
Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
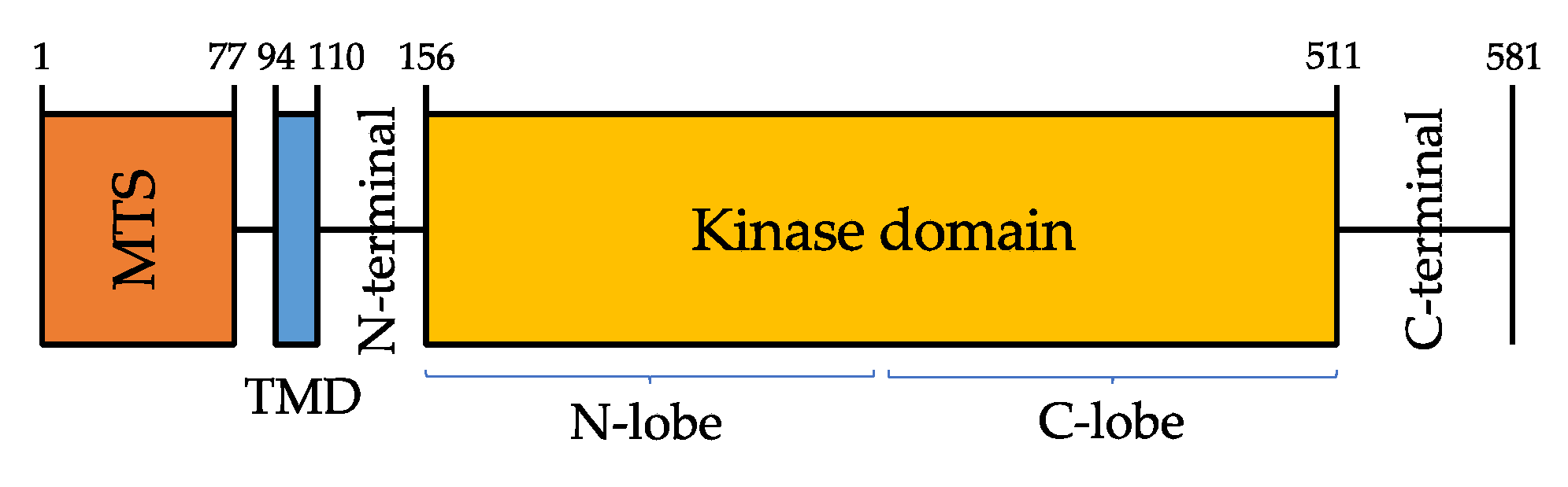
2. PINK1 Gene Structure and Most Common Mutations
3. PINK1 Protein Functions
4. Clinical Features and Genotype-Phenotype Correlation
5. Current Therapeutic Approaches
6. In Vitro Models
7. In Vivo Models
8. Future Therapeutic Targets
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Guzman, R.E.; Schwarz, Y.N.; Rettig, J.; Bruns, D. SNARE Force Synchronizes Synaptic Vesicle Fusion and Controls the Kinetics of Quantal Synaptic Transmission. J. Neurosci. 2010, 30, 10272–10281. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology Of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Matsuda, N.; Tanaka, K. The ubiquitin signal and autophagy: An orchestrated dance leading to mitochondrial degradation. EMBO Rep. 2016, 17, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Schlossmacher, M.G. Parkinson disease, 10 years after its genetic revolution: Multiple clues to a complex disorder. Neurology 2007, 69, 2093–2104. [Google Scholar] [CrossRef]
- Zhang, B.-R.; Hu, Z.-X.; Yin, X.-Z.; Cai, M.; Zhao, G.-H.; Liu, Z.-R.; Luo, W. Mutation analysis of parkin and PINK1 genes in early-onset Parkinson’s disease in China. Neurosci. Lett. 2010, 477, 19–22. [Google Scholar] [CrossRef]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Bentivoglio, A.R.; Dixon, P.H.; Ferraris, A.; Ialongo, T.; Frontali, M.; Albanese, A.; Wood, N.W. Localization of a Novel Locus for Autosomal Recessive Early-Onset Parkinsonism, PARK6, on Human Chromosome 1p35-p36. Am. J. Hum. Genet. 2001, 68, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.M.; Brancati, F.; Ferraris, A.; Graham, E.A.; Davis, M.B.; Breteler, M.M.B.; Gasser, T.; Bonifati, V.; Bentivoglio, A.R.; De Michele, G.; et al. PARK6-Linked Parkinsonism Occurs in Several European Families. Ann. Neurol. 2002, 51, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawajiri, S.; Saiki, S.; Sato, S.; Hattori, N. Genetic mutations and functions of PINK1. Trends Pharmacol. Sci. 2011, 32, 573–580. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.-J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef]
- Djarmati, A.; Hedrich, K.; Svetel, M.; Lohnau, T.; Schwinger, E.; Romac, S.; Pramstaller, P.P.; Kostić, V.; Klein, C. HeterozygousPINK1 mutations: A susceptibility factor for Parkinson disease? Mov. Disord. 2006, 21, 1526–1530. [Google Scholar] [CrossRef] [PubMed]
- Siuda, J.; Jasinska-Myga, B.; Boczarska-Jedynak, M.; Opala, G.; Fiesel, F.C.; Moussaud-Lamodière, E.L.; Scarffe, L.A.; Dawson, V.L.; Ross, O.A.; Springer, W.; et al. Early-onset Parkinson’s disease due to PINK1 p.Q456X mutation—Clinical and functional study. Parkinsonism Relat. Disord. 2014, 20, 1274–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grünewald, A.; Breedveld, G.J.; Lohmann-Hedrich, K.; Rohé, C.F.; König, I.R.; Hagenah, J.; Vanacore, N.; Meco, G.; Antonini, A.; Goldwurm, S.; et al. Biological effects of the PINK1 c.1366C>T mutation: Implications in Parkinson disease pathogenesis. Neurogenetics 2007, 8, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rohe, C.F.; Breedveld, G.J.; Fabrizio, E.; De Mari, M.; Tassorelli, C.; Tavella, A.; Marconi, R.; Nicholl, D.J.; Chien, H.F.; et al. Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 2005, 65, 87–95. [Google Scholar] [CrossRef]
