
The Response of Mitochondrial Respiration and Quantity in Skeletal Muscle and Adipose Tissue to Exercise in Humans with Prediabetes

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
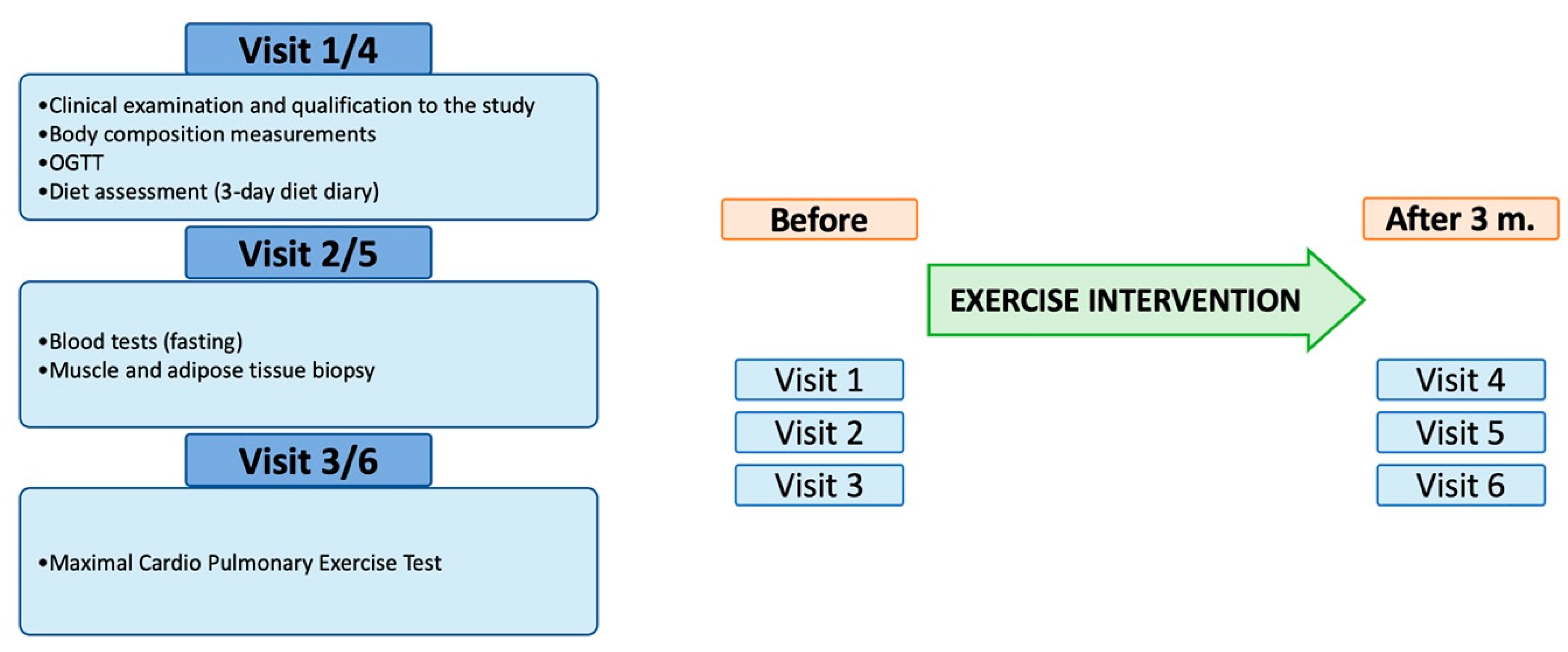
2.1. Studied Population and Study Design
- NG group (n = 19)—subjects with normal fasting glucose and normal glucose tolerance, defined as fasting plasma glucose (FPG) <100 mg/dL and 2-h glucose during OGTT (2h-GLU) <140 mg/dL.
- IFG group (n = 27)—subjects with impaired fasting glucose and normal glucose tolerance, defined as FPG 100-125 mg/dL and 2h-GLU <140 mg/dL.
- IFG + IGT group (n = 8)—subjects with impaired fasting glucose and impaired glucose tolerance, defined as FPG 100–125 mg/dL and 2h-GLU 140–199 mg/dL.
