is difficult and only 50–65% of applied fertilizer is taken up by the plant the same year it is applied [
9,
10]. Applied N fertilizer can be incorporated into soil organic matter, lost due to ammonia volatility, leached, and lost due to denitrification [
11,
12,
13,
14]. Precise control of N in field environments required to develop transgenic improvements in NUE is very difficult and requires controlled environments to complement field experimentation to bridge the gap between what is known biochemically, required to make transgenic modifications, and field performance. Nitrate uptake and assimilation have been studied in controlled environments using the model system,
Arabidopsis thaliana (L.) Heynh. [
8,
15,
16], but
A. thaliana is a rather poor model for maize both genetically [
17] and physiologically. Not only is photosynthetic carbon metabolism different between maize (C4) and
A. thaliana (C3) but nitrogen metabolism is dissimilar [
18,
19].
Recently, Fan et al. [
20] showed transgenic expression of a high affinity nitrate transporter significantly improves NUE in rice. It is difficult to conceive that increasing nitrate uptake would have a significant effect on maize, especially under high N fertility, since maize accumulates significant amounts of nitrate under these conditions. As much as 70% of stalk N is nitrate [
21]. Cliquet et al. [
22] showed that 47% of the N applied after pollination accumulated in the stalk as nitrate. Currently, the main interest in stalk nitrate is in predicting N carryover and overall plant health when soil nitrate is limited by poor fertilization or nitrate leaching [
4,
23,
24] but stalk nitrate is also associated with reduced NUE. Brouder et al. [
25] showed that stalk nitrate and agronomic efficiency, defined identically as NUE, were inversely related. Binford et al. [
4] showed, using 900 crop years of data, that stalk nitrate is linearly related to grain yield up to the linear regression plateau (LRP) inflection point. This would suggest that accumulation of stalk nitrate is symptomatic of reduced NUE as demonstrated by Varvel et al. [
26] and Brouder et al. [
25]. Varvel et al. [
26] showed stalk nitrate linearly increased with increased N fertility past the LRP inflection point while grain yields were unchanged. With no additional grain yield with increased N fertility, NUE dropped as stalk nitrate dramatically increased. Under normal or high N fertility, maize will concentrate nitrate in the stalk 20–100 times the soil nitrate concentration [
24] which would argue that N uptake is not limiting under high N fertility (low NUE).
Linking physiological/biochemical information to relevant field performance is critical in developing transgenic improvements in maize NUE. Though nitrate transporter research is more recent than NA research, NA is likely more relevant in improving NUE in maize. Beevers and Hageman [
27] proposed that NRA (in vitro) is the limiting step in N metabolism in plants. Though grain yields in maize can be significantly improved by N fertility, a direct correlation between NRA (in vitro) and grain yield has never been established. Blackmer et al. [
28] showed >80% of grain yield was related to spring soil nitrate concentrations. Klepper et al. [
29] showed in vivo nitrate reductase activity, NRA (in vivo), or the ability of leaf tissue to generate nitrite from nitrate in the dark, was enhanced by respiratory metabolites. Klepper et al. [
29] also demonstrated that NRA (in vitro) could be supported by the addition of glyceraldehyde-3-phosphate and NAD
+ to cell free plant extracts suggesting glyceraldehyde-3-phosphate dehydrogenase (GA3PDH—EC 1.2.1.12) the source of NADH for nitrate reduction. Later, Gowri and Campbell [
30] showed that NRA (in vitro) and GA3PDH are coincidentally induced by nitrate in etiolated maize. Though extractable levels of GA3PDH are high enough to provide sufficient NADH to support NRA (in vitro) and NRA (in vitro) and GA3PDH are co-induced by nitrate, this is not a proof of in vivo metabolism. Klepper et al. [
29] also noted that NRA (in vitro) was 2.5–20 times higher than the rate of NRA (in vivo) which would suggest that extractable NRA (in vitro), per se, is far in excess of what is required to support NA. Also, since extractable levels of GA3PDH are high enough to support NRA (in vitro) it would follow that GA3PDH is in excess of that required to support NA. Later, Neyra and Hageman [
31] suggested malate could be a substrate for generating NADH to support NA. Neither Klepper et al. [
29] nor Neyra and Hageman [
31] were able to demonstrate enhanced NRA (in vivo), by the addition of malate to the NRA (in vivo) assay medium. Rathnam [
32] showed NA, measured by the disappearance of nitrate, in spinach protoplast could be supported by the addition of phospho-3-glyceric acid and oxaloacetic acid (OAA) in the light and/or by glyceraldehyde-3-phosphate and malate in either light or dark showing GA3PDH and/or malate dehydrogenase (MDH—EC 1.1.1.37) capable of supporting NA. Similar to the observation made by Klepper et al. [
29], Kaiser et al. [
33] also observed higher extractable levels of NRA (in vitro) than NRA (in vivo) and concluded that NADH levels, and not NRA (in vitro), limits NA. Later, increased leaf NADH levels were also reported [
33] to be associated with NA.
