Genomic Selection in Cereal Breeding


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Set-Up of Genomic Selection
2.1. Size of Training Data
2.2. Relatedness between Training and Test Individuals
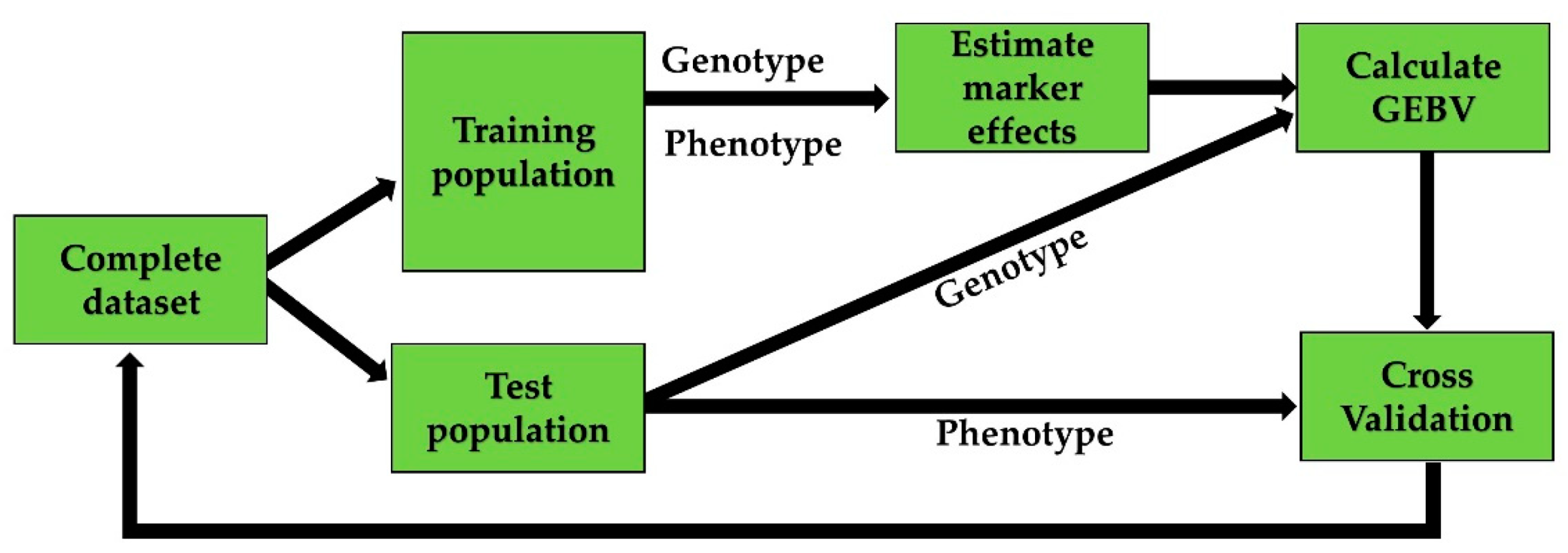
2.3. Cross-Validation Strategies
2.4. Marker Density
2.5. Prediction of Genomic Estimated Breeding Values (GEBV)
3. Strategies for Implementation of Genomic Selection
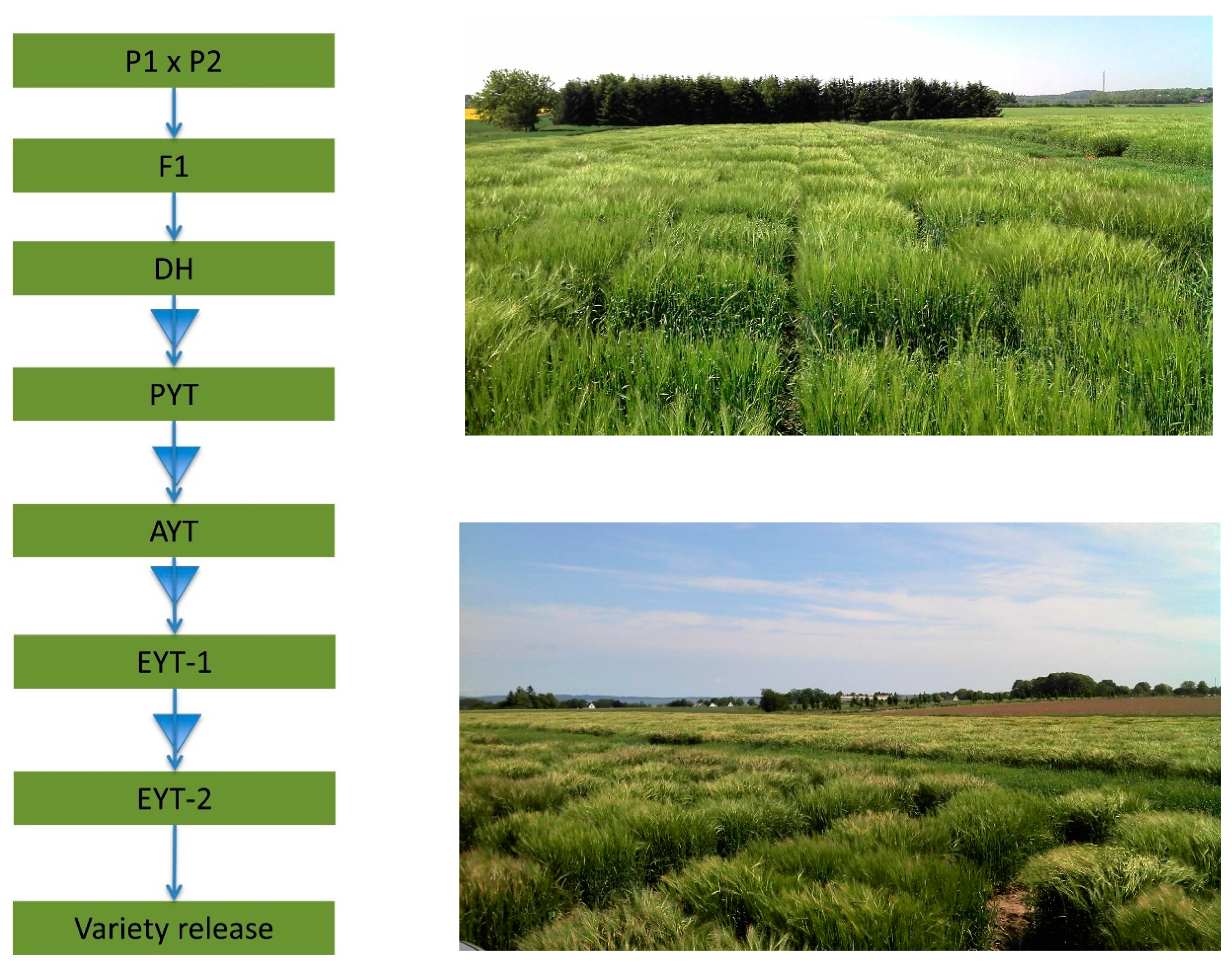
3.1. Basic Breeding Scheme in Cereals
3.2. Across-Breeding Cycle Genomic Selection
3.3. Within-Breeding Cycle Genomic Selection
3.4. Genomic Selection Using Untested Parents for Breeding
4. Pedigree Information
5. Use of Additive and Non-Additive Genetic Effects
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lande, R.; Thompson, R. Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 1990, 124, 743–756. [Google Scholar] [PubMed]
- Heffner, E.L.; Sorrells, M.E.; Jannink, J.L. Genomic selection for crop improvement. Crop Sci. 2009, 49, 1–12. [Google Scholar] [CrossRef]
- Laidig, F.; Piepho, H.P.; Rentel, D.; Drobek, T.; Meyer, U.; Huesken, A. Breeding progress, environmental variation and correlation of winter wheat yield and quality traits in German official variety trials and on-farm during 1983–2014. Theor. Appl. Genet. 2017, 130, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.C.; Crossa, J.; Velu, G.; Huerta-Espino, J.; Vargas, M.; Payne, T.S.; Singh, R.P. Genetic gains for grain yield in CIMMYT spring bread wheat across international environments. Crop Sci. 2012, 52, 1522–1533. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Su, G.; Guldbrandtsen, B.; Gregersen, V.R.; Lund, M.S. Preliminary investigation on reliability of genomic estimated breeding values in the Danish Holstein population. J. Dairy Sci. 2010, 93, 1175–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffner, E.L.; Lorenz, A.J.; Jannink, J.L.; Sorrells, M.E. Plant breeding with genomic selection: Gain per unit time and cost. Crop Sci. 2010, 50, 1681–1690. [Google Scholar] [CrossRef]
- Bernardo, R.; Yu, J.M. Prospects for genomewide selection for quantitative traits in maize. Crop Sci. 2007, 47, 1082–1090. [Google Scholar] [CrossRef]
- Bernardo, R. A model for marker-assisted selection among single crosses with multiple genetic markers. Theor. Appl. Genet. 1998, 97, 473–478. [Google Scholar] [CrossRef]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Perez, P.; Hickey, J.; Burgueno, J.; Ornella, L.; Ceron-Rojas, J.; Zhang, X.; Dreisigacker, S.; Babu, R.; Li, Y.; et al. Genomic prediction in CIMMYT maize and wheat breeding programs. Heredity 2014, 112, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, A.J.; Smith, K.P.; Jannink, J.L. Potential and optimization of genomic selection for fusarium head blight resistance in six-row barley. Crop Sci. 2012, 52, 1609–1621. [Google Scholar] [CrossRef]