- Choi, J.M.; Woo, M.S.; Ma, H.-I.; Kang, S.Y.; Sung, Y.-H.; Yong, S.W.; Chung, S.J.; Kim, J.-S.; Shin, H.-W.; Lyoo, C.H.; et al. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 2008, 9, 263–269. [Google Scholar] [CrossRef]
- Weng, Y.-H.; Chou, Y.-H.W.; Wu, W.-S.; Lin, K.-J.; Chang, H.-C.; Yen, T.-C.; Chen, R.-S.; Wey, S.-P.; Lu, C.-S. PINK1 mutation in Taiwanese early-onset parkinsonism. J. Neurol. 2007, 254, 1347–1355. [Google Scholar] [CrossRef]
- Brooks, J.; Ding, J.; Sánchez, J.S.; Paisan-Ruiz, C.; Singleton, A.B.; Scholz, S. Parkin and PINK1 mutations in early-onset Parkinson’s disease: Comprehensive screening in publicly available cases and control. J. Med. Genet. 2009, 46, 375–381. [Google Scholar] [CrossRef] [PubMed]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef] [Green Version]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Daou, S.; Sicheri, F. Vivid views of the PINK1 protein. Nat. Cell Biol. 2017, 552, 38–39. [Google Scholar] [CrossRef]
- Kumar, A.; Tamjar, J.; Waddell, A.D.; Woodroof, H.I.; Raimi, O.G.; Shaw, A.M.; Peggie, M.; Muqit, M.M.; Van Aalten, D.M. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. eLife 2017, 6, e29985. [Google Scholar] [CrossRef] [Green Version]
- Schubert, A.F.; Gladkova, C.; Pardon, E.; Wagstaff, J.L.; Freund, S.M.V.; Steyaert, J.; Maslen, S.L.; Komander, D. Structure of PINK1 in complex with its substrate ubiquitin. Nat. Cell Biol. 2017, 552, 51–56. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.A. Parkinson’s Disease in a Chemist Working with 1-Methyl-4-Phenyl-L,2,5,6-Tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310. [Google Scholar] [CrossRef]
- Schapira, A.; Cooper, J.; Dexter, D.; Jenner, P.; Clark, J.; Marsden, C. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 333, 1269. [Google Scholar] [CrossRef]
- Greene, A.W.; Grenier, K.; Aguileta, M.A.; Muise, S.; Farazifard, R.; Haque, M.E.; McBride, H.M.; Park, D.S.; Fon, E.A. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 2012, 13, 378–385. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.; Whitworth, A.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2010, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [Green Version]
- Lazarou, M.; Jin, S.M.; Kane, L.A.; Youle, R.J. Role of PINK1 Binding to the TOM Complex and Alternate Intracellular Membranes in Recruitment and Activation of the E3 Ligase Parkin. Dev. Cell 2012, 22, 320–333. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Dorn, G.W., II. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K. The PINK1–Parkin axis: An Overview. Neurosci. Res. 2020, 159, 9–15. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Martinez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK 1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Sauvé, V.; Lilov, A.; Seirafi, M.; Vranas, M.; Rasool, S.; Kozlov, G.; Sprules, T.; Wang, J.; Trempe, J.; Gehring, K. A Ubl/ubiquitin switch in the activation of Parkin. EMBO J. 2015, 34, 2492–2505. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nat. Cell Biol. 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Wang, L.; Cho, Y.-L.; Tang, Y.; Wang, J.; Park, J.-E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1–Parkin-mediated mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Thomas, R.E.; Andrews, L.A.; Burman, J.L.; Lin, W.-Y.; Pallanck, L.J. PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix. PLoS Genet. 2014, 10, e1004279. [Google Scholar] [CrossRef] [Green Version]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The Mitochondrial Fusion-Promoting Factor Mitofusin Is a Substrate of the PINK1/Parkin Pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Voigt, A.; Berlemann, L.A.; Winklhofer, K.F. The mitochondrial kinase PINK1: Functions beyond mitophagy. J. Neurochem. 2016, 139, 232–239. [Google Scholar] [CrossRef] [Green Version]