In order to complete the nitrogen assimilation pathway from nitrate to the formation of glutamate, NADH, ATP, reduced ferredoxin, and α-ketoglutarate (αKG) are required. Mitochondrial or cytoplasmic isocitrate dehydrogenase (ICDH—EC 1.1.1.41, EC 1.1.1.42) produce αKG. Reducing the expression of mitochondrial citrate synthase (CS—EC 2.3.3.1) [
34] or ICDH [
35] in tomato resulted in elevated levels of lamina nitrate. This suggests a direct link between αKG and nitrate reduction. When isocitrate or αKG moves out of the mitochondria, NADH levels drop in the mitochondria and since NADH does not pass through the mitochondrial membrane it cannot be replenished directly. Reestablishing the mitochondrial NADH concentration may be done by importing malate from the cytoplasm. Assuming malate provides significant reduction power for NA the loss of malate from the cytoplasm results in NA becoming limited by low cytoplasmic NADH. Thus, the pathway becomes self-regulated. Cytoplasmic male sterile (CMS) tobacco [
36] and CMS cucumber [
37] have improved NUE under lower N fertility. These CMS mutants with defective Complex I (NADH dehydrogenase—EC 1.6.5.3) have reduced capacity to oxidize mitochondrial NADH which results in increased mitochondrial NADH concentrations, reducing the need to import malate to balance the mitochondrial NADH levels due to the transport of αKG out of the mitochondria. Based on these observations, Foyer et al. [
38] predicted over-expression of ICDH or co-suppression of Complex I would improve NUE. CMS tobacco [
36] and CMS cucumber [
37] might be examples of improved NUE through diminished Complex I activity but these have associated deleterious attributes (slow growth, male sterility) which makes these agronomically unsuitable. The mutant NCS2 in maize also has defective Complex I and expresses undesirable traits [
39] but has not, specifically, been shown to have improved NUE.
In this report, multi-year field experiments of maize grown at different levels of NUE showed reproductive plant parameters are associated with improved NUE under both low and high NUE conditions. The response of these parameters to different forms of N applied at different times of development were used, under controlled environments, to elucidate key physiological factors related to improved NUE. The accumulation of nitrate in lower internodes was investigated between plants grown under high and low NUE conditions and shown to be inversely related to improved NUE. Finally, increasing cytoplasmic NADH using a transgenic co-suppression of Complex I under the control of a tissue preferred promoter increased kernel number plant−1 (KNP) and grain yield under low NUE (high N fertility) conditions. In all, these results suggest that NA limits NUE in maize via reduced availability of cytoplasmic NADH.