- Asoro, F.G.; Newell, M.A.; Beavis, W.D.; Scott, M.P.; Tinker, N.A.; Jannink, J.L. Genomic, marker-assisted, and pedigree-BLUP selection methods for beta-glucan concentration in elite oat. Crop Sci. 2013, 53, 1894–1906. [Google Scholar] [CrossRef]
- Lariepe, A.; Moreau, L.; Laborde, J.; Bauland, C.; Mezmouk, S.; Decousset, L.; Mary-Huard, T.; Fievet, J.B.; Gallais, A.; Dubreuil, P.; et al. General and specific combining abilities in a maize (Zea mays L.) test-cross hybrid panel: Relative importance of population structure and genetic divergence between parents. Theor. Appl. Genet. 2017, 130, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Riedelsheimer, C.; Czedik-Eysenberg, A.; Grieder, C.; Lisec, J.; Technow, F.; Sulpice, R.; Altmann, T.; Stitt, M.; Willmitzer, L.; Melchinger, A.E. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat. Genet. 2012, 44, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Song, J.Y.; Carver, B.F.; Powers, C.; Yan, L.L.; Klapste, J.; El-Kassaby, Y.A.; Chen, C. Practical application of genomic selection in a doubled-haploid winter wheat breeding program. Mol. Breed. 2017, 37, 117. [Google Scholar] [CrossRef] [PubMed]
- De los Campos, G.; Hickey, J.M.; Pong-Wong, R.; Daetwyler, H.D.; Calus, M.P.L. Whole-genome regression and prediction methods applied to plant and animal breeding. Genetics 2013, 193, 327–345. [Google Scholar] [CrossRef]
- Nielsen, N.H.; Jahoor, A.; Jensen, D.; Orabi, J.; Cericola, F.; Edriss, V.; Jensen, J. Genomic prediction of seed quality traits using advanced barley breeding lines. PLoS ONE 2016, 11, e0164494. [Google Scholar] [CrossRef]
- Cericola, F.; Jahoor, A.; Orabi, J.; Andersen, J.R.; Janss, L.L.; Jensen, J. Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information. A case of study in advanced wheat breeding lines. PLoS ONE 2017, 12, e0169606. [Google Scholar] [CrossRef]
- Norman, A.; Taylor, J.; Edwards, J.; Kuchel, H. Optimising genomic selection in wheat: Effect of marker density, population size and population structure on prediction accuracy. G3-Genes Genomes Genet. 2018, 8, 2889–2899. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E. Accuracy of breeding values of ‘unrelated’ individuals predicted by dense SNP genotyping. Genet. Sel. Evol. 2009, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Tetens, J.; Seefried, F.R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet. Sel. Evol. 2010, 42, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidro, J.; Jannink, J.L.; Akdemir, D.; Poland, J.; Heslot, N.; Sorrells, M.E. Training set optimization under population structure in genomic selection. Theor. Appl. Genet. 