- Soubannier, V.; McLelland, G.-L.; Zunino, R.; Braschi, E.; Rippstein, P.; Fon, E.A.; McBride, H.M. A Vesicular Transport Pathway Shuttles Cargo from Mitochondria to Lysosomes. Curr. Biol. 2012, 22, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Braschi, E.; Goyon, V.; Zunino, R.; Mohanty, A.; Xu, L.; McBride, H. Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Curr. Biol. 2010, 20, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilariño-Güell, C.; Wider, C.; Ross, O.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Charman, M.; Kennedy, B.E.; Osborne, N.; Karten, B. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 2010, 51, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Elbaz-Alon, Y.; Rosenfeld-Gur, E.; Shinder, V.; Futerman, A.; Geiger, T.; Schuldiner, M. A Dynamic Interface between Vacuoles and Mitochondria in Yeast. Dev. Cell 2014, 30, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todkar, K.; Chikhi, L.; Germain, M. Mitochondrial interaction with the endosomal compartment in endocytosis and mitochondrial transfer. Mitochondrion 2019, 49, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Kawarai, T.; Paitel, E.; Sanjo, N.; Maj, M.; Scheid, M.; Chen, F.; Gu, Y.; Hasegawa, H.; Salehi-Rad, S.; et al. Wild-Type PINK1 Prevents Basal and Induced Neuronal Apoptosis, a Protective Effect Abrogated by Parkinson Disease-Related Mutations. J. Biol. Chem. 2005, 280, 34025–34032. [Google Scholar] [CrossRef] [Green Version]
- Klinkenberg, M.; Thurow, N.; Gispert, S.; Ricciardi, F.; Eich, F.; Prehn, J.; Auburger, G.; Kögel, D. Enhanced vulnerability of PARK6 patient skin fibroblasts to apoptosis induced by proteasomal stress. Neuroscience 2010, 166, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.E.; Mount, M.P.; Safarpour, F.; Abdel-Messih, E.; Callaghan, S.; Mazerolle, C.; Kitada, T.; Slack, R.; Wallace, V.; Shen, J.; et al. Inactivation of Pink1 Gene in Vivo Sensitizes Dopamine-Producing Neurons to 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and Can Be Rescued by Autosomal Recessive Parkinson Disease Genes, Parkin or DJ-1. J. Biol. Chem. 2012, 287, 23162–23170. [Google Scholar] [CrossRef] [Green Version]
- Geisler, S.; Holmström, K.; Skujat, D.; Fiesel, F.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Ricciardi, L.; Petrucci, S.; Guidubaldi, A.; Ialongo, T.; Serra, L.; Ferraris, A.; Spanò, B.; Bozzali, M.; Valente, E.M.; Bentivoglio, A.R. Phenotypic variability of PINK1 expression: 12 Years’ clinical follow-up of two Italian families. Mov. Disord. 2014, 29, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Piredda, R.; Desmarais, P.; Masellis, M.; Gasca-Salas, C. Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: A systematic review. Eur. J. Neurol. 2019, 27, 229–234. [Google Scholar] [CrossRef]