2. Materials and Methods
2.1. Field NUE Analyses under Normal and Depleted N Conditions
Field plots depleted of N for a minimum of two years were used in NUE experiments over a period of three years in Johnston, Iowa. Non-depleted plots were plots in previous cropping seasons fertilized with N to obtain maximum economic grain yield of maize. In the depleted plots N treatments consisted of 0, 22, 45, 67 kg ha
−1 N applied as urea and in the non-depleted plots N treatments were 0, 34, 67, 101, 135, and 168 kg ha
−1 N applied as urea. All N applications were made at V3. The three DuPont-Pioneer hybrids (33K42, 33W84, and 33T56) used in these studies were selected based on a range of yield responses to N fertility when grown under depleted N conditions in multi-environment trials. The experiments were arranged in a split, split plot experimental design with season as the main plot, N fertility as the subplot and cultivar as the sub, subplot. Sampling dates, R1 and maturity (black layer), were blocked as separate experiments to avoid contamination of the maturity sampling by the R1 sampling. Planting density was 75,000 plants ha
−1, each plot consisted of two 5.1 m rows spaced 0.75 m apart. There were five replicates of all treatments. Soluble leaf amino acids at V9 was the first parameter measured. At R1 chlorophyll measurements (Minolta SPAD, Minolta Camera Co., Ramsey, NJ, USA) were made by averaging five samplings taken down the ear leaf of 10 plants in each plot. Chlorophyll measurements were made in a similar manner at R2 and R3. At R1 10 plants were sampled from each plot and the ears removed and dried (70 °C, 72 h). Ear shoots of plants sampled for R1 ear measurements were bagged prior to silk emergence to avoid pollination. Ear length, ear width and ear dry weight were determined. Total fresh weight of the remaining chopped plants was measured and vegetative biomass was determined by weighing and drying (70 °C, 72 h) a subsample. Samples were ground and total N determined. At maturity, 10 plants were sampled, the ears removed and dried (70 °C, 72 h). Ear weight, grain weight, and kernel density were determined of the dried ears. Vegetative biomass and total N were determined similarly as at R1. Statistical analysis was performed as previously described [