2.2. Exercise Intervention
2.3. Muscle and Fat Biopsy
2.4. Biochemical Measurements of Subjects
2.4.1. Anthropometrics and Body Composition
2.4.2. OGTT and Laboratory Measurements
2.4.3. Glucose Homeostasis Derivates
2.5. Exercise Testing
2.6. Mitochondria Measurements
2.7. Statistical Analyses
3. Results
3.1. Clinical Characteristic of Studied Groups and the Effectiveness of Exercise Intervention
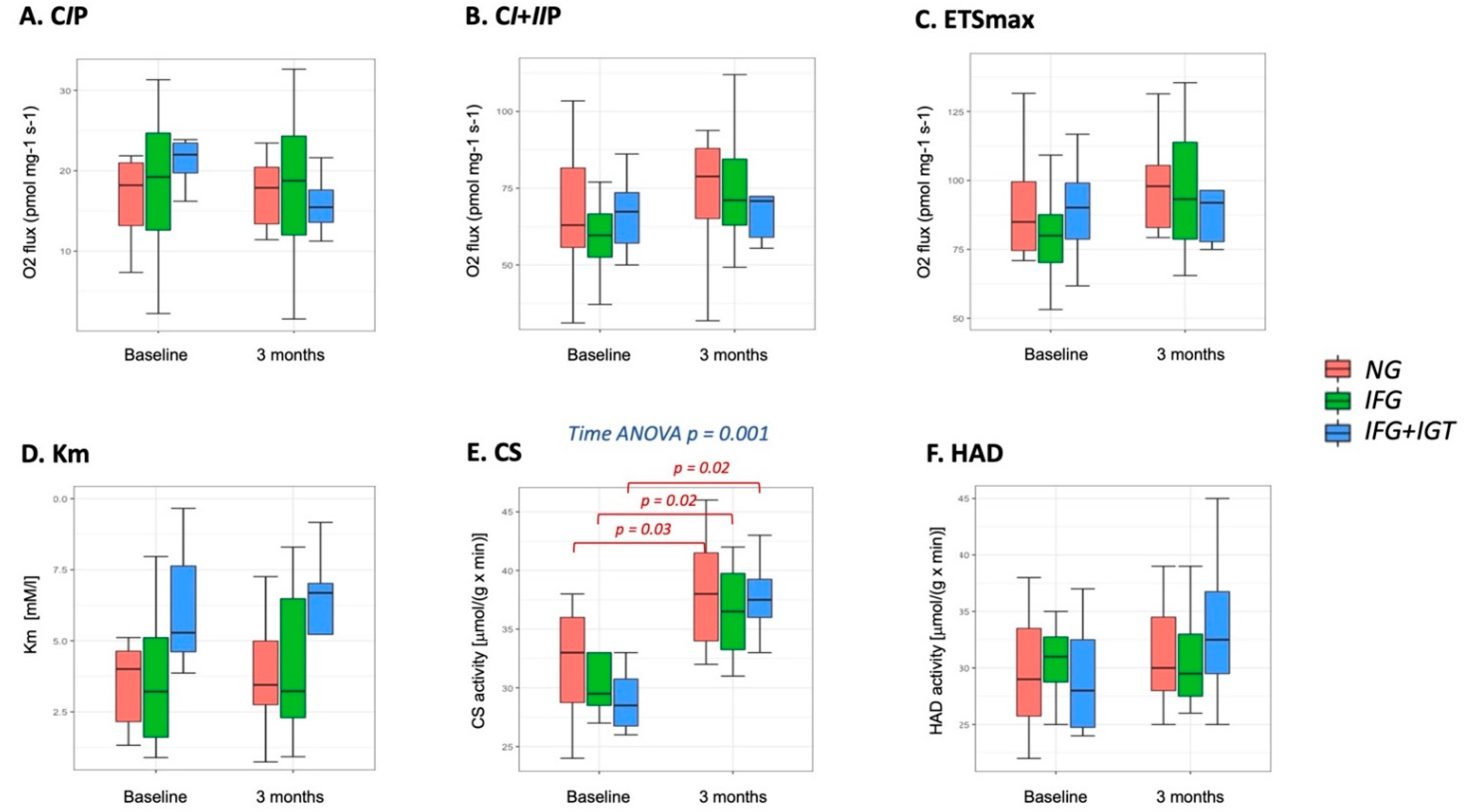
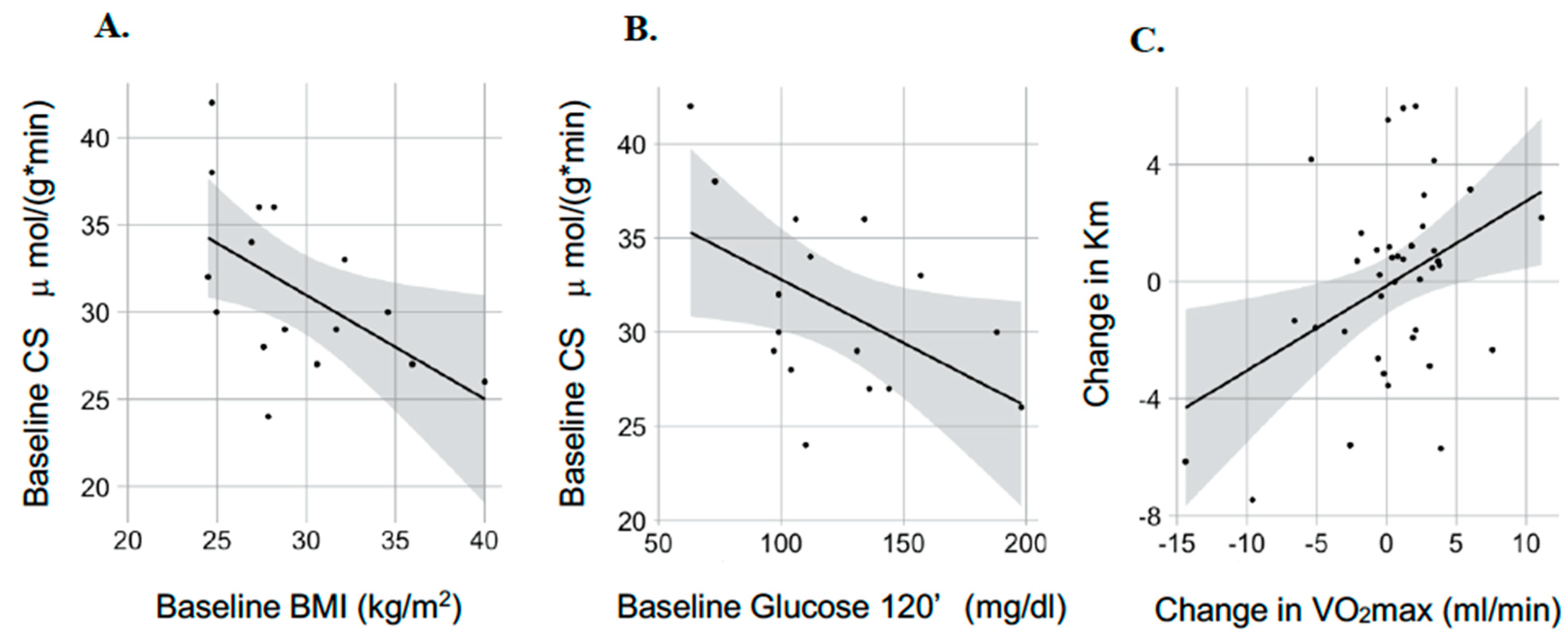
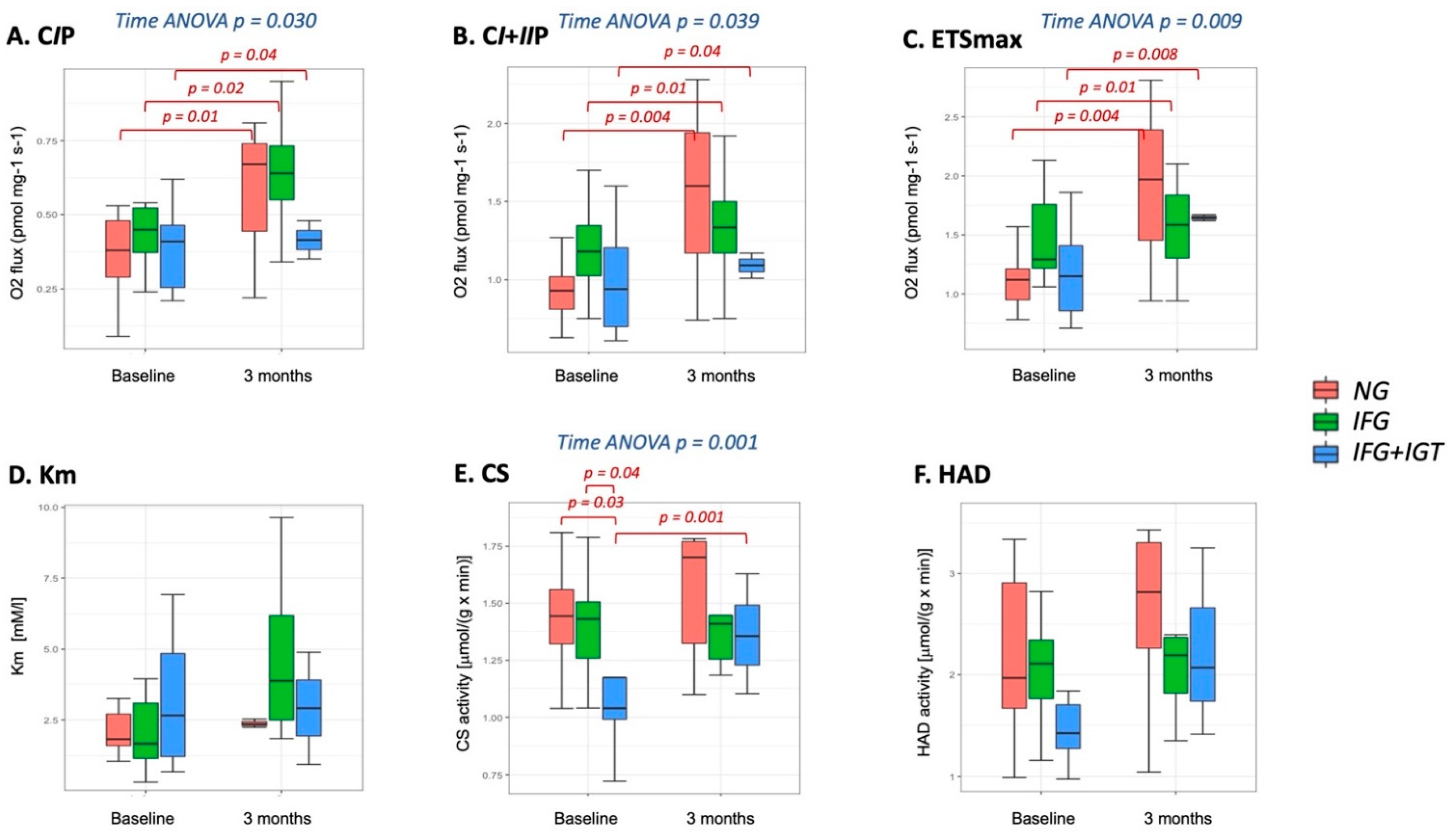
3.2. Mitochondria-Skeletal Muscle
3.3. Mitochondria-Adipose Tissue
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patti, M.E.; Corvera, S. The Role of Mitochondria in the Pathogenesis of Type 2 Diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial Dysfunction in the Elderly: Possible Role in Insulin Resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, M.; Sahlin, K.; Fernstrom, M.; Glintborg, D.; Vind, B.F.; Beck-Nielsen, H.; Hojlund, K. Mitochondrial Respiration Is Decreased in Skeletal Muscle of Patients with Type 2 Diabetes. Diabetes 2007, 56, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Ritov, V.B.; Menshikova, E.V.; He, J.; Ferrell, R.E.; Goodpaster, B.H.; Kelley, D.E. Deficiency of Subsarcolemmal Mitochondria in Obesity and Type 2 Diabetes. Diabetes 2005, 54, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomentowski, P.; Coen, P.M.; Radikova, Z.; Goodpaster, B.H.; Toledo, F.G. Skeletal Muscle Mitochondria in Insulin Resistance: Differences in Intermyofibrillar versus Subsarcolemmal Subpopulations and Relationship to Metabolic Flexibility. J. Clin. Endocrinol. Metab. 2011, 96, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.; Jeneson, J.A.; Backes, W.H.; van Echteld, C.J.; van Engelshoven, J.M.; Mensink, M.; Schrauwen, P. Impaired in Vivo Mitochondrial Function but Similar Intramyocellular Lipid Content in Patients with Type 2 Diabetes Mellitus and BMI-Matched Control Subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Boushel, R.; Gnaiger, E.; Schjerling, P.; Skovbro, M.; Kraunsoe, R.; Dela, F. Patients with Type 2 Diabetes Have Normal Mitochondrial Function in Skeletal Muscle. Diabetologia 2007, 50, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.P.; Hesselink, M.K.; et al. Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies in Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of Insulin on Human Skeletal Muscle Mitochondrial ATP Production, Protein Synthesis, and MRNA Transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Schmid, A.I.; Chmelik, M.; Toth, C.; Brehm, A.; Krssak, M.; Nowotny, P.; Wolzt, M.; Waldhausl, W.; Roden, M. Muscle Mitochondrial ATP Synthesis and Glucose Transport/Phosphorylation in Type 2 Diabetes. PLoS Med. 2007, 4, e154. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Stride, N.; Hey-Mogensen, M.; Hansen, C.N.; Andersen, J.L.; Madsbad, S.; Worm, D.; Helge, J.W.; Dela, F. Increased Mitochondrial Substrate Sensitivity in Skeletal Muscle of Patients with Type 2 Diabetes. Diabetologia 2011, 54, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodpaster, B.H.; Sparks, L.M. Review Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Simoneau, J.A.; Kelley, D.E. Altered Glycolytic and Oxidative Capacities of Skeletal Muscle Contribute to Insulin Resistance in NIDDM. J. Appl. Physiol. 1997, 83, 166–171. [Google Scholar] [CrossRef]
