Complement as a Therapeutic Target in Systemic Autoimmune Diseases


Abstract
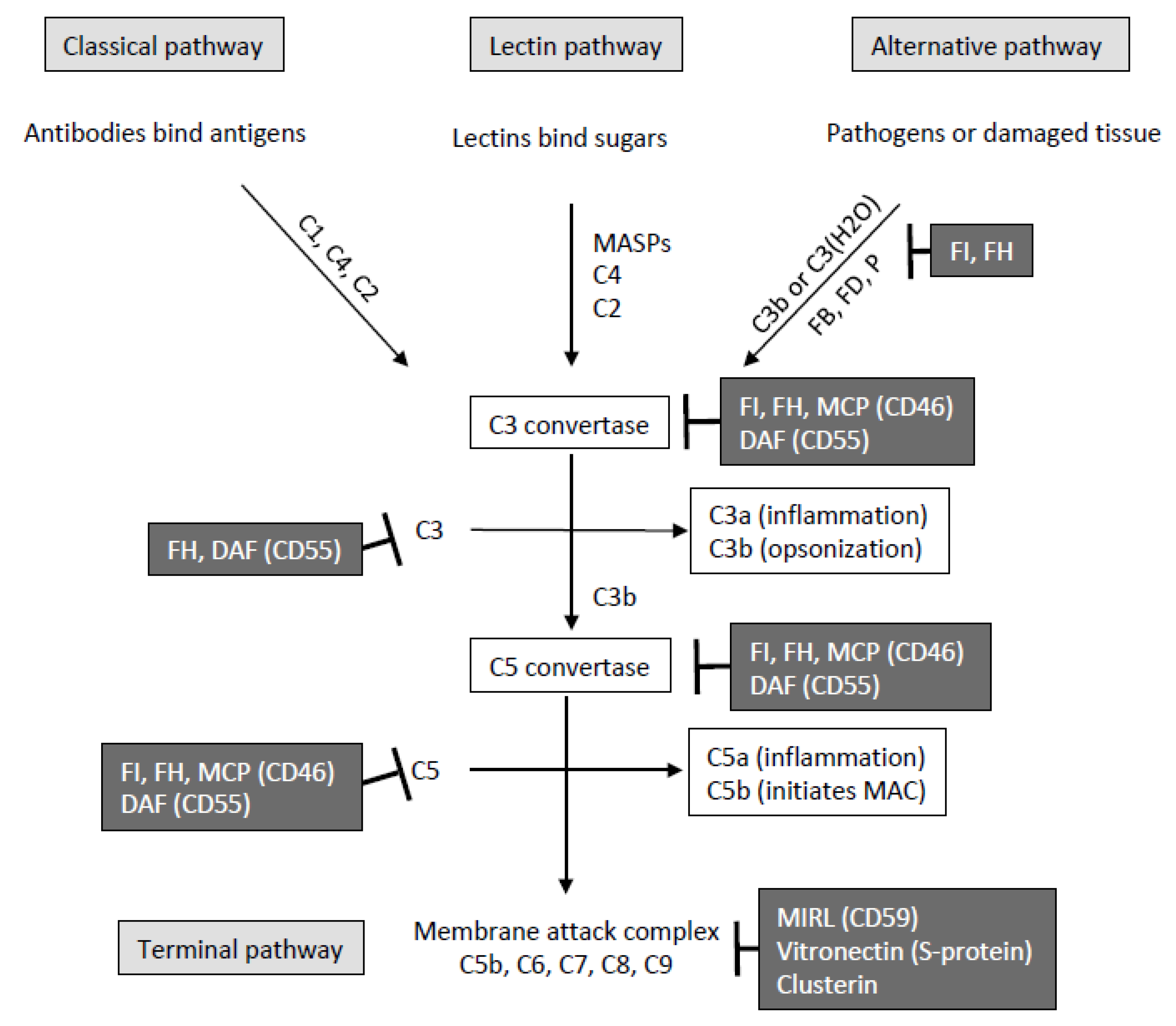
:1. Introduction
2. Systemic Lupus Erythematosus
3. Antiphospholipid Syndrome
4. Sjögren’s Syndrome
5. Rheumatoid Arthritis
6. ANCA Associated Vasculitis
7. Other
8. The Complement System as a Therapeutic Target
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement Activation, Regulation, and Molecular Basis for Complement-Related Diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Reis, E.S.; Lambris, J.D. Complement in Disease: A Defence System Turning Offensive. Nat. Rev. Nephrol. 2016, 12, 383–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.-H.; Tan, L.A.; Carroll, M.V.; Gentle, M.E.; Sim, R.B. Target Pattern Recognition by Complement Proteins of the Classical and Alternative Pathways. Adv. Exp. Med. Biol. 2009, 653, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Dodds, A.W.; Sim, R.B.; Porter, R.R.; Kerr, M.A. Activation of the First Component of Human Complement (C1) by Antibody-Antigen Aggregates. Biochem. J. 1978, 175, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlaud, G.J.; Gaboriaud, C.; Thielens, N.M.; Rossi, V. Structural Biology of C1. Biochem. Soc. Trans. 2002, 30, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, T.R.; Thiel, S.; Andersen, G.R. Toward a Structure-Based Comprehension of the Lectin Pathway of Complement. Mol. Immunol. 2013, 56, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobó, J.; Kocsis, A.; Gál, P. Be on Target: Strategies of Targeting Alternative and Lectin Pathway Components in Complement-Mediated Diseases. Front. Immunol. 2018, 9, 1851. [Google Scholar] [CrossRef] [PubMed]
- Troldborg, A.; Hansen, A.; Hansen, S.W.K.; Jensenius, J.C.; Stengaard-Pedersen, K.; Thiel, S. Lectin Complement Pathway Proteins in Healthy Individuals. Clin. Exp. Immunol. 2017, 188, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Holers, V.M. The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. Baltim. Md. 1950 2006, 176, 1305–1310. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, P.J. The Amplification Loop of the Complement Pathways. Adv. Immunol. 2009, 104, 115–149. [Google Scholar] [CrossRef]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The Role of the Anaphylatoxins in Health and Disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef] [Green Version]
- Markiewski, M.M.; Lambris, J.D. The Role of Complement in Inflammatory Diseases from behind the Scenes into the Spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipfel, P.F.; Skerka, C. Complement Regulators and Inhibitory Proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Lesher, A.M.; Song, W.-C. Review: Complement and Its Regulatory Proteins in Kidney Diseases. Nephrol. Carlton Vic. 2010, 15, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A Key System for Immune Surveillance and Homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaboriaud, C.; Ling, W.L.; Thielens, N.M.; Bally, I.; Rossi, V. Deciphering the Fine Details of C1 Assembly and Activation Mechanisms: “Mission Impossible”? Front. Immunol. 2014, 5, 565. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Esparza, A.; Kalchishkova, N.; Kurbasic, E.; Jiang, W.G.; Blom, A.M. The Novel Complement Inhibitor Human CUB and Sushi Multiple Domains 1 (CSMD1) Protein Promotes Factor I-Mediated Degradation of C4b and C3b and Inhibits the Membrane Attack Complex Assembly. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 5083–5093. [Google Scholar] [CrossRef] [PubMed]
- Serna, M.; Giles, J.L.; Morgan, B.P.; Bubeck, D. Structural Basis of Complement Membrane Attack Complex Formation. Nat. Commun. 2016, 7, 10587. [Google Scholar] [CrossRef] [Green Version]
- Tabib, A.; Karbian, N.; Mevorach, D. Demyelination, Strokes, and Eculizumab: Lessons from the Congenital CD59 Gene Mutations. Mol. Immunol. 2017, 89, 69–72. [Google Scholar] [CrossRef]
- Kemper, C.; Köhl, J. Novel Roles for Complement Receptors in T Cell Regulation and Beyond. Mol. Immunol. 2013, 56, 181–190. [Google Scholar] [CrossRef]
- Holers, V.M. Complement and Its Receptors: New Insights into Human Disease. Annu. Rev. Immunol. 2014, 32, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 2016, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botto, M.; Dell’Agnola, C.; Bygrave, A.E.; Thompson, E.M.; Cook, H.T.; Petry, F.; Loos, M.; Pandolfi, P.P.; Walport, M.J. Homozygous C1q Deficiency Causes Glomerulonephritis Associated with Multiple Apoptotic Bodies. Nat. Genet. 1998, 19, 56–59. [Google Scholar] [CrossRef]
- Ling, G.S.; Crawford, G.; Buang, N.; Bartok, I.; Tian, K.; Thielens, N.M.; Bally, I.; Harker, J.A.; Ashton-Rickardt, P.G.; Rutschmann, S.; et al. C1q Restrains Autoimmunity and Viral Infection by Regulating CD8+ T Cell Metabolism. Science 2018, 360, 558–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; Norsworthy, P.J.; Hall, A.E.; Kelly, S.J.; Walport, M.J.; Botto, M.; Pickering, M.C. SLE with C1q Deficiency Treated with Fresh Frozen Plasma: A 10-Year Experience. Rheumatol. Oxf. Engl. 2010, 49, 823–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macedo, A.C.L.; Isaac, L. Systemic Lupus Erythematosus and Deficiencies of Early Components of the Complement Classical Pathway. Front. Immunol. 2016, 7, 55. [Google Scholar] [CrossRef] [Green Version]
- Degn, S.E.; Jensenius, J.C.; Thiel, S. Disease-Causing Mutations in Genes of the Complement System. Am. J. Hum. Genet. 2011, 88, 689–705. [Google Scholar] [CrossRef] [Green Version]
- Mayilyan, K.R. Complement Genetics, Deficiencies, and Disease Associations. Protein Cell 2012, 3, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Skare, T.L.; Nisihara, R.; Cieslinski, J.Z.; Zeni, J.O.; Rasera, H.N.; Messias-Reason, I.; Utiyama, S.R.R. Mannose-Binding Lectin Deficiency in Brazilian Patients with Spondyloarthritis. Immunol. Investig. 2017, 46, 183–189. [Google Scholar] [CrossRef]
- Xu, W.-D.; Peng, H.; Zhou, M.; Zhang, M.; Li, B.-Z.; Pan, H.-F.; Ye, D.-Q. Association of RANTES and MBL Gene Polymorphisms with Systemic Lupus Erythematosus: A Meta-Analysis. Mol. Biol. Rep. 2013, 40, 941–948. [Google Scholar] [CrossRef]