40].
2.2. Field Stalk Nitrate Experiments
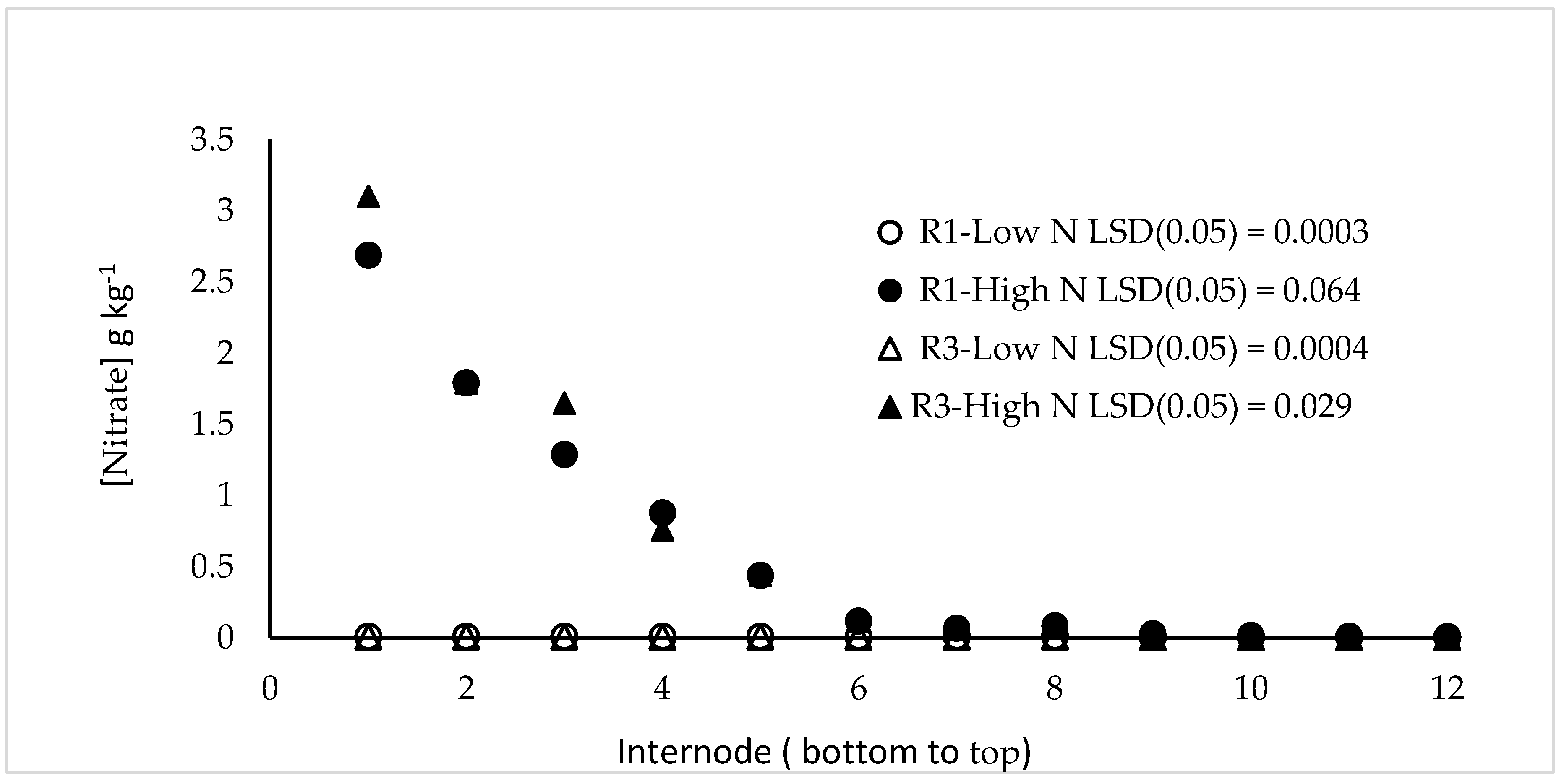
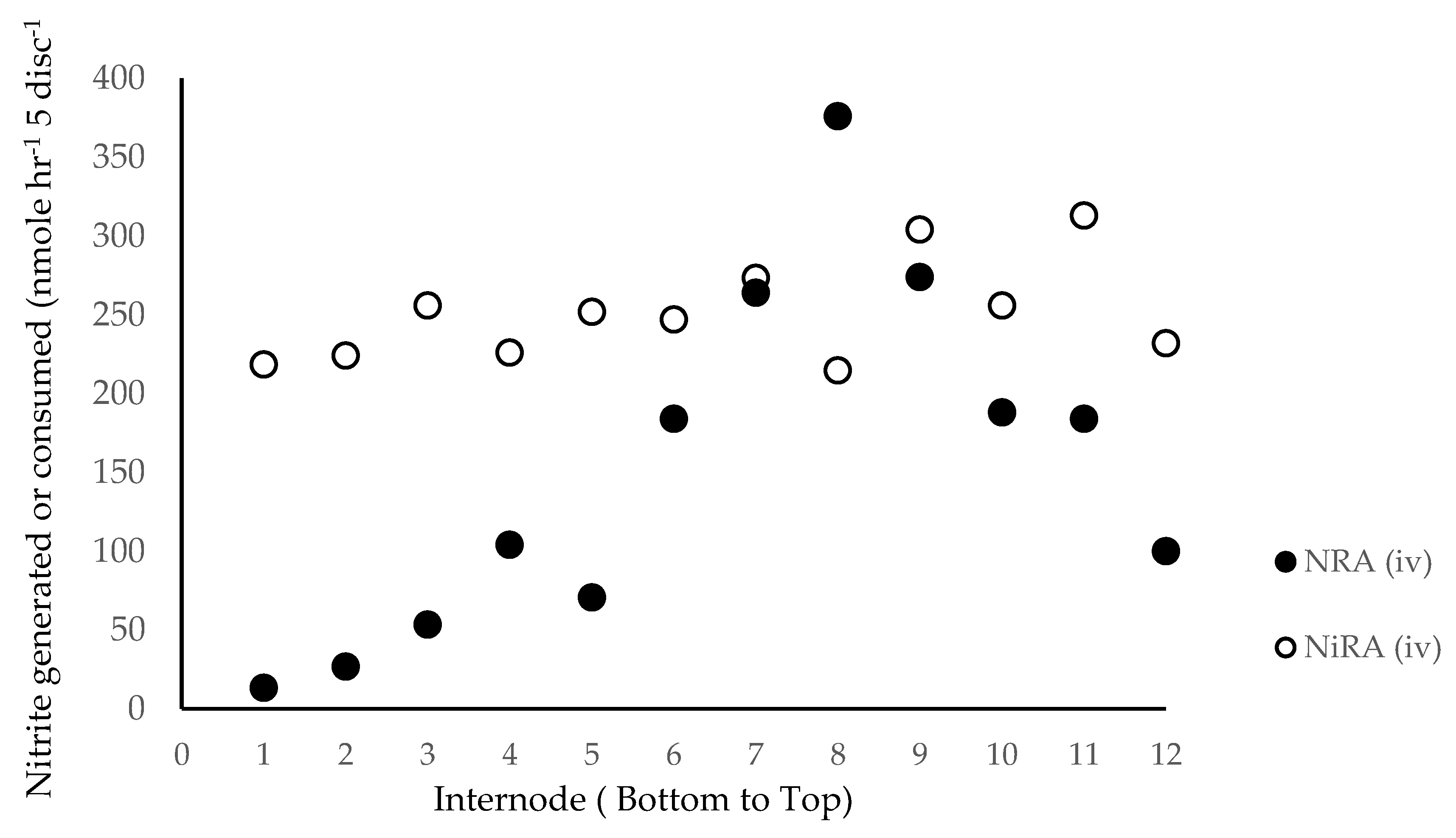
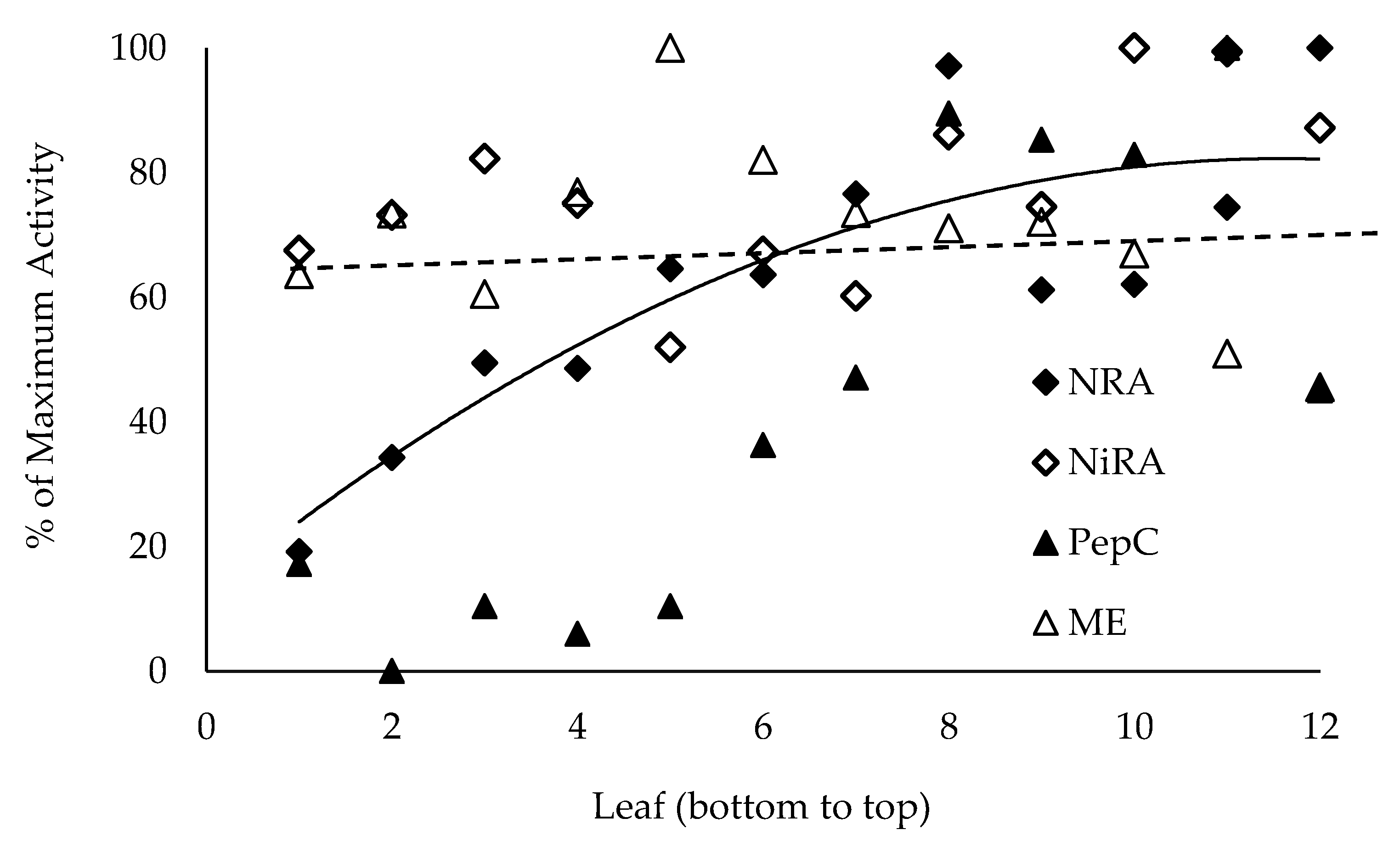
The DuPont-Pioneer cultivar 33W84 was grown in field plots in Johnston, IA either fertilized for optimum grain yield (224 kg N ha−1) or in plots depleted for N for at least two years and fertilized with 77 kg N ha−1 immediately after planting. Plants were sampled for stalk nitrate at V11, VT, R1, R2 and R3. Leaves associated with each internode were punched (10, 5 mm diameter) and used for metabolic analysis. Leaves, including leaf sheath, were removed and stalk internodes were numbered and cut at the top of each node, dried (70 °C, 72 h), then ground to a fine powder. When physiological measurements were made, plants were cut at ground level and transported to the laboratory in buckets filled with water.
2.3. Controlled Environment Experiments
A Conviron PGR15 growth chamber set at 30 °C, 60% RH, 16 h light/25 °C, 50% RH, 8 h dark was used to grow the maize model system plant, Gaspe Flint-3 (GF3) (manufacturer, city, state, country) [
41] under controlled environmental conditions. A semi-hydroponics irrigation system [
41], similar to that described [
42], was used in growth chamber and field hydroponics to attain a high level of control of N input. When ammonia was used as an N source 1 mM NH
4Cl was substituted for KNO
3 but KCl was maintained at 4 mM. GF3 was grown in 1.74 L pots and field hydroponics plants were grown in 15.14 L pots.
2.4. Controlled Environment Plant Samplings
When plants were sampled, the Turface™ (Turface/Profile Products, LLC, Buffalo Grove, IL, USA) was washed off the roots and the plant separated into shoot, roots, leaves, midribs, tassel, tillers, husk, and ear. Plant parts were dried (70 °C, 72 h) but when metabolic profiling was performed, plant parts were frozen in dry ice and lyophilized. Individual plant parts were weighed, ground to a fine powder, and a sample (30–60 mg) weighed for extraction. Fresh tissue was also extracted as leaf punches (10, 5 mm diameter). In either case, duplicate samples were extracted by Genogrinder (SPEX SamplePrep, Metuchen, NJ, USAin 500 µL acid and 500 µL base as described by Queval and Noctor [
43]. When NAD
+/NADH was quantified a small aliquot of the acid extract was heat-treated for the quantification of NADH and the remaining acid extract was quenched and used for metabolic analysis.