- Ukropcova, B.; Sereda, O.; de Jonge, L.; Bogacka, I.; Nguyen, T.; Xie, H.; Bray, G.A.; Smith, S.R. Family History of Diabetes Links Impaired Substrate Switching and Reduced Mitochondrial Content in Skeletal Muscle. Diabetes 2007, 56, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Meex, R.C.; Schrauwen-Hinderling, V.B.; Moonen-Kornips, E.; Schaart, G.; Mensink, M.; Phielix, E.; van de Weijer, T.; Sels, J.P.; Schrauwen, P.; Hesselink, M.K. Restoration of Muscle Mitochondrial Function and Metabolic Flexibility in Type 2 Diabetes by Exercise Training Is Paralleled by Increased Myocellular Fat Storage and Improved Insulin Sensitivity. Diabetes 2010, 59, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Song, W. Exercise and Mitochondrial Remodeling in Skeletal Muscle in Type 2 Diabetes. J. Obes. Metab. Syndr. 2018, 27, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: A High-Risk State for Diabetes Development. Lancet 2012, 379, 2279–2790. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43 (Suppl. 1), S14–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillett, M.; Royle, P.; Snaith, A.; Scotland, G.; Poobalan, A.; Imamura, M.; Black, C.; Boroujerdi, M.; Jick, S.; Wyness, L.; et al. Non-Pharmacological Interventions to Reduce the Risk of Diabetes in People with Impaired Glucose Regulation: A Systematic Review and Economic Evaluation. Health Technol. Assess. 2012, 16, 1–236, iii–iv. [Google Scholar] [CrossRef] [Green Version]
- Balk, E.M.; Earley, A.; Raman, G.; Avendano, E.A.; Pittas, A.G.; Remington, P.L. Combined Diet and Physical Activity Promotion Programs to Prevent Type 2 Diabetes Among Persons at Increased Risk: A Systematic Review for the Community Preventive Services Task Force. Ann. Intern. Med. 2015, 163, 437–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmingsen, B.; Gimenez-Perez, G.; Mauricio, D.; Roque, I.F.M.; Metzendorf, M.I.; Richter, B. Diet, Physical Activity or Both for Prevention or Delay of Type 2 Diabetes Mellitus and Its Associated Complications in People at Increased Risk of Developing Type 2 Diabetes Mellitus. Cochrane Database Syst. Rev. 2017, 12, Cd003054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phielix, E.; Meex, R.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Exercise Training Increases Mitochondrial Content and Ex Vivo Mitochondrial Function Similarly in Patients with Type 2 Diabetes and in Control Individuals. Diabetologia 2010, 53, 1714–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevellin, E.; Scorzeto, M.; Olivieri, M.; Granzotto, M.; Valerio, A.; Tedesco, L.; Fabris, R.; Serra, R.; Quarta, M.; Reggiani, C.; et al. Exercise Training Induces Mitochondrial Biogenesis and Glucose Uptake in Subcutaneous Adipose Tissue through ENOS-Dependent Mechanisms. Diabetes 2014, 63, 2800–2811. [Google Scholar] [CrossRef] [Green Version]