- Troldborg, A.; Thiel, S.; Trendelenburg, M.; Friebus-Kardash, J.; Nehring, J.; Steffensen, R.; Hansen, S.W.K.; Laska, M.J.; Deleuran, B.; Jensenius, J.C.; et al. The Lectin Pathway of Complement Activation in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2018, 45, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- S Reis, E.; Falcão, D.A.; Isaac, L. Clinical Aspects and Molecular Basis of Primary Deficiencies of Complement Component C3 and Its Regulatory Proteins Factor I and Factor H. Scand. J. Immunol. 2006, 63, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.H.; Bayles, T.B.; Favour, C.B. The Response of Serum Gamma Globulin Level and Complement Titer to Adrenocorticotropic Hormone Therapy in Lupus Erythematosus Disseminatus. J. Lab. Clin. Med. 1951, 37, 698–702. [Google Scholar] [PubMed]
- Schur, P.H.; Sandson, J. Immunologic Factors and Clinical Activity in Systemic Lupus Erythematosus. N. Engl. J. Med. 1968, 278, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Valentijn, R.M.; van Overhagen, H.; Hazevoet, H.M.; Hermans, J.; Cats, A.; Daha, M.R.; van ES, L.A. The Value of Complement and Immune Complex Determinations in Monitoring Disease Activity in Patients with Systemic Lupus Erythematosus. Arthritis Rheum. 1985, 28, 904–913. [Google Scholar] [CrossRef]
- Dalmasso, A.P. Complement in the Pathophysiology and Diagnosis of Human Diseases. Crit. Rev. Clin. Lab. Sci. 1986, 24, 123–183. [Google Scholar] [CrossRef]
- Manzi, S.; Rairie, J.E.; Carpenter, A.B.; Kelly, R.H.; Jagarlapudi, S.P.; Sereika, S.M.; Medsger, T.A.; Ramsey-Goldman, R. Sensitivity and Specificity of Plasma and Urine Complement Split Products as Indicators of Lupus Disease Activity. Arthritis Rheum. 1996, 39, 1178–1188. [Google Scholar] [CrossRef]
- Stojan, G.; Petri, M. Anti-C1q in Systemic Lupus Erythematosus. Lupus 2016, 25, 873–877. [Google Scholar] [CrossRef] [Green Version]
- Mahler, M.; van Schaarenburg, R.A.; Trouw, L.A. Anti-C1q Autoantibodies, Novel Tests, and Clinical Consequences. Front. Immunol. 2013, 4, 117. [Google Scholar] [CrossRef] [Green Version]
- Seelen, M.A.; Trouw, L.A.; van der Hoorn, J.W.A.; Fallaux-van den Houten, F.C.; Huizinga, T.W.J.; Daha, M.R.; Roos, A. Autoantibodies against Mannose-Binding Lectin in Systemic Lupus Erythematosus. Clin. Exp. Immunol. 2003, 134, 335–343. [Google Scholar] [CrossRef]
- Plawecki, M.; Lheritier, E.; Clavarino, G.; Jourde-Chiche, N.; Ouili, S.; Paul, S.; Gout, E.; Sarrot-Reynauld, F.; Bardin, N.; Boëlle, P.-Y.; et al. Association between the Presence of Autoantibodies Targeting Ficolin-3 and Active Nephritis in Patients with Systemic Lupus Erythematosus. PLoS ONE 2016, 11, e0160879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Wu, X.; Shan, G.; Zhang, X. Diagnostic Value of Serum Anti-C1q Antibodies in Patients with Lupus Nephritis: A Meta-Analysis. Lupus 2012, 21, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.T.; Østergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H.H. Increased IgG on Cell-Derived Plasma Microparticles in Systemic Lupus Erythematosus Is Associated with Autoantibodies and Complement Activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.G.; Burge, J.J. A New Enzyme-Linked Immunosorbent Assay (ELISA) for Measuring Immunoconglutinins Directed against the Third Component of Human Complement. Findings in Systemic Lupus Erythematosus. J. Immunol. Methods 1984, 73, 57–66. [Google Scholar] [CrossRef]
- Kenyon, K.D.; Cole, C.; Crawford, F.; Kappler, J.W.; Thurman, J.M.; Bratton, D.L.; Boackle, S.A.; Henson, P.M. IgG Autoantibodies against Deposited C3 Inhibit Macrophage-Mediated Apoptotic Cell Engulfment in Systemic Autoimmunity. J. Immunol. Baltim. Md. 1950 2011, 187, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Birmingham, D.J.; Bitter, J.E.; Ndukwe, E.G.; Dials, S.; Gullo, T.R.; Conroy, S.; Nagaraja, H.N.; Rovin, B.H.; Hebert, L.A. Relationship of Circulating Anti-C3b and Anti-C1q IgG to Lupus Nephritis and Its Flare. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Birmingham, D.J.; Merchant, M.; Waikar, S.S.; Nagaraja, H.; Klein, J.B.; Rovin, B.H. Biomarkers of Lupus Nephritis Histology and Flare: Deciphering the Relevant amidst the Noise. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2017, 32, i71–i79. [Google Scholar] [CrossRef]
- Vasilev, V.V.; Noe, R.; Dragon-Durey, M.-A.; Chauvet, S.; Lazarov, V.J.; Deliyska, B.P.; Fremeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. Functional Characterization of Autoantibodies against Complement Component C3 in Patients with Lupus Nephritis. J. Biol. Chem. 2015, 290, 25343–25355. [Google Scholar] [CrossRef] [Green Version]
- Mészáros, T.; Füst, G.; Farkas, H.; Jakab, L.; Temesszentandrási, G.; Nagy, G.; Kiss, E.; Gergely, P.; Zeher, M.; Griger, Z.; et al. C1-Inhibitor Autoantibodies in SLE. Lupus 2010, 19, 634–638. [Google Scholar] [CrossRef]
- Kavai, M. Immune Complex Clearance by Complement Receptor Type 1 in SLE. Autoimmun. Rev. 2008, 8, 160–164. [Google Scholar] [CrossRef]
- Richaud-Patin, Y.; Pérez-Romano, B.; Carrillo-Maravilla, E.; Rodriguez, A.B.; Simon, A.J.; Cabiedes, J.; Jakez-Ocampo, J.; Llorente, L.; Ruiz-Argüelles, A. Deficiency of Red Cell Bound CD55 and CD59 in Patients with Systemic Lupus Erythematosus. Immunol. Lett. 2003, 88, 95–99. [Google Scholar] [CrossRef]
- Alegretti, A.P.; Mucenic, T.; Merzoni, J.; Faulhaber, G.A.; Silla, L.M.; Xavier, R.M. Expression of CD55 and CD59 on Peripheral Blood Cells from Systemic Lupus Erythematosus (SLE) Patients. Cell. Immunol. 2010, 265, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Seya, T.; Koni, I.; Mabuchi, H. Elevated Serum Levels of Soluble Membrane Cofactor Protein (CD46, MCP) in Patients with Systemic Lupus Erythematosus (SLE). Clin. Exp. Immunol. 1999, 116, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Tseng, M.-H.; Lin, S.-H.; Wu, C.-Y.; Chien, H.-P.; Yang, H.-Y.; Chen, Y.-C.; Chou, Y.-C.; Huang, J.-L. Serum Complement Factor I Is Associated with Disease Activity of Systemic Lupus Erythematosus. Oncotarget 2018, 9, 8502–8511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, E.; Nielsen, L.A.; Nielsen, C.T.; Munthe-Fog, L.; Skjoedt, M.-O.; Jacobsen, S.; Garred, P. Ficolins and the Lectin Pathway of Complement in Patients with Systemic Lupus Erythematosus. Mol. Immunol. 2015, 63, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B.; Given, W.P.; Edelson, H.S.; Weissmann, G. Neutrophil Aggregation Induced by Sera from Patients with Active Systemic Lupus Erythematosus. Arthritis Rheum. 1983, 26, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Karpman, D.; Kahn, R. The Contact/Kinin and Complement Systems in Vasculitis. APMIS. Suppl. 2009, 48–54. [Google Scholar] [CrossRef]