2015, 128, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Gowda, M.; Zhao, Y.; Wuerschum, T.; Longin, C.F.H.; Miedaner, T.; Ebmeyer, E.; Schachschneider, R.; Kazman, E.; Schacht, J.; Martinant, J.P.; et al. Relatedness severely impacts accuracy of marker-assisted selection for disease resistance in hybrid wheat. Heredity 2014, 112, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, A.J.; Smith, K.P. Adding genetically distant individuals to training populations reduces genomic prediction accuracy in barley. Crop Sci. 2015, 55, 2657–2667. [Google Scholar] [CrossRef]
- Kristensen, P.S.; Jahoor, A.; Andersen, J.R.; Cericola, F.; Orabi, J.; Janss, L.L.; Jensen, J. Genome-wide association studies and comparison of models and cross-validation strategies for genomic prediction of quality traits in advanced winter wheat breeding lines. Front. Plant Sci. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.R. Best linear unbiased estimation and prediction under a selection model. Biometrics 1975, 31, 423–447. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Gianola, D. Priors in whole-genome regression: The Bayesian alphabet returns. Genetics 2013, 194, 573–596. [Google Scholar] [CrossRef]
- Heslot, N.; Yang, H.P.; Sorrells, M.E.; Jannink, J.L. Genomic selection in plant breeding: A comparison of models. Crop Sci. 2012, 52, 146–160. [Google Scholar] [CrossRef]
- Maltecca, C.; Parker, K.L.; Cassady, J.P. Application of multiple shrinkage methods to genomic predictions. J. Anim. Sci. 2012, 90, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.B.E.; Cuevas, J.; Couto, E.G.D.; Perez-Rodriguez, P.; Jarquin, D.; Fritsche-Neto, R.; Burgueno, J.; Crossa, J. Genomic-enabled prediction in maize using kernel models with genotype x environment interaction. G3-Genes Genomes Genet. 2017, 7, 1995–2014. [Google Scholar] [CrossRef]
- Cuevas, J.; Granato, I.; Fritsche-Neto, R.; Montesinos-Lopez, O.A.; Burgueno, J.; Bandeira, M.B.E.; Crossa, J. Genomic-enabled prediction kernel models with random intercepts for multi-environment trials. G3-Genes Genomes Genet. 2018, 8, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; de los Campos, G.; Perez-Enciso, M. Can deep learning improve genomic prediction of complex human traits? Genetics 2018, 210, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Christensen, O.F.; Janss, L.; Lund, M.S. Comparison of genomic predictions using genomic relationship matrices built with different weighting factors to account for locus-specific variances. J. Dairy Sci. 2014, 97, 6547–6559. [Google Scholar] [CrossRef] [Green Version]
- Park, T.; Casella, G. The Bayesian LASSO. J. Am. Stat. Assoc. 2008, 103, 681–686. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef]