- Boyle, K.E.; Friedman, J.E.; Janssen, R.C.; Underkofler, C.; Houmard, J.A.; Rasouli, N. Metabolic Inflexibility with Obesity and the Effects of Fenofibrate on Skeletal Muscle Fatty Acid Oxidation. Horm. Metab. Res. 2017, 49, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Van Tienen, F.H.J.; Praet, S.F.E.; De Feyter, H.M.; Van Den Broek, N.M.; Lindsey, P.J.; Schoonderwoerd, K.G.C.; De Coo, I.F.M.; Nicolay, K.; Prompers, J.J.; Smeets, H.J.M.; et al. Physical Activity Is the Key Determinant of Skeletal Muscle Mitochondrial Function in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 3261–3269. [Google Scholar] [CrossRef]
- Fabbri, E.; Chia, C.W.; Spencer, R.G.; Fishbein, K.W.; Reiter, D.A.; Cameron, D.; Zane, A.C.; Moore, Z.A.; Gonzalez-Freire, M.; Zoli, M.; et al. Insulin Resistance Is Associated with Reduced Mitochondrial Oxidative Capacity Measured by 31P-Magnetic Resonance Spectroscopy in Participants without Diabetes from the Baltimore Longitudinal Study of Aging. Diabetes 2017. [Google Scholar] [CrossRef] [Green Version]
- Montero, D.; Lundby, C. Refuting the Myth of Non-Response to Exercise Training: ‘Non-Responders’ Do Respond to Higher Dose of Training. J. Physiol. 2017, 595, 3377–3387. [Google Scholar] [CrossRef]
- Hoppeler, H.; Howald, H.; Conley, K.; Lindstedt, S.L.; Claassen, H.; Vock, P.; Weibel, E.R. Endurance Training in Humans: Aerobic Capacity and Structure of Skeletal Muscle. J. Appl. Physiol. 1985, 59, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Daussin, F.N.; Zoll, J.; Dufour, S.P.; Ponsot, E.; Lonsdorfer-Wolf, E.; Doutreleau, S.; Mettauer, B.; Piquard, F.; Geny, B.; Richard, R. Effect of Interval versus Continuous Training on Cardiorespiratory and Mitochondrial Functions: Relationship to Aerobic Performance Improvements in Sedentary Subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkonogi, M.; Walsh, B.; Svensson, M.; Sahlin, K. Mitochondrial Function and Antioxidative Defence in Human Muscle: Effects of Endurance Training and Oxidative Stress. J. Physiol. 2000, 528, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of Mitochondrial Content in Skeletal Muscle of Healthy Young Human Subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Granata, C.; Jamnick, N.A.; Bishop, D.J. Training-Induced Changes in Mitochondrial Content and Respiratory Function in Human Skeletal Muscle. Sports Med. 2018, 48, 1809–1828. [Google Scholar] [CrossRef]
- Granata, C.; Oliveira, R.S.F.; Little, J.P.; Renner, K.; Bishop, D.J. Mitochondrial Adaptations to High-Volume Exercise Training Are Rapidly Reversed after a Reduction in Training Volume in Human Skeletal Muscle. FASEB J. 2016, 30, 3413–3423. [Google Scholar] [CrossRef] [Green Version]
- Granata, C.; Oliveira, R.S.F.; Little, J.P.; Renner, K.; Bishop, D.J. Training Intensity Modulates Changes in PGC-1α and P53 Protein Content and Mitochondrial Respiration, but Not Markers of Mitochondrial Content in Human Skeletal Muscle. FASEB J. 2016, 30, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Montero, D.; Cathomen, A.; Jacobs, R.A.; Flück, D.; de Leur, J.; Keiser, S.; Bonne, T.; Kirk, N.; Lundby, A.K.; Lundby, C. Haematological Rather than Skeletal Muscle Adaptations Contribute to the Increase in Peak Oxygen Uptake Induced by Moderate Endurance Training. J. Physiol. 2015, 593, 4677–4688. [Google Scholar] [CrossRef]