- Parra, S.; Vives, G.; Ferré, R.; González, M.; Guardiola, M.; Ribalta, J.; Castro, A. Complement System and Small HDL Particles Are Associated with Subclinical Atherosclerosis in SLE Patients. Atherosclerosis 2012, 225, 224–230. [Google Scholar] [CrossRef]
- Holers, V.M.; Girardi, G.; Mo, L.; Guthridge, J.M.; Molina, H.; Pierangeli, S.S.; Espinola, R.; Xiaowei, L.E.; Mao, D.; Vialpando, C.G.; et al. Complement C3 Activation Is Required for Antiphospholipid Antibody-Induced Fetal Loss. J. Exp. Med. 2002, 195, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Girardi, G.; Berman, J.; Redecha, P.; Spruce, L.; Thurman, J.M.; Kraus, D.; Hollmann, T.J.; Casali, P.; Caroll, M.C.; Wetsel, R.A.; et al. Complement C5a Receptors and Neutrophils Mediate Fetal Injury in the Antiphospholipid Syndrome. J. Clin. Investig. 2003, 112, 1644–1654. [Google Scholar] [CrossRef] [Green Version]
- Fischetti, F.; Durigutto, P.; Pellis, V.; Debeus, A.; Macor, P.; Bulla, R.; Bossi, F.; Ziller, F.; Sblattero, D.; Meroni, P.; et al. Thrombus Formation Induced by Antibodies to Beta2-Glycoprotein I Is Complement Dependent and Requires a Priming Factor. Blood 2005, 106, 2340–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, A.M.; Gibbs, R.S.; Murphy, J.R.; Giclas, P.C.; Salmon, J.E.; Holers, V.M. Early Elevations of the Complement Activation Fragment C3a and Adverse Pregnancy Outcomes. Obstet. Gynecol. 2011, 117, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M.; Kraus, D.M.; Girardi, G.; Hourcade, D.; Kang, H.J.; Royer, P.A.; Mitchell, L.M.; Giclas, P.C.; Salmon, J.; Gilkeson, G.; et al. A Novel Inhibitor of the Alternative Complement Pathway Prevents Antiphospholipid Antibody-Induced Pregnancy Loss in Mice. Mol. Immunol. 2005, 42, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Alpers, C.E.; Pippin, J.; Shankland, S.J.; Kurokawa, K.; Adler, S.; Morgan, B.P.; Johnson, R.J.; Couser, W.G. CD59 Protects Glomerular Endothelial Cells from Immune-Mediated Thrombotic Microangiopathy in Rats. J. Am. Soc. Nephrol. JASN 1998, 9, 590–597. [Google Scholar]
- Pierangeli, S.S.; Girardi, G.; Vega-Ostertag, M.; Liu, X.; Espinola, R.G.; Salmon, J. Requirement of Activation of Complement C3 and C5 for Antiphospholipid Antibody-Mediated Thrombophilia. Arthritis Rheum. 2005, 52, 2120–2124. [Google Scholar] [CrossRef]
- Davis, W.D.; Brey, R.L. Antiphospholipid Antibodies and Complement Activation in Patients with Cerebral Ischemia. Clin. Exp. Rheumatol. 1992, 10, 455–460. [Google Scholar]
- Breen, K.A.; Seed, P.; Parmar, K.; Moore, G.W.; Stuart-Smith, S.E.; Hunt, B.J. Complement Activation in Patients with Isolated Antiphospholipid Antibodies or Primary Antiphospholipid Syndrome. Thromb. Haemost. 2012, 107, 423–429. [Google Scholar] [CrossRef]
- Meroni, P.L.; Macor, P.; Durigutto, P.; De Maso, L.; Gerosa, M.; Ferraresso, M.; Borghi, M.O.; Mollnes, T.E.; Tedesco, F. Complement Activation in Antiphospholipid Syndrome and Its Inhibition to Prevent Rethrombosis after Arterial Surgery. Blood 2016, 127, 365–367. [Google Scholar] [CrossRef]
- Gropp, K.; Weber, N.; Reuter, M.; Micklisch, S.; Kopka, I.; Hallström, T.; Skerka, C. Β₂-Glycoprotein I, the Major Target in Antiphospholipid Syndrome, Is a Special Human Complement Regulator. Blood 2011, 118, 2774–2783. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Brodsky, R.A.; McCrae, K.R. Complement in the Pathophysiology of the Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 449. [Google Scholar] [CrossRef] [Green Version]
- Salmon, J.E.; Girardi, G. Antiphospholipid Antibodies and Pregnancy Loss: A Disorder of Inflammation. J. Reprod. Immunol. 2008, 77, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Oku, K.; Atsumi, T.; Bohgaki, M.; Amengual, O.; Kataoka, H.; Horita, T.; Yasuda, S.; Koike, T. Complement Activation in Patients with Primary Antiphospholipid Syndrome. Ann. Rheum. Dis. 2009, 68, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Watson, R.; Winkelstein, J.A.; McLean, R.H. Clinical Expression of Systemic Lupus Erythematosus in Patients with C4A Deficiency. Med. (Baltim.) 1993, 72, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Sturfelt, G.; Sjöholm, A.G.; Svensson, B. Complement Components, C1 Activation and Disease Activity in SLE. Int. Arch. Allergy Appl. Immunol. 1983, 70, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brito-Zerón, P.; Yagüe, J.; Akasbi, M.; Bautista, R.; Ruano, M.; Claver, G.; Gil, V.; Font, J. Hypocomplementaemia as an Immunological Marker of Morbidity and Mortality in Patients with Primary Sjogren’s Syndrome. Rheumatol. Oxf. Engl. 2005, 44, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Moffat, G.J.; Lappin, D.; Birnie, G.D.; Whaley, K. Complement Biosynthesis in Human Synovial Tissue. Clin. Exp. Immunol. 1989, 78, 54–60. [Google Scholar]
- Linton, S.M.; Morgan, B.P. Complement Activation and Inhibition in Experimental Models of Arthritis. Mol. Immunol. 1999, 36, 905–914. [Google Scholar] [CrossRef]
- Neumann, E.; Barnum, S.R.; Tarner, I.H.; Echols, J.; Fleck, M.; Judex, M.; Kullmann, F.; Mountz, J.D.; Schölmerich, J.; Gay, S.; et al. Local Production of Complement Proteins in Rheumatoid Arthritis Synovium. Arthritis Rheum. 2002, 46, 934–945. [Google Scholar] [CrossRef]
- Inman, R.D.; Harpel, P.C. C1 Inactivator-C1s Complexes in Inflammatory Joint Disease. Clin. Exp. Immunol. 1983, 53, 521–528. [Google Scholar]