2.5. NAD+/NADH Measurements
The method of Queval and Noctor [
43] was used.
2.6. Total Amino Acids
An aliquot of each tissue extract was suspended in a total volume of 100 μL water and 50 μL of a solution containing 350 mM Borate buffer pH 9.5, 1% SDS, 0.5% β-mercaptoethanol (ME), and 200 μg o-phthadialdehyde was added to each well. Blank samples were treated similarly but without o-phthadialdehyde. Fluorescence (Excitation (Ex) 360 nm Emission (Em) 520 nm) was determined immediately and each complete sample was corrected with the respective blank sample. Alanine from 0 to 5 nmole in 0.5 nmole increments, were used as standards.
2.7. Total N
N of ground plant samples was converted to (NO)n equivalents by oxidation using a FlashEA 1112 series combustion analyzer (Thermo Fisher Scientific, Waltham, MA, USA) applying Association of Official Analytical Chemists (AOCS) method Ba 4e-93
2.8. Nitrate Quantification
The method of Miranda et al. [
44] was used with slight modification to correct for background anthocyanins that absorb at 540 nm in acid, and for nitrite. An equal volume of 1% sulfanilamide, 0.01% naphthalene ethylene diamine in 2 M H
3PO
4 was added and optical density at 540 nm (OD
540) was determined followed by the addition of a sample volume of saturated VCl
2 in 1 M HCl then incubated at 37 °C for 1 h. The OD
540 was again measured and the previous absorption readings used to correct for anthocyanin and nitrite after correction for differences in path lengths.
2.9. Malate Quantification
An aliquot was suspended in a total volume of 100 μL with water. 10 μL of 10 mM NADP, 5 mM MgCl2, 1 M Tris-HCl pH 7.5 was added to each well followed by 10 μL containing 0.05 units of malic enzyme (EC 1.1.1.40). Blank plates were prepared in the same fashion but 10 μL of water was added instead of malic enzyme. These were incubated for 1 h at room temperature and fluorescence (Ex 345, Em 460) was measured subtracting the blank from the sample plate. Malate standards from 0 to 5 nmoles, in 0.5 nmole increments, were used.
2.10. In Vivo Nitrate Reductase Activity—NRA (In Vivo)
In vivo nitrate reductase activity was measured similar to the method of Reed and Hageman [
45]. Leaf punches (10, 5 mm diameter) were submerged in 400 µL 50 mM KH
2PO
4-KHPO
4 pH 6.0, 300 mM sorbitol, 0.04% Trition X-100 then vacuum infiltrated. A 100 µL aliquot of a 100 mM KNO
3, 50 mM KHCO
3, 300 mM sorbitol, 50 mM KH
2PO
4-KHPO
4 pH 6.0, 0.04% Trition X-100 solution was added to each tube so the final assay concentration was 300 mM sorbitol, 50 mM KH
2PO
4-KHPO
4 pH 6.0, 0.04% Trition X-100, 20 mM KNO
3, and 10 mM KHCO
3. Tubes were incubated in the dark at 30 °C and 50 µL aliquots were remove every 30 min for 2 h. The production of nitrite was determined by adding 100 µL of 1% sulfanilamide, 2 M H
3PO
4, 0.02% naphthalene ethylene diamine, and 50 µL acetonitrile and measuring OD
540. The assay was linear for at least 3 h.
2.11. In Vivo Nitrite Reductase Activity—NiRA (In Vivo)
In vivo nitrite reductase activity was measure by the loss of nitrite from the medium. Leaf punches (5, 5 mm diameter) were submerged in 200 µL 50 mM KH2PO4-KHPO4 pH 6.0, 300 mM sorbitol, 0.04% Trition X-100 then vacuum infiltrated. A 50 µL aliquot of a 1 mM KNO2, 50 mM KHCO3, 300 mM sorbitol, 50 mM KH2PO4-KHPO4 pH 6.0, 0.04% Trition X-100 solution was added to each tube so the final assay concentration was 300 mM sorbitol, 50 mM KH2PO4-KHPO4 pH 6.0, 0.04% Trition X-100, 200 µM KNO2, and 10 mM KHCO3. Tubes were incubated at 30 °C under a bank of light emitting diodes of photosynthetic quality (Quantum Devices, Model # SL1515-470-670, manufacturer, Barneveld, WI, USA). Duplicate 10 µL aliquots were removed every 30 min for 2 h and treated with 150 µL of 1% sulfanilamide, 2 M H3PO4, 0.02% naphthalene ethylene diamine, and 50 µL acetonitrile and OD540 determined. The assay was linear for at least 3 h.
2.12. Enzyme (In Vitro) Extraction
Extraction tubes (1.2) mL were filled with 500 µL of a 1% slurry of insoluble polyvinyl polypyrrolidone (PVPP), reduced to dryness by Speedvac (SPD131DDA-115 Thermo Fisher Scientific, Waltham, MA, USA), then stored at room temperature until use. Extraction tubes were filled with 500 µL extraction medium (100 mM Tris-HCl pH 8, 10 mM cysteine, 10 µM leupeptin, 4 °C) and leaf punches (10, 5 mm diameter) were delivered to the tubes. The tubes were arranged in a 96-well plate such that surrounding tubes contained ice and were vacuum infiltrated. Tubes were ground by Genogrinder for 2, 1 min intervals. Plates were centrifuged 4000× g for 20 min at 4 °C and a 200 µL aliquot removed and place on a 2 mL bed of Sephadex G-25 previously equilibrated with 50 mM Tris-HCl pH 7.5 with the void volume removed by a 1 min 500× g centrifugation. Small molecular weight metabolites were removed from the plant extract without dilution by a second low speed centrifugation with the receiving wells containing concentrated cysteine and leupeptin to maintain 10 mM and 10 µM concentrations, respectively.