- Jacobs, R.A.; Lundby, C. Mitochondria Express Enhanced Quality as Well as Quantity in Association with Aerobic Fitness across Recreationally Active Individuals up to Elite Athletes. J. Appl. Physiol. 2013, 114, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Craig, C.L.; Marshall, A.L.; Sjöström, M.; Bauman, A.E.; Booth, M.L.; Ainsworth, B.E.; Pratt, M.; Ekelund, U.; Yngve, A.; Sallis, J.F.; et al. International Physical Activity Questionnaire: 12-Country Reliability and Validity. Med. Sci. Sports Exerc. 2003, 35, 1381–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grontved, A.; Rimm, E.B.; Willett, W.C.; Andersen, L.B.; Hu, F.B. A Prospective Study of Weight Training and Risk of Type 2 Diabetes Mellitus in Men. Arch. Intern. Med. 2012, 172, 1306–1312. [Google Scholar] [CrossRef] [Green Version]
- Cuff, D.J.; Meneilly, G.S.; Martin, A.; Ignaszewski, A.; Tildesley, H.D.; Frohlich, J.J. Effective Exercise Modality to Reduce Insulin Resistance in Women with Type 2 Diabetes. Diabetes Care 2003, 26, 2977–2982. [Google Scholar] [CrossRef] [Green Version]
- Sigal, R.J.; Kenny, G.P.; Boule, N.G.; Wells, G.A.; Prud’homme, D.; Fortier, M.; Reid, R.D.; Tulloch, H.; Coyle, D.; Phillips, P.; et al. Effects of Aerobic Training, Resistance Training, or Both on Glycemic Control in Type 2 Diabetes: A Randomized Trial. Ann. Intern. Med. 2007, 147, 357–369. [Google Scholar] [CrossRef]
- Shanely, R.A.; Zwetsloot, K.A.; Triplett, N.T.; Meaney, M.P.; Farris, G.E.; Nieman, D.C. Human Skeletal Muscle Biopsy Procedures Using the Modified Bergstrom Technique. J. Vis. Exp. 2014, 51812. [Google Scholar] [CrossRef]
- Chachopoulos, V.; Dinas, P.C.; Chasioti, M.; Jamurtas, A.Z.; Koutedakis, Y.; Flouris, A.D. A Technique for Subcutaneous Abdominal Adipose Tissue Biopsy via a Nondiathermy Method. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis Model Assessment: Insulin Resistance and Beta-Cell Function from Fasting Plasma Glucose and Insulin Concentrations in Man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Balke, B.; Ware, R.W. An Experimental Study of Physical Fitness of Air Force Personnel. US Armed Forces Med. J. 1959, 10, 675–688. [Google Scholar]
- Larsen, S.; Danielsen, J.H.; Søndergård, S.D.; Søgaard, D.; Vigelsoe, A.; Dybboe, R.; Skaaby, S.; Dela, F.; Helge, J.W. The Effect of High-Intensity Training on Mitochondrial Fat Oxidation in Skeletal Muscle and Subcutaneous Adipose Tissue. Scand. J. Med. Sci. Sports 2015, 25, e59–e69. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar] [CrossRef] [PubMed]
- Kraunsøe, R.; Boushel, R.; Hansen, C.N.; Schjerling, P.; Qvortrup, K.; Støckel, M.; Mikines, K.J.; Dela, F. Mitochondrial Respiration in Subcutaneous and Visceral Adipose Tissue from Patients with Morbid Obesity. J. Physiol. 2010, 588 Pt 12, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- R Core Team R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://Www.R-Project.Org/ (accessed on 18 October 2020).