- Moxley, G.; Ruddy, S. Elevated C3 Anaphylatoxin Levels in Synovial Fluids from Patients with Rheumatoid Arthritis. Arthritis Rheum. 1985, 28, 1089–1095. [Google Scholar] [CrossRef]
- Mollnes, T.E.; Paus, A. Complement Activation in Synovial Fluid and Tissue from Patients with Juvenile Rheumatoid Arthritis. Arthritis Rheum. 1986, 29, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- Swaak, A.J.; Van Rooyen, A.; Planten, O.; Han, H.; Hattink, O.; Hack, E. An Analysis of the Levels of Complement Components in the Synovial Fluid in Rheumatic Diseases. Clin. Rheumatol. 1987, 6, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, J.P.; Ruddy, S.; Schwartz, L.B.; Moxley, G. Synovial Fluid Levels of Complement SC5b-9 and Fragment Bb Are Elevated in Patients with Rheumatoid Arthritis. Arthritis Rheum. 1991, 34, 1531–1537. [Google Scholar] [CrossRef]
- Shingu, M.; Watanabe, Y.; Tomooka, K.; Yoshioka, K.; Ohtsuka, E.; Nobunaga, M. Complement Degradation Products in Rheumatoid Arthritis Synovial Fluid. Br. J. Rheumatol. 1994, 33, 299–300. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.; Richards, N.; Hornby, J.; Powell, R. Relation between Synovial Fluid C3 Degradation Products and Local Joint Inflammation in Rheumatoid Arthritis, Osteoarthritis, and Crystal Associated Arthropathy. Ann. Rheum. Dis. 1988, 47, 190–197. [Google Scholar] [CrossRef]
- Jose, P.J.; Moss, I.K.; Maini, R.N.; Williams, T.J. Measurement of the Chemotactic Complement Fragment C5a in Rheumatoid Synovial Fluids by Radioimmunoassay: Role of C5a in the Acute Inflammatory Phase. Ann. Rheum. Dis. 1990, 49, 747–752. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Ceponis, A.; Meri, S.; Vuorikoski, A.; Kortekangas, P.; Sorsa, T.; Sukura, A.; Santavirta, S. Complement in Acute and Chronic Arthritides: Assessment of C3c, C9, and Protectin (CD59) in Synovial Membrane. Ann. Rheum. Dis. 1996, 55, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Sjöholm, A.G.; Berglund, K.; Johnson, U.; Laurell, A.B.; Sturfelt, G. C1 Activation, with C1q in Excess of Functional C1 in Synovial Fluid from Patients with Rheumatoid Arthritis. Int. Arch. Allergy Appl. Immunol. 1986, 79, 113–119. [Google Scholar] [CrossRef]
- Okroj, M.; Heinegård, D.; Holmdahl, R.; Blom, A.M. Rheumatoid Arthritis and the Complement System. Ann. Med. 2007, 39, 517–530. [Google Scholar] [CrossRef]
- Wouters, D.; Voskuyl, A.E.; Molenaar, E.T.H.; Dijkmans, B.A.C.; Hack, C.E. Evaluation of Classical Complement Pathway Activation in Rheumatoid Arthritis: Measurement of C1q-C4 Complexes as Novel Activation Products. Arthritis Rheum. 2006, 54, 1143–1150. [Google Scholar] [CrossRef]
- Sjöberg, A.; Onnerfjord, P.; Mörgelin, M.; Heinegård, D.; Blom, A.M. The Extracellular Matrix and Inflammation: Fibromodulin Activates the Classical Pathway of Complement by Directly Binding C1q. J. Biol. Chem. 2005, 280, 32301–32308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöberg, A.P.; Manderson, G.A.; Mörgelin, M.; Day, A.J.; Heinegård, D.; Blom, A.M. Short Leucine-Rich Glycoproteins of the Extracellular Matrix Display Diverse Patterns of Complement Interaction and Activation. Mol. Immunol. 2009, 46, 830–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happonen, K.E.; Saxne, T.; Aspberg, A.; Mörgelin, M.; Heinegård, D.; Blom, A.M. Regulation of Complement by Cartilage Oligomeric Matrix Protein Allows for a Novel Molecular Diagnostic Principle in Rheumatoid Arthritis. Arthritis Rheum. 2010, 62, 3574–3583. [Google Scholar] [CrossRef] [Green Version]
- Melin Fürst, C.; Mörgelin, M.; Vadstrup, K.; Heinegård, D.; Aspberg, A.; Blom, A.M. The C-Type Lectin of the Aggrecan G3 Domain Activates Complement. PLoS ONE 2013, 8, e61407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banda, N.K.; Kraus, D.; Vondracek, A.; Huynh, L.H.; Bendele, A.; Holers, V.M.; Arend, W.P. Mechanisms of Effects of Complement Inhibition in Murine Collagen-Induced Arthritis. Arthritis Rheum. 2002, 46, 3065–3075. [Google Scholar] [CrossRef]
- Hietala, M.A.; Jonsson, I.-M.; Tarkowski, A.; Kleinau, S.; Pekna, M. Complement Deficiency Ameliorates Collagen-Induced Arthritis in Mice. J. Immunol. Baltim. Md. 1950 2002, 169, 454–459. [Google Scholar] [CrossRef] [Green Version]
- Hietala, M.A.; Nandakumar, K.S.; Persson, L.; Fahlén, S.; Holmdahl, R.; Pekna, M. Complement Activation by Both Classical and Alternative Pathways Is Critical for the Effector Phase of Arthritis. Eur. J. Immunol. 2004, 34, 1208–1216. [Google Scholar] [CrossRef]
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057. [Google Scholar] [CrossRef]
- Ji, H.; Ohmura, K.; Mahmood, U.; Lee, D.M.; Hofhuis, F.M.A.; Boackle, S.A.; Takahashi, K.; Holers, V.M.; Walport, M.; Gerard, C.; et al. Arthritis Critically Dependent on Innate Immune System Players. Immunity 2002, 16, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Banda, N.K.; Thurman, J.M.; Kraus, D.; Wood, A.; Carroll, M.C.; Arend, W.P.; Holers, V.M. Alternative Complement Pathway Activation Is Essential for Inflammation and Joint Destruction in the Passive Transfer Model of Collagen-Induced Arthritis. J. Immunol. Baltim. Md. 1950 2006, 177, 1904–1912. [Google Scholar] [CrossRef] [Green Version]
- Banda, N.K.; Takahashi, K.; Wood, A.K.; Holers, V.M.; Arend, W.P. Pathogenic Complement Activation in Collagen Antibody-Induced Arthritis in Mice Requires Amplification by the Alternative Pathway. J. Immunol. Baltim. Md. 1950 2007, 179, 4101–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferluga, J.; Kouser, L.; Murugaiah, V.; Sim, R.B.; Kishore, U. Potential Influences of Complement Factor H in Autoimmune Inflammatory and Thrombotic Disorders. Mol. Immunol. 2017, 84, 84–106. [Google Scholar] [CrossRef] [PubMed]
- Banda, N.K.; Levitt, B.; Glogowska, M.J.; Thurman, J.M.; Takahashi, K.; Stahl, G.L.; Tomlinson, S.; Arend, W.P.; Holers, V.M. Targeted Inhibition of the Complement Alternative Pathway with Complement Receptor 2 and Factor H Attenuates Collagen Antibody-Induced Arthritis in Mice. J. Immunol. Baltim. Md. 1950 2009, 183, 5928–5937. [Google Scholar] [CrossRef] [Green Version]