- Erbe, M.; Hayes, B.J.; Matukumalli, L.K.; Goswami, S.; Bowman, P.J.; Reich, C.M.; Mason, B.A.; Goddard, M.E. Improving accuracy of genomic predictions within and between dairy cattle breeds with imputed high-density single nucleotide polymorphism panels. J. Dairy Sci. 2012, 95, 4114–4129. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.; Jiang, D.; Li, D.D.; Yang, R.Q.; Fu, W.X.; Pu, L.J.; Gao, H.J.; Wang, G.H.; Yu, L.Y. Improved LASSO priors for shrinkage quantitative trait loci mapping. Theor. Appl. Genet. 2012, 124, 1315–1324. [Google Scholar] [CrossRef]
- Hoggart, C.J.; Whittaker, J.C.; De Iorio, M.; Balding, D.J. Simultaneous analysis of all SNPs in genome-wide and re-Sequencing association studies. PLoS Genet. 2008, 4, e1000130. [Google Scholar] [CrossRef]
- Gao, H.; Su, G.; Janss, L.; Zhang, Y.; Lund, M.S. Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J. Dairy Sci. 2013, 96, 4678–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbyla, K.L.; Hayes, B.J.; Bowman, P.J.; Goddard, M.E. Accuracy of genomic selection using stochastic search variable selection in Australian Holstein Friesian dairy cattle. Genet. Res. 2009, 91, 307–311. [Google Scholar] [CrossRef] [PubMed]
- George, E.I.; McCulloch, R.E. Variable selection via Gibbs sampling. J. Am. Stat. Assoc. 1993, 88, 881–889. [Google Scholar] [CrossRef]
- Kapell, D.; Sorensen, D.; Su, G.S.; Janss, L.L.G.; Ashworth, C.J.; Roehe, R. Efficiency of genomic selection using Bayesian multi-marker models for traits selected to reflect a wide range of heritabilities and frequencies of detected quantitative traits loci in mice. BMC Genet. 2012, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Abraham, G.; Tye-Din, J.A.; Bhalala, O.G.; Kowalczyk, A.; Zobel, J.; Inouye, M. Accurate and robust genomic prediction of celiac disease using statistical learning. PLoS Genet. 2014, 10, e1004137. [Google Scholar] [CrossRef]
- Piepho, H.P. Ridge regression and extensions for genomewide selection in maize. Crop Sci. 2009, 49, 1165–1176. [Google Scholar] [CrossRef]
- Endelman, J.B. Ridge regression and other kernels for genomic selection with R package rrBLUP. Plant Genome 2011, 4, 250–255. [Google Scholar] [CrossRef]
- Waldmann, P. Genome-wide prediction using Bayesian additive regression trees. Genet. Sel. Evol. 2016, 48, 42. [Google Scholar] [CrossRef]
- Gianola, D.; van Kaam, J. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of quantitative traits. Genetics 2008, 178, 2289–2303. [Google Scholar] [CrossRef]