- Nathan, D.M.; Davidson, M.B.; DeFronzo, R.A.; Heine, R.J.; Henry, R.R.; Pratley, R.; Zinman, B.; American Diabetes, A. Impaired Fasting Glucose and Impaired Glucose Tolerance: Implications for Care. Diabetes Care 2007, 30, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; Jenkinson, C.P.; Richardson, D.K.; Tripathy, D.; DeFronzo, R.A. Insulin Secretion and Action in Subjects with Impaired Fasting Glucose and Impaired Glucose Tolerance: Results from the Veterans Administration Genetic Epidemiology Study. Diabetes 2006, 55, 1430–1435. [Google Scholar] [CrossRef] [Green Version]
- Faerch, K.; Vaag, A.; Holst, J.J.; Glumer, C.; Pedersen, O.; Borch-Johnsen, K. Impaired Fasting Glycaemia vs Impaired Glucose Tolerance: Similar Impairment of Pancreatic Alpha and Beta Cell Function but Differential Roles of Incretin Hormones and Insulin Action. Diabetologia 2008, 51, 853–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.; Pimenta, W.; Woerle, H.J.; Van Haeften, T.; Szoke, E.; Mitrakou, A.; Gerich, J. Different Mechanisms for Impaired Fasting Glucose and Impaired Postprandial Glucose Tolerance in Humans. Diabetes Care 2006, 29, 1909–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festa, A.; D’Agostino, R., Jr.; Hanley, A.J.; Karter, A.J.; Saad, M.F.; Haffner, S.M. Differences in Insulin Resistance in Nondiabetic Subjects with Isolated Impaired Glucose Tolerance or Isolated Impaired Fasting Glucose. Diabetes 2004, 53, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Chanseaume, E.; Barquissau, V.; Salles, J.; Aucouturier, J.; Patrac, V.; Giraudet, C.; Gryson, C.; Duché, P.; Boirie, Y.; Chardigny, J.M.; et al. Muscle Mitochondrial Oxidative Phosphorylation Activity, but Not Content, Is Altered with Abdominal Obesity in Sedentary Men: Synergism with Changes in Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.; Ara, I.; Rabøl, R.; Andersen, J.L.; Boushel, R.; Dela, F.; Helge, J.W. Are Substrate Use during Exercise and Mitochondrial Respiratory Capacity Decreased in Arm and Leg Muscle in Type 2 Diabetes? Diabetologia 2009. [Google Scholar] [CrossRef] [Green Version]
- Hey-Mogensen, M.; Højlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernström, M.; Sahlin, K. Effect of Physical Training on Mitochondrial Respiration and Reactive Oxygen Species Release in Skeletal Muscle in Patients with Obesity and Type 2 Diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Lund, M.T.; Gregers, E.; Kraunsøe, R.; Van Hall, G.; Helge, J.W.; Dela, F. Adipose Tissue Mitochondrial Respiration and Lipolysis before and after a Weight Loss by Diet and RYGB. Obesity 2015. [Google Scholar] [CrossRef]
- Camera, D.M.; Anderson, M.J.; Hawley, J.A.; Carey, A.L. Short-Term Endurance Training Does Not Alter the Oxidative Capacity of Human Subcutaneous Adipose Tissue. Eur. J. Appl. Physiol. 2010, 109, 307–316. [Google Scholar] [CrossRef]
- Otero-Díaz, B.; Rodríguez-Flores, M.; Sánchez-Muñoz, V.; Monraz-Preciado, F.; Ordoñez-Ortega, S.; Becerril-Elias, V.; Baay-Guzmán, G.; Obando-Monge, R.; García-García, E.; Palacios-González, B.; et al. Exercise Induces White Adipose Tissue Browning Across the Weight Spectrum in Humans. Front. Physiol. 2018, 9, 1781. [Google Scholar] [CrossRef]
- Meinild Lundby, A.K.; Jacobs, R.A.; Gehrig, S.; de Leur, J.; Hauser, M.; Bonne, T.C.; Flück, D.; Dandanell, S.; Kirk, N.; Kaech, A.; et al. Exercise Training Increases Skeletal Muscle Mitochondrial Volume Density by Enlargement of Existing Mitochondria and Not de Novo Biogenesis. Acta Physiol. 2018, 222. [Google Scholar] [CrossRef] [PubMed]