- Foltyn Zadura, A.; Zipfel, P.F.; Bokarewa, M.I.; Sturfelt, G.; Jönsen, A.; Nilsson, S.C.; Hillarp, A.; Saxne, T.; Trouw, L.A.; Blom, A.M. Factor H Autoantibodies and Deletion of Complement Factor H-Related Protein-1 in Rheumatic Diseases in Comparison to Atypical Hemolytic Uremic Syndrome. Arthritis Res. Ther. 2012, 14, R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, J.; Wishaupt, J.O.; van Lier, R.A.; Smeets, T.J.; Breedveld, F.C.; Tak, P.P. Expression of the Activation Antigen CD97 and Its Ligand CD55 in Rheumatoid Synovial Tissue. Arthritis Rheum. 1999, 42, 650–658. [Google Scholar] [CrossRef]
- Hoek, R.M.; de Launay, D.; Kop, E.N.; Yilmaz-Elis, A.S.; Lin, F.; Reedquist, K.A.; Verbeek, J.S.; Medof, M.E.; Tak, P.P.; Hamann, J. Deletion of Either CD55 or CD97 Ameliorates Arthritis in Mouse Models. Arthritis Rheum. 2010, 62, 1036–1042. [Google Scholar] [CrossRef]
- Kouskoff, V.; Korganow, A.S.; Duchatelle, V.; Degott, C.; Benoist, C.; Mathis, D. Organ-Specific Disease Provoked by Systemic Autoimmunity. Cell 1996, 87, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Carrasco-Marin, E.; Shimizu, J.; Kanagawa, O.; Unanue, E.R. The Class II MHC I-Ag7 Molecules from Non-Obese Diabetic Mice Are Poor Peptide Binders. J. Immunol. Baltim. Md. 1950 1996, 156, 450–458. [Google Scholar]
- Matsumoto, I.; Staub, A.; Benoist, C.; Mathis, D. Arthritis Provoked by Linked T and B Cell Recognition of a Glycolytic Enzyme. Science 1999, 286, 1732–1735. [Google Scholar] [CrossRef] [Green Version]
- Basu, D.; Horvath, S.; Matsumoto, I.; Fremont, D.H.; Allen, P.M. Molecular Basis for Recognition of an Arthritic Peptide and a Foreign Epitope on Distinct MHC Molecules by a Single TCR. J. Immunol. Baltim. Md. 1950 2000, 164, 5788–5796. [Google Scholar] [CrossRef] [Green Version]
- Korganow, A.S.; Ji, H.; Mangialaio, S.; Duchatelle, V.; Pelanda, R.; Martin, T.; Degott, C.; Kikutani, H.; Rajewsky, K.; Pasquali, J.L.; et al. From Systemic T Cell Self-Reactivity to Organ-Specific Autoimmune Disease via Immunoglobulins. Immunity 1999, 10, 451–461. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Gauguier, D.; Ohmura, K.; Gonzalez, A.; Duchatelle, V.; Danoy, P.; Garchon, H.J.; Degott, C.; Lathrop, M.; Benoist, C.; et al. Genetic Influences on the End-Stage Effector Phase of Arthritis. J. Exp. Med. 2001, 194, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, I.; Maccioni, M.; Lee, D.M.; Maurice, M.; Simmons, B.; Brenner, M.; Mathis, D.; Benoist, C. How Antibodies to a Ubiquitous Cytoplasmic Enzyme May Provoke Joint-Specific Autoimmune Disease. Nat. Immunol. 2002, 3, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Monach, P.A.; Verschoor, A.; Jacobs, J.P.; Carroll, M.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Circulating C3 Is Necessary and Sufficient for Induction of Autoantibody-Mediated Arthritis in a Mouse Model. Arthritis Rheum. 2007, 56, 2968–2974. [Google Scholar] [CrossRef] [PubMed]
- Tsao, P.Y.; Arora, V.; Ji, M.Q.; Wright, A.C.; Eisenberg, R.A. KRN/I-Ag7 Mouse Arthritis Is Independent of Complement C3. J. Clin. Immunol. 2011, 31, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, S.; Kolb, C.; Mohanty, S.; Jeisy-Walder, E.; Preyer, R.; Schöllhorn, V.; Illges, H. Transmission of Antibody-Induced Arthritis Is Independent of Complement Component 4 (C4) and the Complement Receptors 1 and 2 (CD21/35). Eur. J. Immunol. 2002, 32, 644–651. [Google Scholar] [CrossRef]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.-M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-Molecule Factor B Inhibitor for the Treatment of Complement-Mediated Diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef] [Green Version]
- Hornum, L.; Hansen, A.J.; Tornehave, D.; Fjording, M.S.; Colmenero, P.; Wätjen, I.F.; Søe Nielsen, N.H.; Bliddal, H.; Bartels, E.M. C5a and C5aR Are Elevated in Joints of Rheumatoid and Psoriatic Arthritis Patients, and C5aR Blockade Attenuates Leukocyte Migration to Synovial Fluid. PLoS ONE 2017, 12, e0189017. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Jayne, D.R.W.; Zhao, M.-H. Complement in ANCA-Associated Vasculitis: Mechanisms and Implications for Management. Nat. Rev. Nephrol. 2017, 13, 359–367. [Google Scholar] [CrossRef]
- Arnaud, L.; Haroche, J.; Mathian, A.; Gorochov, G.; Amoura, Z. Pathogenesis of Takayasu’s Arteritis: A 2011 Update. Autoimmun. Rev. 2011, 11, 61–67. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Ballanti, E.; Triggianese, P.; Perricone, R. Vasculitides and the Complement System: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2015, 49, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative Complement Pathway in the Pathogenesis of Disease Mediated by Anti-Neutrophil Cytoplasmic Autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil Cytoplasmic Autoantibodies Specific for Myeloperoxidase Cause Glomerulonephritis and Vasculitis in Mice. J. Clin. Investig. 2002, 110, 955–963. [Google Scholar] [CrossRef]
- Xiao, H.; Dairaghi, D.J.; Powers, J.P.; Ertl, L.S.; Baumgart, T.; Wang, Y.; Seitz, L.C.; Penfold, M.E.T.; Gan, L.; Hu, P.; et al. C5a Receptor (CD88) Blockade Protects against MPO-ANCA GN. J. Am. Soc. Nephrol. JASN 2014, 25, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, G.; Chen, M.; Liu, G.; Heeringa, P.; Zhang, J.; Zheng, X.; Jie, E.; Kallenberg, C.G.M.; Zhao, M. Complement Activation Is Involved in Renal Damage in Human Antineutrophil Cytoplasmic Autoantibody Associated Pauci-Immune Vasculitis. J. Clin. Immunol. 2009, 29, 282–291. [Google Scholar] [CrossRef]
- Miao, D.; Li, D.-Y.; Chen, M.; Zhao, M.-H. Platelets Are Activated in ANCA-Associated Vasculitis via Thrombin-PARs Pathway and Can Activate the Alternative Complement Pathway. Arthritis Res. Ther. 2017, 19, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-F.; Wang, F.-M.; Li, Z.-Y.; Yu, F.; Zhao, M.-H.; Chen, M. Plasma Complement Factor H Is Associated with Disease Activity of Patients with ANCA-Associated Vasculitis. Arthritis Res. Ther. 2015, 17, 129. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-F.; Wang, F.-M.; Li, Z.-Y.; Yu, F.; Chen, M.; Zhao, M.-H. The Functional Activities of Complement Factor H Are Impaired in Patients with ANCA-Positive Vasculitis. Clin. Immunol. Orlando Fla. 2017, 175, 41–50. [Google Scholar] [CrossRef]