2.13. Nitrate Reductase Activity (NRA (In Vitro)) EC 1.6.6.1
Cyclic renewal of NADH was used to avoid excess NADH from interfering with color development. The assay was performed in a 20 µL volume in 384-well plates with the following component concentrations: enzyme extract; 100 mM Tris-HCl pH 7.5; 10 mM cysteine; 10 mM KNO
3; 20 µM NAD
+; 1 mM glucose-6-phosphate; and, 1 unit glucose-6-phosphate dehydrogenase-NAD
+ (EC 1.1.1.388). The reaction was started by the addition of enzyme extract and stopped at 30 min intervals by adding two volumes of 1% sulfanilamide, 2 M H
3PO
4, 0.02% naphthalene ethylene diamine. Activation state of NRA (in vitro) was estimated by including either 5 mM EDTA or 10 mM MgCl
2 in the assay. The ratio of MgCl
2 to EDTA enzyme activities was used as estimate of the activation state of NRA (in vitro) [
46].
Since the concentration of NADH was maintained at 20 µM, inhibition of color development was not statistically (p ≤ 0.05) significant, relative to the inherent variability of the experiments. If there was interference of color development it would be constant across all samples since the concentration of NADH was maintained constant. In an assay development experiment where the concentration of NAD+ was varied 0, 20, 50, 100, and 200 µM in the assay media across a range of nitrite concentrations from 0 to 100 µM, in 10 µM increments, NAD+ (NADH) significantly (p ≤ 0.05) inhibited color development starting at 50 µM NAD+ but the absorption of varying NAD+ concentrations across nitrite concentrations were parallel (equal slopes (p ≤ 0.05)), demonstrating equal levels of inhibition of color development across nitrite concentrations. This would negate the effect of variable leftover concentrations NADH on the assay since color reduction would be equal at the end of the assay irrespective of the intensity of NRA (in vitro). Also, since NAD(P)H inhibits color development by reducing the diazonium salt formed between sulfanilamide and nitrite to a hydrazone such that the azo-compound is not formed, combining sulfanilamide and naphthalene ethylene diamine increases the chance the diazonium salt reacts with the diamine rather than with NAD(P)H. Finally, the assay was further improved by removing small molecular weight molecules including oxaloacetic acid (OAA) which reduces the level of detectable nitrite over time (not shown), presumably by forming an oxime.
2.14. Nitrite Reductase Activity (NiRA (In Vitro)) EC 1.7.7.1
NiRA (in vitro) was assayed in a 200 µL volume containing enzyme extract, 100 mM KH2PO4-KHPO4 pH 6.9, 400 µM KNO2, 10 mM methyl viologen, 30 mM Na2S2O4 and 30 mM KHCO3. The assay was started by addition of Na2S2O4 + KHCO3 and incubated at 30 °C. Aliquots (50 µL) were removed after 0, 10 and 20 min and methyl viologen oxidized by shaking. The loss of nitrite was determined after methyl viologen oxidation by adding 150 µL of 1% sulfanilamide, 2 M H3PO4, 0.02% naphthalene ethylene diamine and 50 µL acetonitrile and determining OD540.
2.15. Phosphoenolpyruvate Carboxylase (PEP Carboxylase) EC 4.1.1.31
PEP carboxylase activity was measured by the loss of NADH coupled to the conversion of OAA, formed, to malate via malate dehydrogenase (EC 1.1.1.37). The extract was diluted 10× with water and 10 µL was used in a 200 µL assay volume which was 25 mM HEPES pH 7.5, 20% ethylene glycol, 5 mM MgCl2, 10 mM KHCO3, 2 mM phosphoenolpyruvate (PEP), 40 μM NADH, and 1 unit malate dehydrogenase (EC 1.1.1.37). Blanks contained water instead of PEP. The reaction was started by the addition of PEP and fluorescence (Ex 345, Em 440) was measure every minute for 10 min. The linear portion of the reduction in fluorescence was used to determine enzyme activity. Removal of OAA from the enzyme extract was crucial to maintain a constant baseline for this assay.
2.16. Malate Enzyme (ME) EC 1.1.1.40
Malate enzyme activity was determined by measuring NADPH formed as a result of malate conversion to pyruvate and HCO3−. The extract was diluted 10× with water and 10 µL was used in a 200 µL assay volume which was 50 mM HEPES pH 7.0, 1 mM NADP+, 5 mM MgCl2 and 2 mM malate. The reaction was started by adding malate; the blanks had water instead of malate. Fluorescence (Ex 345, Em 440) was measured every minute for 10 min. The linear portion of the increase in fluorescence was used to determine enzyme activity.
2.17. Protein
The method of Bradford [
47] was used.