- Savikj, M.; Zierath, J.R. Train like an Athlete: Applying Exercise Interventions to Manage Type 2 Diabetes. Diabetologia 2020, 63, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Holten, M.K.; Zacho, M.; Gaster, M.; Juel, C.; Wojtaszewski, J.F.P.; Dela, F. Strength Training Increases Insulin-Mediated Glucose Uptake, GLUT4 Content, and Insulin Signaling in Skeletal Muscle in Patients with Type 2 Diabetes. Diabetes 2004, 53, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Pesta, D.H.; Goncalves, R.L.S.; Madiraju, A.K.; Strasser, B.; Sparks, L.M. Resistance Training to Improve Type 2 Diabetes: Working toward a Prescription for the Future. Nutr. Metab. 2017, 14, 24. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Plasma Glucose Concentration and Prediction of Future Risk of Type 2 Diabetes. Diabetes Care 2009, 32, S194–S198. [Google Scholar] [CrossRef] [Green Version]
- Chait, A.; den Hartigh, L.J. Adipose Tissue Distribution, Inflammation and Its Metabolic Consequences, Including Diabetes and Cardiovascular Disease. Front. Cardiovasc. Med. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of Human Skeletal Muscle in Health and Disease: Contribution of Proteomics. J. Proteom. 2011, 74, 774–795. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria | Exclusion Criteria |
|---|---|
|
|
| Parameter (Unit) | Normoglycemia n = 19 | IFG n = 27 | IFG + IGT n = 8 | p-Value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Before | After 3 m. | Before | After 3 m. | Before | After 3 m. | Baseline | Time | Time × Diagnosis | |
| Weight (kg) | 89.43 | 89.30 | 92.80 | 91.69 | 95.60 | 93.57 | 0.557 | 0.020 | 0.272 |
| (83.84–95.02) | (83.70–94.89) | (88.20–97.40) | (87.09–96.30) | (87.07–104.14) | (85.01–102.13) | ||||
| BMI (kg/m2) | 27.62 | 27.55 | 29.37 | 29.00 | 31.98 | 31.31 | 0.021 | 0.012 | 0.288 |
| (26.08–29.17) | (26.00–29.09) | (28.09–30.64) | (27.73–30.28) | (29.62–34.34) | (28.94–33.69) | ||||
| HbA1c (%) | 5.24 | 5.12 | 5.46 | 5.31 | 5.76 | 5.61 | 0.008 | <0.001 | 0.881 |
| (5.06–5.42) | (4.94–5.30) | (5.32–5.61) | (5.16–5.46) | (5.49–6.03) | (5.33–5.88) | ||||
| Glucose 0′ (mg/dL) | 96.14 | 101.19 | 109.50 | 105.26 | 120.19 | 110.70 | <0.001 | 0.148 | <0.001 |
| (99.15–93.32) | (104.60–98) | (112.72–106.46) | (108.29–102.40) | (127.59–113.60) | (117.36–104.76) | ||||
| Glucose 120′ (mg/dL) | 85.76 | 98.79 | 96.10 | 92.09 | 165.88 | 116.15 | <0.001 | 0.006 | <0.001 |
| (75.14–97.08) | (87.19–111.11) | (86.74–105.93) | (82.82–101.86) | (143.36–190.04) | (96.61–137.47) | ||||
| Insulin 0′ (μU/mL) | 9.44 | 9.28 | 12.32 | 10.37 | 21.07 | 15.60 | <0.001 | <0.001 | 0.015 |
| (7.23–11.66) | (7.06–11.49) | (10.48–14.15) | (8.54–12.21) | (17.28–24.87) | (11.62–19.57) | ||||
| Insulin 120′ (μU/mL) | 18.23 | 19.79 | 36.36 | 22.69 | 178.29 | 82.96 | <0.001 | 0.003 | 0.014 |
| (12.13–27.40) | (13.09–29.91) | (25.99–50.87) | (16.16–31.87) | (88.88–357.62) | (39.9–172.50) | ||||
| VAT mass (kg) | 1.55 | 1.46 | 1.80 | 1.57 | 2.59 | 2.19 | 0.030 | <0.001 | 0.075 |
| (1.24–1.88) | (1.16–1.79) | (1.52–2.09) | (1.32–1.85) | (2.00–3.26) | (1.65–2.82) | ||||