- Yuan, J.; Gou, S.-J.; Huang, J.; Hao, J.; Chen, M.; Zhao, M.-H. C5a and Its Receptors in Human Anti-Neutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Arthritis Res. Ther. 2012, 14, R140. [Google Scholar] [CrossRef] [Green Version]
- Gou, S.-J.; Yuan, J.; Wang, C.; Zhao, M.-H.; Chen, M. Alternative Complement Pathway Activation Products in Urine and Kidneys of Patients with ANCA-Associated GN. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1884–1891. [Google Scholar] [CrossRef] [Green Version]
- Hilhorst, M.; van Paassen, P.; van Rie, H.; Bijnens, N.; Heerings-Rewinkel, P.; van Breda Vriesman, P.; Cohen Tervaert, J.W. Limburg Renal Registry Complement in ANCA-Associated Glomerulonephritis. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2017, 32, 1302–1313. [Google Scholar] [CrossRef]
- Hudson, M.; Walker, J.G.; Fritzler, M.; Taillefer, S.; Baron, M. Hypocomplementemia in Systemic Sclerosis--Clinical and Serological Correlations. J. Rheumatol. 2007, 34, 2218–2223. [Google Scholar] [PubMed]
- Devresse, A.; Aydin, S.; Le Quintrec, M.; Demoulin, N.; Stordeur, P.; Lambert, C.; Gastoldi, S.; Pirson, Y.; Jadoul, M.; Morelle, J. Complement Activation and Effect of Eculizumab in Scleroderma Renal Crisis. Med. (Baltim.) 2016, 95, e4459. [Google Scholar] [CrossRef] [PubMed]
- Kissel, J.T.; Mendell, J.R.; Rammohan, K.W. Microvascular Deposition of Complement Membrane Attack Complex in Dermatomyositis. N. Engl. J. Med. 1986, 314, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Leddy, J.P.; Griggs, R.C.; Klemperer, M.R.; Frank, M.M. Hereditary Complement (C2) Deficiency with Dermatomyositis. Am. J. Med. 1975, 58, 83–91. [Google Scholar] [CrossRef]
- Mascaró, J.M.; Hausmann, G.; Herrero, C.; Grau, J.M.; Cid, M.C.; Palou, J.; Mascaró, J.M. Membrane Attack Complex Deposits in Cutaneous Lesions of Dermatomyositis. Arch. Dermatol. 1995, 131, 1386–1392. [Google Scholar] [CrossRef]
- Ichikawa, E.; Furuta, J.; Kawachi, Y.; Imakado, S.; Otsuka, F. Hereditary Complement (C9) Deficiency Associated with Dermatomyositis. Br. J. Dermatol. 2001, 144, 1080–1083. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic-Uremic Syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.J.; Hill, A.; Arnold, L.M.; Brooksbank, G.L.; Richards, S.J.; Cullen, M.; Mitchell, L.D.; Cohen, D.R.; Gregory, W.M.; Hillmen, P. Long-Term Treatment with Eculizumab in Paroxysmal Nocturnal Hemoglobinuria: Sustained Efficacy and Improved Survival. Blood 2011, 117, 6786–6792. [Google Scholar] [CrossRef] [Green Version]
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.; Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The Complement Inhibitor Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef]
- Riedl, M.A.; Grivcheva-Panovska, V.; Moldovan, D.; Baker, J.; Yang, W.H.; Giannetti, B.M.; Reshef, A.; Andrejevic, S.; Lockey, R.F.; Hakl, R.; et al. Recombinant Human C1 Esterase Inhibitor for Prophylaxis of Hereditary Angio-Oedema: A Phase 2, Multicentre, Randomised, Double-Blind, Placebo-Controlled Crossover Trial. Lancet Lond. Engl. 2017, 390, 1595–1602. [Google Scholar] [CrossRef]
- Howard, J.F.; Utsugisawa, K.; Benatar, M.; Murai, H.; Barohn, R.J.; Illa, I.; Jacob, S.; Vissing, J.; Burns, T.M.; Kissel, J.T.; et al. Safety and Efficacy of Eculizumab in Anti-Acetylcholine Receptor Antibody-Positive Refractory Generalised Myasthenia Gravis (REGAIN): A Phase 3, Randomised, Double-Blind, Placebo-Controlled, Multicentre Study. Lancet Neurol. 2017, 16, 976–986. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Petersen, B.H.; Lee, T.J.; Snyderman, R.; Brooks, G.F. Neisseria Meningitidis and Neisseria Gonorrhoeae Bacteremia Associated with C6, C7, or C8 Deficiency. Ann. Intern. Med. 1979, 90, 917–920. [Google Scholar] [CrossRef]
- Ducret, F.; Decoux, M.; Pointet, P.; Lambert, C.; Grosperrin, E.; Sédaillan, A. [Hereditary C5 deficiency and recurrent Neisseria meningitidis meningitis]. Rev. Med. Interne 1988, 9, 534–537. [Google Scholar] [CrossRef]
- West, C.D. The Complement Profile in Clinical Medicine. Inherited and Acquired Conditions Lowering the Serum Concentrations of Complement Component and Control Proteins. Complement Inflamm. 1989, 6, 49–64. [Google Scholar] [CrossRef]
- Ricklin, D.; Lambris, J.D. New Milestones Ahead in Complement-Targeted Therapy. Semin. Immunol. 2016, 28, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and Development of the Complement Inhibitor Eculizumab for the Treatment of Paroxysmal Nocturnal Hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef]
- Mahaffey, K.W.; Granger, C.B.; Nicolau, J.C.; Ruzyllo, W.; Weaver, W.D.; Theroux, P.; Hochman, J.S.; Filloon, T.G.; Mojcik, C.F.; Todaro, T.G.; et al. Effect of Pexelizumab, an Anti-C5 Complement Antibody, as Adjunctive Therapy to Fibrinolysis in Acute Myocardial Infarction: The COMPlement Inhibition in Myocardial Infarction Treated with ThromboLYtics (COMPLY) Trial. Circulation 2003, 108, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Granger, C.B.; Mahaffey, K.W.; Weaver, W.D.; Theroux, P.; Hochman, J.S.; Filloon, T.G.; Rollins, S.; Todaro, T.G.; Nicolau, J.C.; Ruzyllo, W.; et al. Pexelizumab, an Anti-C5 Complement Antibody, as Adjunctive Therapy to Primary Percutaneous Coronary Intervention in Acute Myocardial Infarction: The COMplement Inhibition in Myocardial Infarction Treated with Angioplasty (COMMA) Trial. Circulation 2003, 108, 1184–1190. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, R.A. Paroxysmal Nocturnal Hemoglobinuria. Blood 2014, 124, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Hall, C.; Marsh, J.C.W.; Elebute, M.; Bombara, M.P.; Petro, B.E.; Cullen, M.J.; Richards, S.J.; Rollins, S.A.; Mojcik, C.F.; et al. Effect of Eculizumab on Hemolysis and Transfusion Requirements in Patients with Paroxysmal Nocturnal Hemoglobinuria. N. Engl. J. Med. 2004, 350, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, R.A.; Young, N.S.; Antonioli, E.; Risitano, A.M.; Schrezenmeier, H.; Schubert, J.; Gaya, A.; Coyle, L.; de Castro, C.; Fu, C.