- Morota, G.; Koyama, M.; Rosa, G.J.M.; Weigel, K.A.; Gianola, D. Predicting complex traits using a diffusion kernel on genetic markers with an application to dairy cattle and wheat data. Genet. Sel. Evol. 2013, 45, 17. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Reif, J.C. Modeling epistasis in genomic selection. Genetics 2015, 201, 759–768. [Google Scholar] [CrossRef]
- Martini, J.W.R.; Wimmer, V.; Erbe, M.; Simianer, H. Epistasis and covariance: How gene interaction translates into genomic relationship. Theor. Appl. Genet. 2016, 129, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.; Carriquiry, A.L.; Beavis, W.D. Parametric and nonparametric statistical methods for genomic selection of traits with additive and epistatic genetic architectures. G3-Genes Genomes Genet. 2014, 4, 1027–1046. [Google Scholar] [CrossRef]
- Du, C.; Wei, J.L.; Wang, S.B.; Jia, Z.Y. Genomic selection using principal component regression. Heredity 2018, 121, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Bassi, F.M.; Bentley, A.R.; Charmet, G.; Ortiz, R.; Crossa, J. Breeding schemes for the implementation of genomic selection in wheat (Triticum spp.). Plant Sci. 2016, 242, 23–36. [Google Scholar] [CrossRef]
- Gaynor, R.C.; Gorjanc, G.; Bentley, A.R.; Ober, E.S.; Howell, P.; Jackson, R.; Mackay, I.J.; Hickey, J.M. A two-part strategy for using genomic selection to develop inbred lines. Crop Sci. 2017, 57, 2372–2386. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Accelerating improvement of livestock with genomic selection. Annu. Rev. Anim. Biosci. 2013, 1, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Kollers, S.; Maasberg-Prelle, A.; Grosser, J.; Schinkel, B.; Tomerius, A.; Graner, A.; Korzun, V. Prediction of malting quality traits in barley based on genome-wide marker data to assess the potential of genomic selection. Theor. Appl. Genet. 2016, 129, 203–213. [Google Scholar] [CrossRef]
- Michel, S.; Kummer, C.; Gallee, M.; Hellinger, J.; Ametz, C.; Akgol, B.; Epure, D.; Gungor, H.; Loschenberger, F.; Buerstmayr, H. Improving the baking quality of bread wheat by genomic selection in early generations. Theor. Appl. Genet. 2018, 131, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Ametz, C.; Gungor, H.; Akgol, B.; Epure, D.; Grausgruber, H.; Loschenberger, F.; Buerstmayr, H. Genomic assisted selection for enhancing line breeding: Merging genomic and phenotypic selection in winter wheat breeding programs with preliminary yield trials. Theor. Appl. Genet. 2017, 130, 363–376. [Google Scholar] [CrossRef]
- Michel, S.; Ametz, C.; Gungor, H.; Epure, D.; Grausgruber, H.; Loschenberger, F.; Buerstmayr, H. Genomic selection across multiple breeding cycles in applied bread wheat breeding. Theor. Appl. Genet. 2016, 129, 1179–1189. [Google Scholar] [CrossRef] [Green Version]
- Schaeffer, L.R. Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 2006, 123, 218–223. [Google Scholar] [CrossRef]