2.18. Ear Growth Response to Switching Nitrate Concentrations at Different Stages of Development
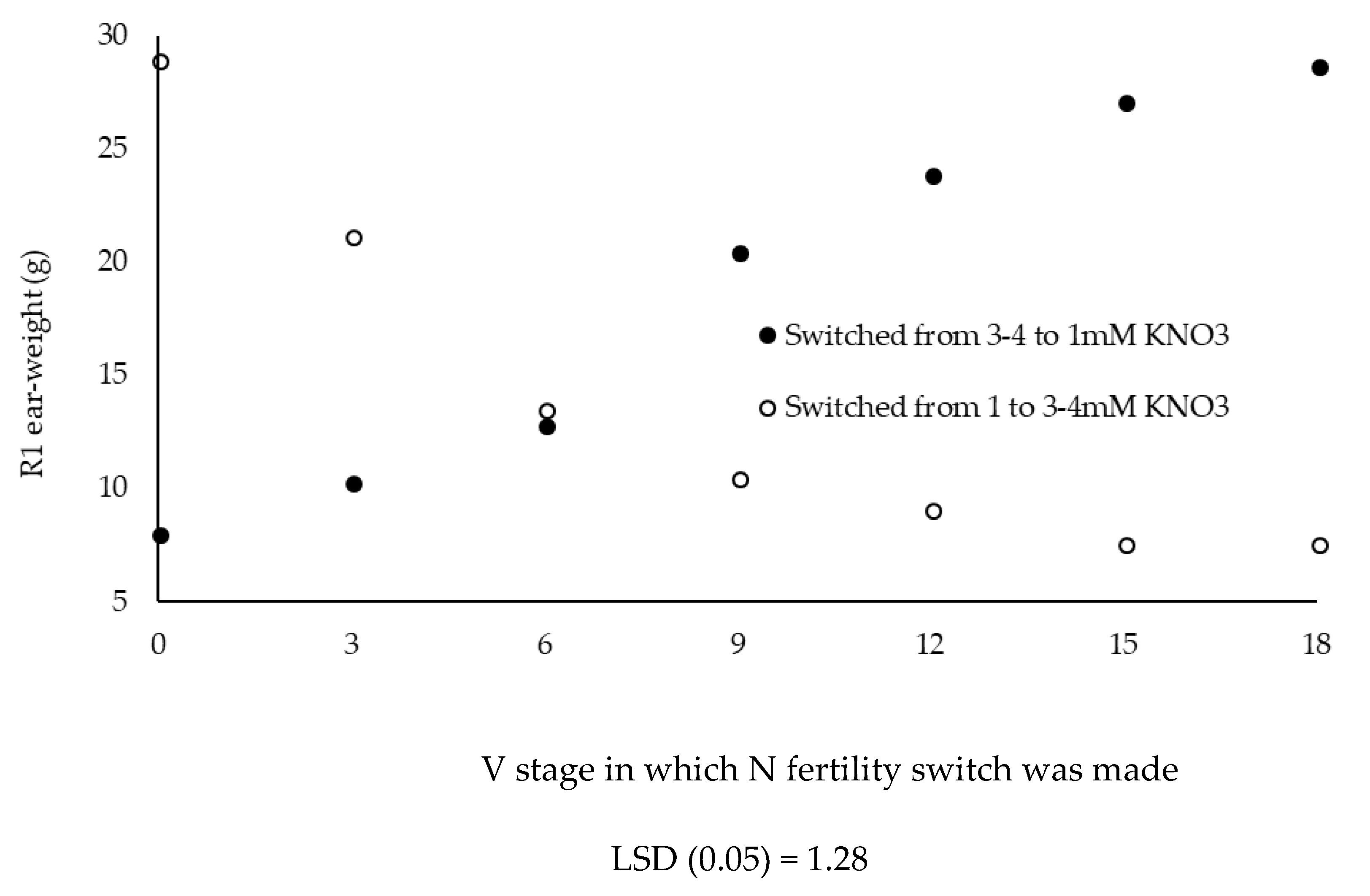
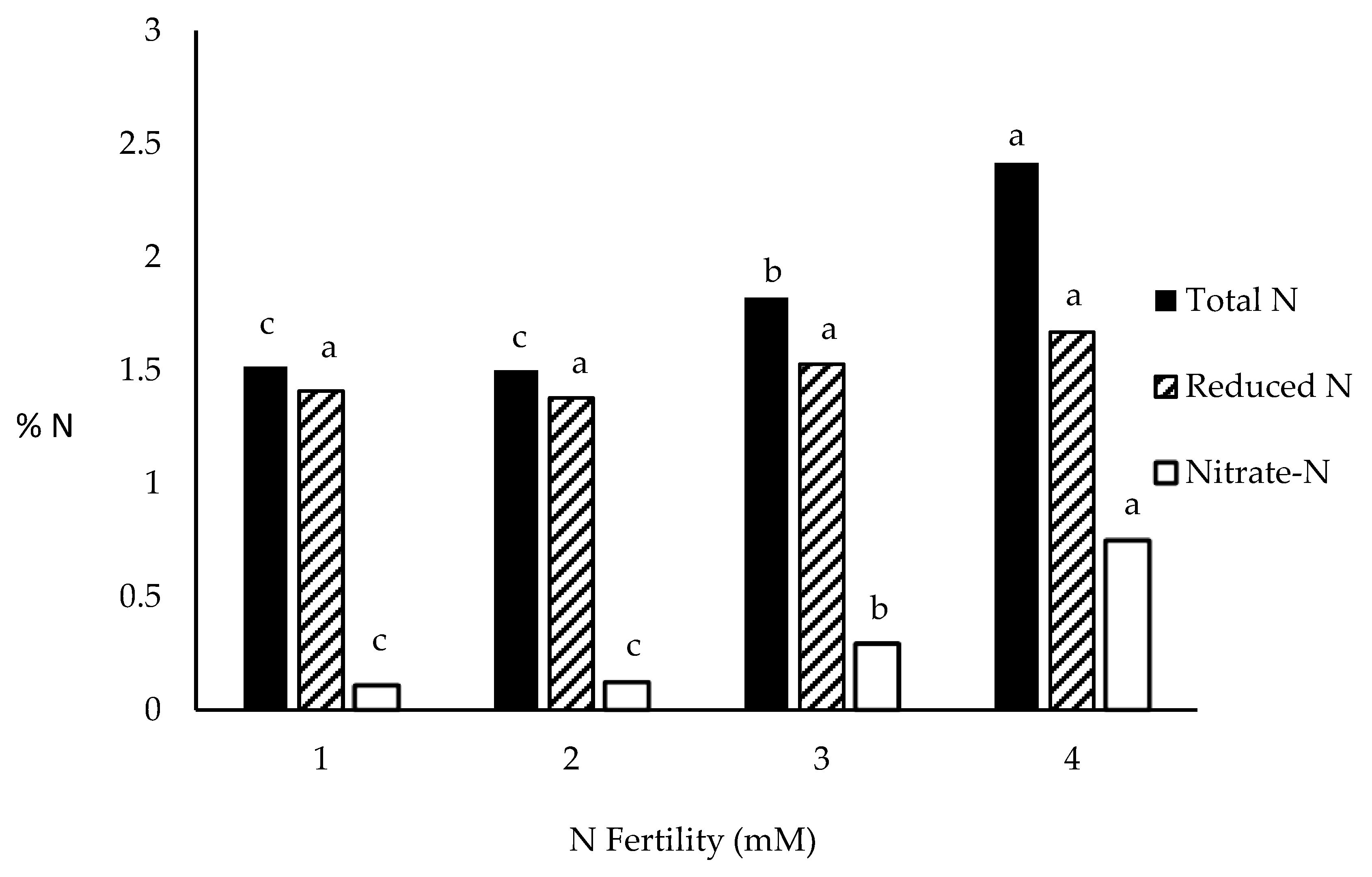
The DuPont-Pioneer hybrid, 33W84, was grown in a field semi-hydroponics system previously described. Plants were grown in either 1 or 3 mM KNO3 as the N source and at V0 (control), V3, V6, V9, V12, V15, and V18 (tassel emergence) plants growing in 1 mM KNO3 were switched to 3 mM KNO3 and plants growing in 3 mM KNO3 switched to 1 mM KNO3. Ear shoots were bagged as the shoots emerged to avoid seed set. Plants were sampled at R1 and separated into ear and remaining vegetative plant then dried (70 °C, 72 h). There were 10 replicates of each treatment combination. The experiment was repeated a second season but the concentrations of KNO3 used were 1 and 4 mM KNO3.
2.19. Response to Nitrate and Ammonia Nutrition during Later Stages of Ear Development
The DuPont-Pioneer hybrid, 33W84, was grown in a field semi-hydroponics system and ear dry weight response to ammonia tested for two consecutive growing seasons. In the first season plants were grown in nutrient medium containing 1 and 3 mM KNO3 and converted to 1 mM NH4Cl at V12 and V15 or maintained at 1 and 3 mM KNO3. The plants were treated similarly in the subsequent year, except the 3 mM KNO3 treatments were replaced by 4 mM KNO3 treatments. Ear shoots were bagged as the shoots emerged to avoid seed set. Plants were harvested at R1 and separated into ear and remaining vegetative plant then dried (70 °C, 72 h). There were 10 replicates of each treatment combination.
GF3 was grown semi-hydroponically in either 1 or 2 mM KNO3 as the sole N source. At 23 days after emergence (DAE), plants growing in 1 mM KNO3 fertility were switched to 2 mM KNO3, to 1 mM NH4Cl, or maintained at 1 mM KNO3. Plants growing in 2 mM KNO3 were switched to 1 mM KNO3, to 1 mM NH4Cl, or maintained at 2 mM KNO3. At 30 DAE (R1) plants were separated into ears and remaining plant biomass then dried by lyophilization. Ear weight, remaining plant biomass, and total N were determined.
GF3 was grown semi-hydroponically under controlled environment in 1 mM KNO3 up to 22 DAE and the plants were switched to 1 mM NH4Cl, 10 mM KNO3, or maintained at 1 mM KNO3. Ear and plant biomass was determined of all treatments at 23, 25, and 27 DAE along with total N.