-L.; et al. Multicenter Phase 3 Study of the Complement Inhibitor Eculizumab for the Treatment of Patients with Paroxysmal Nocturnal Hemoglobinuria. Blood 2008, 111, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Muus, P.; Röth, A.; Elebute, M.O.; Risitano, A.M.; Schrezenmeier, H.; Szer, J.; Browne, P.; Maciejewski, J.P.; Schubert, J.; et al. Long-Term Safety and Efficacy of Sustained Eculizumab Treatment in Patients with Paroxysmal Nocturnal Haemoglobinuria. Br. J. Haematol. 2013, 162, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Muus, P.; Dührsen, U.; Risitano, A.M.; Schubert, J.; Luzzatto, L.; Schrezenmeier, H.; Szer, J.; Brodsky, R.A.; Hill, A.; et al. Effect of the Complement Inhibitor Eculizumab on Thromboembolism in Patients with Paroxysmal Nocturnal Hemoglobinuria. Blood 2007, 110, 4123–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loschi, M.; Porcher, R.; Barraco, F.; Terriou, L.; Mohty, M.; de Guibert, S.; Mahe, B.; Lemal, R.; Dumas, P.-Y.; Etienne, G.; et al. Impact of Eculizumab Treatment on Paroxysmal Nocturnal Hemoglobinuria: A Treatment versus No-Treatment Study. Am. J. Hematol. 2016, 91, 366–370. [Google Scholar] [CrossRef]
- Jokiranta, T.S. HUS and Atypical HUS. Blood 2017, 129, 2847–2856. [Google Scholar] [CrossRef] [Green Version]
- Licht, C.; Greenbaum, L.A.; Muus, P.; Babu, S.; Bedrosian, C.L.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Furman, R.R.; Gaber, O.A.; et al. Efficacy and Safety of Eculizumab in Atypical Hemolytic Uremic Syndrome from 2-Year Extensions of Phase 2 Studies. Kidney Int. 2015, 87, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Fakhouri, F.; Hourmant, M.; Campistol, J.M.; Cataland, S.R.; Espinosa, M.; Gaber, A.O.; Menne, J.; Minetti, E.E.; Provôt, F.; Rondeau, E.; et al. Terminal Complement Inhibitor Eculizumab in Adult Patients With Atypical Hemolytic Uremic Syndrome: A Single-Arm, Open-Label Trial. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2016, 68, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, L.A.; Fila, M.; Ardissino, G.; Al-Akash, S.I.; Evans, J.; Henning, P.; Lieberman, K.V.; Maringhini, S.; Pape, L.; Rees, L.; et al. Eculizumab Is a Safe and Effective Treatment in Pediatric Patients with Atypical Hemolytic Uremic Syndrome. Kidney Int. 2016, 89, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Vivarelli, M.; Emma, F. Treatment of C3 Glomerulopathy with Complement Blockers. Semin. Thromb. Hemost. 2014, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Röth, A.; Rottinghaus, S.T.; Hill, A.; Bachman, E.S.; Kim, J.S.; Schrezenmeier, H.; Terriou, L.; Urbano-Ispizua, Á.; Wells, R.A.; Jang, J.H.; et al. Ravulizumab (ALXN1210) in Patients with Paroxysmal Nocturnal Hemoglobinuria: Results of 2 Phase 1b/2 Studies. Blood Adv. 2018, 2, 2176–2185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Sicre de Fontbrune, F.; Wong Lee Lee, L.; Pessoa, V.; Gualandro, S.; Füreder, W.; Ptushkin, V.; Rottinghaus, S.T.; Volles, L.; Shafner, L.; et al. Ravulizumab (ALXN1210) vs. Eculizumab in Adult Patients with PNH Naive to Complement Inhibitors: The 301 Study. Blood 2019, 133, 530–539. [Google Scholar] [CrossRef] [Green Version]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The Long-Acting C5 Inhibitor, Ravulizumab, Is Effective and Safe in Adult Patients with Atypical Hemolytic Uremic Syndrome Naïve to Complement Inhibitor Treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Steinsson, K.; Erlendsson, K.; Valdimarsson, H. Successful Plasma Infusion Treatment of a Patient with C2 Deficiency and Systemic Lupus Erythematosus: Clinical Experience over Forty-Five Months. Arthritis Rheum. 1989, 32, 906–913. [Google Scholar] [PubMed]
- Wang, Y.; Hu, Q.; Madri, J.A.; Rollins, S.A.; Chodera, A.; Matis, L.A. Amelioration of Lupus-like Autoimmune Disease in NZB/WF1 Mice after Treatment with a Blocking Monoclonal Antibody Specific for Complement Component C5. Proc. Natl. Acad. Sci. USA 1996, 93, 8563–8568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Caselman, N.; Ulmer, S.; Weitz, I.C. Complement-Mediated Thrombotic Microangiopathy Associated with Lupus Nephritis. Blood Adv. 2018, 2, 2090–2094. [Google Scholar] [CrossRef] [Green Version]
- Pickering, M.C.; Ismajli, M.; Condon, M.B.; McKenna, N.; Hall, A.E.; Lightstone, L.; Terence Cook, H.; Cairns, T.D. Eculizumab as Rescue Therapy in Severe Resistant Lupus Nephritis. Rheumatol. Oxf. Engl. 2015, 54, 2286–2288. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R.; Peruzzi, L.; Amore, A.; Martino, S.; Vergano, L.; Lastauka, I.; Schieppati, A.; Noris, M.; Tovo, P.A.; Remuzzi, G. Dramatic Effects of Eculizumab in a Child with Diffuse Proliferative Lupus Nephritis Resistant to Conventional Therapy. Pediatr. Nephrol. Berl. Ger. 2015, 30, 167–172. [Google Scholar] [CrossRef]
- Sciascia, S.; Radin, M.; Yazdany, J.; Tektonidou, M.; Cecchi, I.; Roccatello, D.; Dall’Era, M. Expanding the Therapeutic Options for Renal Involvement in Lupus: Eculizumab, Available Evidence. Rheumatol. Int. 2017, 37, 1249–1255. [Google Scholar] [CrossRef]
- Kello, N.; Khoury, L.E.; Marder, G.; Furie, R.; Zapantis, E.; Horowitz, D.L. Secondary Thrombotic Microangiopathy in Systemic Lupus Erythematosus and Antiphospholipid Syndrome, the Role of Complement and Use of Eculizumab: Case Series and Review of Literature. Semin. Arthritis Rheum. 2019, 49, 74–83. [Google Scholar] [CrossRef] [PubMed]
- El-Husseini, A.; Hannan, S.; Awad, A.; Jennings, S.; Cornea, V.; Sawaya, B.P. Thrombotic Microangiopathy in Systemic Lupus Erythematosus: Efficacy of Eculizumab. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2015, 65, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Hadaya, K.; Ferrari-Lacraz, S.; Fumeaux, D.; Boehlen, F.; Toso, C.; Moll, S.; Martin, P.-Y.; Villard, J. Eculizumab in Acute Recurrence of Thrombotic Microangiopathy after Renal Transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2011, 11, 2523–2527. [Google Scholar] [CrossRef]