| Total Chol. (mg/dL) | 193.29 | 194.11 | 194.64 | 200.66 | 206.29 | 196.01 | 0.879 | 0.762 | 0.258 |
| (175.80–210.79) | (176.48–211.74) | (180.22–209.06) | (186.15–215.18) | (179.56–233.02) | (168.67–223.35) | ||||
| TG (mg/dL) | 98.44 | 89.79 | 102.07 | 104.72 | 151.66 | 125.88 | 0.166 | 0.086 | 0.184 |
| (78.50–123.45) | (71.48–112.79) | (84.70–123.01) | (86.80–126.35) | (107.32–214.33) | (88.39–179.28) | ||||
| HDL (mg/dL) | 56.20 | 57.19 | 50.54 | 50.27 | 53.33 | 52.38 | 0.258 | 0.933 | 0.745 |
| (50.25–62.49) | (51.16–63.57) | (45.87–55.43) | (45.60–55.17) | (44.62–62.82) | (43.62–61.94) | ||||
| LDL (mg/dL) | 122.09 | 124.88 | 131.03 | 131.84 | 130.72 | 134.24 | 0.684 | 0.509 | 0.936 |
| (106.35–137.82) | (109.01–140.75) | (118.06–144.00) | (118.77–144.90) | (106.67–154.77) | (109.59–158.89) | ||||
| VO2max (mL/kg/min) | 32.33 | 32.65 | 30.44 | 32.20 | 29.07 | 31.26 | 0.427 | 0.221 | 0.009 |
| (31.18–35.48) | (31.46–35.83) | (28.66–32.22) | (30.40–34.00) | (25.78–32.36) | (27.85–34.68) | ||||
| HOMA-IR | 2.07 | 2.13 | 3.13 | 2.46 | 5.95 | 4.25 | <0.001 | 0.006 | 0.030 |
| (1.70–2.53) | (1.75–2.60) | (2.66–3.69) | (2.09–2.90) | (4.23–8.37) | (2.96–6.09) | ||||
| HOMA-b (%) | 95.75 | 80.84 | 90.42 | 81.05 | 130.24 | 105.04 | 0.146 | 0.007 | 0.729 |
| (79.92–114.71) | (67.47–96.85) | (77.88–105.00) | (69.80–94.11) | (95.58–177.46) | (75.69–145.77) | ||||
| Fat mass (kg) | 25.17 | 24.49 | 28.01 | 26.02 | 31.33 | 28.62 | 0.237 | <0.001 | 0.136 |
| (21.84–28.51) | (21.14–27.83) | (25.26–30.76) | (23.26–28.77) | (26.24–36.43) | (23.49–33.75) | ||||
| Lean mass (kg) | 60.46 | 61.64 | 61.18 | 62.39 | 59.78 | 60.74 | 0.835 | <0.001 | 0.883 |
| (57.30–63.61) | (58.48–64.80) | (58.58–63.78) | (59.79–65.00) | (54.96–64.61) | (55.91–65.57) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szczerbinski, L.; Taylor, M.A.; Puchta, U.; Konopka, P.; Paszko, A.; Citko, A.; Szczerbinski, K.; Goscik, J.; Gorska, M.; Larsen, S.; et al. The Response of Mitochondrial Respiration and Quantity in Skeletal Muscle and Adipose Tissue to Exercise in Humans with Prediabetes. Cells 2021, 10, 3013. https://doi.org/10.3390/cells10113013
Szczerbinski L, Taylor MA, Puchta U, Konopka P, Paszko A, Citko A, Szczerbinski K, Goscik J, Gorska M, Larsen S, et al. The Response of Mitochondrial Respiration and Quantity in Skeletal Muscle and Adipose Tissue to Exercise in Humans with Prediabetes. Cells. 2021; 10(11):3013. https://doi.org/10.3390/cells10113013
Chicago/Turabian StyleSzczerbinski, Lukasz, Mark Alan Taylor, Urszula Puchta, Paulina Konopka, Adam Paszko, Anna Citko, Karol Szczerbinski, Joanna Goscik, Maria Gorska, Steen Larsen, and et al. 2021. "The Response of Mitochondrial Respiration and Quantity in Skeletal Muscle and Adipose Tissue to Exercise in Humans with Prediabetes" Cells 10, no. 11: 3013. https://doi.org/10.3390/cells10113013
APA StyleSzczerbinski, L., Taylor, M. A., Puchta, U., Konopka, P., Paszko, A., Citko, A., Szczerbinski, K., Goscik, J., Gorska, M., Larsen, S., & Kretowski, A. (2021). The Response of Mitochondrial Respiration and Quantity in Skeletal Muscle and Adipose Tissue to Exercise in Humans with Prediabetes. Cells, 10(11), 3013. https://doi.org/10.3390/cells10113013