2.20. Vector Construction, Plant Transformation and Transgene Expression Analysis
A 230-base pair (bp) fragment of maize NADH ubiquinone oxidoreductase (GRMZM2G024484) including the 5’-untranslated region (UTR) and part of the coding sequence was PCR-cloned to make an RNAi construct. An intron from ST-LS1 was added between the two inverted repeats of NADH ubiquinone oxidoreductase. Maize PEP carboxylase gene promoter and sorghum actin gene terminator were used to silence NADH ubiquinone oxidoreductase in mesophyll cells. This cassette was linked to LTP2:DS-RED2:PIN II TERM as a seed marker for transgenic seed as described [
48]. The vector construction and maize transformation were carried out as described previously [
48,
49]. Multiple lines were generated. Single-copy T-DNA integration lines that expressed the transgene were selected for advancement to greenhouse or field test.
2.21. NADH-Ubiquinone Oxidoreductase 51 kDa Subunit (Complex I) Expression
RNA was isolated using a Qiagen RNeasy kit followed by Invitrogen Turbo DNA free kit to remove contaminating DNA. Transcript quantification was performed using Biorad’s iTaq™ Universal One-Step RT-qPCR (Bio-Rad, Hercules, CA. USA) on a Biorad CFX96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA. USA). NADH-ubiquinone oxidoreductase 51 kDa subunit (Complex I) was quantified using the forward primer CAACTCTGGAACCAAGCTCTAT and the reverse primer GAGCAACTCCTTCAGAGGTATG. Transcriptional corepressor LEUNIG was used as a reference using CATCGACACCTTCCACTCATAC as the forward and TCCGTCAGAGCCAAACATTAC as the reverse primer.
2.22. Multi-Location Field Yield Trails
Transgenic Complex I RNAi events and the corresponding null were tested in 5 locations managed for optimal grain yields with four replicates at Johnston IA, Woodland CA, Plainview TX, and Corning AR, and three replicates in Garden City, KS. The field trial and statistical analysis were conducted as described previously [
48]. Grain yield was calculated and adjusted to a standard moisture of 155 g kg
−1. Yield was predicted using Best Linear Unbiased Predictor (BLUP).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}