- Shapira, I.; Andrade, D.; Allen, S.L.; Salmon, J.E. Brief Report: Induction of Sustained Remission in Recurrent Catastrophic Antiphospholipid Syndrome via Inhibition of Terminal Complement with Eculizumab. Arthritis Rheum. 2012, 64, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, R.M.; Williams, A.S.; Levin, J.L.; Williams, B.D.; Morgan, B.P. Soluble Complement Receptor One (SCR1) Inhibits the Development and Progression of Rat Collagen-Induced Arthritis. Clin. Exp. Immunol. 2000, 119, 210–216. [Google Scholar] [CrossRef]
- Ames, R.S.; Lee, D.; Foley, J.J.; Jurewicz, A.J.; Tornetta, M.A.; Bautsch, W.; Settmacher, B.; Klos, A.; Erhard, K.F.; Cousins, R.D.; et al. Identification of a Selective Nonpeptide Antagonist of the Anaphylatoxin C3a Receptor That Demonstrates Antiinflammatory Activity in Animal Models. J. Immunol. Baltim. Md. 1950 2001, 166, 6341–6348. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, T.M.; Strachan, A.J.; Dryburgh, N.; Shiels, I.A.; Reid, R.C.; Fairlie, D.P.; Taylor, S.M. Antiarthritic Activity of an Orally Active C5a Receptor Antagonist against Antigen-Induced Monarticular Arthritis in the Rat. Arthritis Rheum. 2002, 46, 2476–2485. [Google Scholar] [CrossRef]
- Fraser, D.A.; Harris, C.L.; Williams, A.S.; Mizuno, M.; Gallagher, S.; Smith, R.A.G.; Morgan, B.P. Generation of a Recombinant, Membrane-Targeted Form of the Complement Regulator CD59: Activity in Vitro and in Vivo. J. Biol. Chem. 2003, 278, 48921–48927. [Google Scholar] [CrossRef] [Green Version]
- Macor, P.; Durigutto, P.; De Maso, L.; Garrovo, C.; Biffi, S.; Cortini, A.; Fischetti, F.; Sblattero, D.; Pitzalis, C.; Marzari, R.; et al. Treatment of Experimental Arthritis by Targeting Synovial Endothelium with a Neutralizing Recombinant Antibody to C5. Arthritis Rheum. 2012, 64, 2559–2567. [Google Scholar] [CrossRef]
- Durigutto, P.; Macor, P.; Ziller, F.; De Maso, L.; Fischetti, F.; Marzari, R.; Sblattero, D.; Tedesco, F. Prevention of Arthritis by Locally Synthesized Recombinant Antibody Neutralizing Complement Component C5. PLoS ONE 2013, 8, e58696. [Google Scholar] [CrossRef] [Green Version]
- Vergunst, C.E.; Gerlag, D.M.; Dinant, H.; Schulz, L.; Vinkenoog, M.; Smeets, T.J.M.; Sanders, M.E.; Reedquist, K.A.; Tak, P.P. Blocking the Receptor for C5a in Patients with Rheumatoid Arthritis Does Not Reduce Synovial Inflammation. Rheumatol. Oxf. Engl. 2007, 46, 1773–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilla-Labarca, M.-L.; Toder, K.; Furie, R. Targeting the Complement System in Systemic Lupus Erythematosus and Other Diseases. Clin. Immunol. Orlando Fla. 2013, 148, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Vanoni, F.; Jorgensen, C.; Parvex, P.; Chizzolini, C.; Hofer, M. A Difficult Case of Juvenile Dermatomyositis Complicated by Thrombotic Microangiopathy and Purtscher-like Retinopathy. Pediatr. Rheumatol. 2014, 12, P275. [Google Scholar] [CrossRef] [Green Version]
- Bekker, P.; Dairaghi, D.; Seitz, L.; Leleti, M.; Wang, Y.; Ertl, L.; Baumgart, T.; Shugarts, S.; Lohr, L.; Dang, T.; et al. Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study. PLoS ONE 2016, 11, e0164646. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. JASN 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [Green Version]
- Merkel, P.A.; Niles, J.; Jimenez, R.; Spiera, R.F.; Rovin, B.H.; Bomback, A.; Pagnoux, C.; Potarca, A.; Schall, T.J.; Bekker, P.; et al. Adjunctive Treatment With Avacopan, an Oral C5a Receptor Inhibitor, in Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. ACR Open Rheumatol. 2020, 2, 662–671. [Google Scholar] [CrossRef]
- Zelek, W.M.; Xie, L.; Morgan, B.P.; Harris, C.L. Compendium of Current Complement Therapeutics. Mol. Immunol. 2019, 114, 341–352. [Google Scholar] [CrossRef]
- Ballow, M. The IgG Molecule as a Biological Immune Response Modifier: Mechanisms of Action of Intravenous Immune Serum Globulin in Autoimmune and Inflammatory Disorders. J. Allergy Clin. Immunol. 2011, 127, 315–323, quiz 324–325. [Google Scholar] [CrossRef]


{kind=link}
| Model Disease | Complement Deregulation | Therapeutic Management | Clinical Consequences |
|---|---|---|---|
| Antiphospholipid syndrome | C3 or C5 deficiency | C5b-9 blockade Factor B blockade C5 blockade | No fetal losses No thrombosis No fetal losses No fetal losses No fetal losses, no thrombosis |
| Rheumatoid arthritis CIA/CAIA CIA/CAIA KRN/I-Ag7 KRN/I-Ag7 KRN/I-Ag7 | Alternative pathway Alternative pathway Factor B absence | C3 or C5 blockade Factor B blockade | Arthritis development No arthritis development Arthritis development No arthritis development No arthritis development |
| AAV Anti-MPO induced vasculitis | C5 deficiency C5aR deficiency | C5aR blockade | No vasculitis development No vasculitis development No vasculitis development |
| Agent | Disease | Type of Study | Therapeutic Effect |
|---|---|---|---|
| Eculizumab (C5a inhibitor) | SLE SLE and TMA CAPS RA DM | Clinical Trial (phase I) Case Report Case Report Clinical Trial (phase IIb) Clinical Trial (placebo-controlled double blind pilot study) Case Report | No evidence Improvement Improvement No evidence No evidence Improvement |
| PMX53 (C5aR inhibitor) | RA | Clinical trial (placebo-controlled double blind pilot study) | No evidence |
| CCX168 (C5aR inhibitor) | AAV | Clinical Trial (phase II, III) | Improvement |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galindo-Izquierdo, M.; Pablos Alvarez, J.L. Complement as a Therapeutic Target in Systemic Autoimmune Diseases. Cells 2021, 10, 148. https://doi.org/10.3390/cells10010148
Galindo-Izquierdo M, Pablos Alvarez JL. Complement as a Therapeutic Target in Systemic Autoimmune Diseases. Cells. 2021; 10(1):148. https://doi.org/10.3390/cells10010148
Chicago/Turabian StyleGalindo-Izquierdo, María, and José Luis Pablos Alvarez. 2021. "Complement as a Therapeutic Target in Systemic Autoimmune Diseases" Cells 10, no. 1: 148. https://doi.org/10.3390/cells10010148
APA StyleGalindo-Izquierdo, M., & Pablos Alvarez, J. L. (2021). Complement as a Therapeutic Target in Systemic Autoimmune Diseases. Cells, 10(1), 148. https://doi.org/10.3390/cells10010148