- Longin, C.F.H.; Mi, X.F.; Wurschum, T. Genomic selection in wheat: Optimum allocation of test resources and comparison of breeding strategies for line and hybrid breeding. Theor. Appl. Genet. 2015, 128, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D.; et al. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.A.; VanRaden, P.M.; Norman, H.D.; Grosu, H. A 100-Year Review: Methods and impact of genetic selection in dairy cattle—From daughter–dam comparisons to deep learning algorithms. J. Dairy Sci. 2017, 100, 10234–10250. [Google Scholar] [CrossRef] [PubMed]
- Juliana, P.; Singh, R.P.; Singh, P.K.; Crossa, J.; Huerta-Espino, J.; Lan, C.X.; Bhavani, S.; Rutkoski, J.E.; Poland, J.A.; Bergstrom, G.C.; et al. Genomic and pedigree-based prediction for leaf, stem, and stripe rust resistance in wheat. Theor. Appl. Genet. 2017, 130, 1415–1430. [Google Scholar] [CrossRef]
- Burgueno, J.; de los Campos, G.; Weigel, K.; Crossa, J. Genomic prediction of breeding values when modeling genotype x environment interaction using pedigree and dense molecular markers. Crop Sci. 2012, 52, 707–719. [Google Scholar] [CrossRef]
- Legarra, A.; Aguilar, I.; Misztal, I. a relationship matrix including full pedigree and genomic information. J. Dairy Sci. 20009, 92, 4656–4663. [Google Scholar] [CrossRef]
- Christensen, O.F.; Lund, M.S. Genomic prediction when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef] [Green Version]
- Perez-Rodriguez, P.; Crossa, J.; Rutkoski, J.; Poland, J.; Singh, R.; Legarra, A.; Autrique, E.; de los Campos, G.; Burgueno, J.; Dreisigacker, S. Single-step genomic and pedigree genotype x environment interaction models for predicting wheat lines in international environments. Plant Genome 2017, 10. [Google Scholar] [CrossRef]
- Bouvet, J.M.; Makouanzi, G.; Cros, D.; Vigneron, P. Modeling additive and non-additive effects in a hybrid population using genome-wide genotyping: Prediction accuracy implications. Heredity 2016, 116, 146–157. [Google Scholar] [CrossRef] [PubMed]
- El-Dien, O.G.; Ratcliffe, B.; Klapste, J.; Porth, I.; Chen, C.; El-Kassaby, Y.A. Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3-Genes Genomes Genet. 2016, 6, 743–753. [Google Scholar] [CrossRef]
- Perez-Rodriguez, P.; Gianola, D.; Gonzalez-Camacho, J.M.; Crossa, J.; Manes, Y.; Dreisigacker, S. Comparison between linear and non-parametric regression models for genome-enabled prediction in wheat. G3-Genes Genomes Genet. 2012, 2, 1595–1605. [Google Scholar] [CrossRef] [PubMed]