- Nishioka, K.; Kefi, M.; Jasinska-Myga, B.; Wider, C.; Vilarino-Guell, C.; Ross, O.; Heckman, M.G.; Middleton, L.T.; Ishihara-Paul, L.; Gibson, R.; et al. A comparative study of LRRK2, PINK1 and genetically undefined familial Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2009, 81, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, K.; Hagenah, J.; Djarmati, A.; Hiller, A.; Lohnau, T.; Lasek, K.; Grünewald, A.; Hilker, R.; Steinlechner, S.; Boston, H.; et al. Clinical Spectrum of Homozygous and Heterozygous PINK1 Mutations in a Large German Family with Parkinson Disease. Arch. Neurol. 2006, 63, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Puschmann, A.; Fiesel, F.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Krohn, L.; Grenn, F.P.; Makarious, M.B.; Kim, J.J.; Bandres-Ciga, S.; Roosen, D.A.; Gan-Or, Z.; Nalls, M.A.; Singleton, A.B.; Blauwendraat, C. Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 2020, 91, 168.e1–168.e5. [Google Scholar] [CrossRef]
- Schneider, S.A.; Klein, C. PINK1 Type of Young-Onset Parkinson Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Samaranch, L.; Lorenzo-Betancor, O.; Arbelo, J.M.; Ferrer, I.; Lorenzo, E.; Irigoyen, J.; Pastor, M.A.; Marrero, C.; Isla, C.; Herrera-Henriquez, J.; et al. PINK1-Linked Parkinsonism Is Associated with Lewy Body Pathology. Brain 2010, 133, 1128–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; Ferraris, A.; Brusa, L.; Romano, F.; Lombardi, F.; Barzaghi, C.; Stanzione, P.; Garavaglia, B.; Dallapiccola, B.; Valente, E.M. Late Onset Sporadic Parkinson’s Disease Caused by PINK1 Mutations: Clinical and Functional Study: Late Onset Sporadic PD Due to PINK1 Mutations. Mov. Disord. 2008, 23, 881–885. [Google Scholar] [CrossRef]
- Ephraty, L.; Porat, O.; Israeli, D.; Cohen, O.S.; Tunkel, O.; Yael, S.; Hatano, Y.; Hattori, N.; Hassin-Baer, S. Neuropsychiatric and Cognitive Features in Autosomal-Recessive Early Parkinsonism Due to PINK1 Mutations. Mov. Disord. 2007, 22, 566–569. [Google Scholar] [CrossRef]
- Albanese, A.; Valente, E.M.; Romito, L.M.; Bellacchio, E.; Elia, A.E.; Dallapiccola, B. The PINK1 Phenotype Can Be Indistinguishable from Idiopathic Parkinson Disease. Neurology 2005, 64, 1958–1960. [Google Scholar] [CrossRef]
- Savettieri, G.; Annesi, G.; Civitelli, D.; Cirò Candiano, I.C.; Salemi, G.; Ragonese, P.; Annesi, F.; Tarantino, P.; Terruso, V.; D’Amelio, M.; et al. Identification of the Novel D297fsX318 PINK1 Mutation and Phenotype Variation in a Family with Early-Onset Parkinson’s Disease. Parkinsonism Relat. Disord. 2008, 14, 509–512. [Google Scholar] [CrossRef]
- Doostzadeh, J.; Tetrud, J.W.; Allen-Auerbach, M.; Langston, J.W.; Schüle, B. Novel Features in a Patient Homozygous for the L347P Mutation in the PINK1 Gene. Parkinsonism Relat. Disord. 2007, 13, 359–361. [Google Scholar] [CrossRef]
- Tuin, I.; Voss, U.; Kessler, K.; Krakow, K.; Hilker, R.; Morales, B.; Steinmetz, H.; Auburger, G. Sleep Quality in a Family with Hereditary Parkinsonism (PARK6). Sleep Med. 2008, 9, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Hiller, A.; Hagenah, J.M.; Djarmati, A.; Hedrich, K.; Reetz, K.; Schneider-Gold, C.; Kress, W.; Münchau, A.; Klein, C. Phenotypic Spectrum of PINK1-Associated Parkinsonism in 15 Mutation Carriers from 1 Family. Mov. Disord. 2007, 22, 145–147. [Google Scholar] [CrossRef]