- Jarquin, D.; Crossa, J.; Lacaze, X.; Du Cheyron, P.; Daucourt, J.; Lorgeou, J.; Piraux, F.; Guerreiro, L.; Perez, P.; Calus, M.; et al. A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor. Appl. Genet. 2014, 127, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.; Crossa, J.; Montesinos-Lopez, O.A.; Burgueno, J.; Perez-Rodriguez, P.; de los Campos, G. Bayesian genomic prediction with genotype x environment interaction kernel models. G3-Genes Genomes Genet. 2017, 7, 41–53. [Google Scholar] [CrossRef]
- Jarquin, D.; da Silva, C.L.; Gaynor, R.C.; Poland, J.; Fritz, A.; Howard, R.; Battenfield, S.; Crossa, J. Increasing genomic-enabled predictionaccuracy by modeling genotype x environment interactions in Kansas wheat. Plant Genome 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.; Jarquin, D.; Crossa, J.; Reynolds, M. Genomic-enabled prediction accuracies increased by modeling genotype x environment interaction in durum wheat. Plant Genome 2018, 11. [Google Scholar] [CrossRef]
- Lopez-Cruz, M.; Crossa, J.; Bonnett, D.; Dreisigacker, S.; Poland, J.; Jannink, J.L.; Singh, R.P.; Autrique, E.; de los Campos, G. Increased prediction accuracy in wheat breeding trials using a marker x environment interaction genomic selection model. G3-Genes Genomes Genet. 2015, 5, 569–582. [Google Scholar] [CrossRef]
- Sukumaran, S.; Crossa, J.; Jarquin, D.; Reynolds, M. Pedigree-based prediction models with genotype x environment interaction in multienvironment trials of CIMMYT wheat. Crop Sci. 2017, 57, 1865–1880. [Google Scholar] [CrossRef]
- Zhong, S.Q.; Dekkers, J.C.M.; Fernando, R.L.; Jannink, J.L. Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines: A barley case study. Genetics 2009, 182, 355–364. [Google Scholar] [CrossRef]
- Wu, X.; Lund, M.S.; Sun, D.; Zhang, Q.; Su, G. Impact of relationships between test and training animals and among training animals on reliability of genomic prediction. J. Anim. Breed. Genet. 2015, 132, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Thavamanikumar, S.; Dolferus, R.; Thumma, B.R. Comparison of genomic selection models to predict flowering time and spike grain number in two hexaploid wheat doubled haploid populations. G3-Genes Genomes Genet. 2015, 5, 1991–1998. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertsen, C.D.; Hjortshøj, R.L.; Janss, L.L. Genomic Selection in Cereal Breeding. Agronomy 2019, 9, 95. https://doi.org/10.3390/agronomy9020095
Robertsen CD, Hjortshøj RL, Janss LL. Genomic Selection in Cereal Breeding. Agronomy. 2019; 9(2):95. https://doi.org/10.3390/agronomy9020095
Chicago/Turabian StyleRobertsen, Charlotte D., Rasmus L. Hjortshøj, and Luc L. Janss. 2019. "Genomic Selection in Cereal Breeding" Agronomy 9, no. 2: 95. https://doi.org/10.3390/agronomy9020095
APA StyleRobertsen, C. D., Hjortshøj, R. L., & Janss, L. L. (2019). Genomic Selection in Cereal Breeding. Agronomy, 9(2), 95. https://doi.org/10.3390/agronomy9020095