- Leutenegger, A.-L.; Salih, M.A.M.; Ibanez, P.; Mukhtar, M.M.; Lesage, S.; Arabi, A.; Lohmann, E.; Durr, A.; Ahmed, A.E.M.; Brice, A. Juvenile-Onset Parkinsonism as a Result of the First Mutation in the Adenosine Triphosphate Orientation Domain of PINK1. Arch. Neurol. 2006, 63, 1257–1261. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Zamora, A.; Ostrem, J.L. Globus Pallidus Interna or Subthalamic Nucleus Deep Brain Stimulation for Parkinson Disease. JAMA Neurol. 2018, 75, 367–372. [Google Scholar] [CrossRef]
- Moro, E.; Volkmann, J.; König, I.; Winkler, S.; Hiller, A.; Hassin-Baer, S.; Herzog, J.; Schnitzler, A.; Lohmann, K.; Pinsker, M.O.; et al. Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology 2008, 70, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Borellini, L.; Cogiamanian, F.; Carrabba, G.; Locatelli, M.; Rampini, P.; Di Fonzo, A.; Bana, C.; Barbieri, S.; Ardolino, G. Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: A case report. Parkinsonism Relat. Disord. 2017, 38, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Borellini, L.; Cogiamanian, F.; Ardolino, G. Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: An update. Parkinsonism Relat. Disord. 2021, 82, 104–105. [Google Scholar] [CrossRef]
- Johansen, K.K.; Jørgensen, J.V.; White, L.R.; Farrer, M.; Aasly, J.O. Parkinson-related genetics in patients treated with deep brain stimulation. Acta Neurol. Scand. 2011, 123, 201–206. [Google Scholar] [CrossRef]
- Nakahara, K.; Ueda, M.; Yamada, K.; Koide, T.; Yoshimochi, G.; Funayama, M.; Kim, J.-H.; Yamakawa, S.; Mori, A.; Misumi, Y.; et al. Juvenile-onset parkinsonism with digenic parkin and PINK1 mutations treated with subthalamic nucleus stimulation at 45years after disease onset. J. Neurol. Sci. 2014, 345, 276–277. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Koike, M.; Kuzumaki, N.; Hayakawa, H.; Nihira, T.; Kobayashi, T.; Ohyama, M.; Sato, S.; et al. Mitochondrial dysfunction associated with increased oxidative stress and α-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol. Brain 2012, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.Y.; Kishinevsky, S.; Mazzulli, J.R.; Graziotto, J.; Mrejeru, A.; Mosharov, E.V.; Puspita, L.; Valiulahi, P.; Sulzer, D.; Milner, T.A.; et al. Parkin and PINK1 Patient iPSC-Derived Midbrain Dopamine Neurons Exhibit Mitochondrial Dysfunction and α-Synuclein Accumulation. Stem Cell Rep. 2016, 7, 664–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Key, J.; Sen, N.E.; Arsović, A.; Krämer, S.; Hülse, R.; Khan, N.N.; Meierhofer, D.; Gispert, S.; Koepf, G.; Auburger, G. Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells 2020, 9, 2229. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Ishikawa, K.-I.; Inoshita, T.; Shiba-Fukushima, K.; Saiki, S.; Hatano, T.; Mori, A.; Oji, Y.; Okuzumi, A.; Li, Y.; et al. Identifying Therapeutic Agents for Amelioration of Mitochondrial Clearance Disorder in Neurons of Familial Parkinson Disease. Stem Cell Rep. 2020, 14, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.; Götz, J. Shedding light on mitophagy in neurons: What is the evidence for PINK1/Parkin mitophagy in vivo? Cell. Mol. Life Sci. 2017, 75, 1151–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration—What do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53. [Google Scholar] [CrossRef]
- Fiesel, F.; Ando, M.; Hudec, R.; Hill, A.R.; Castanedes-Casey, M.; Caulfield, T.R.; Moussaud-Lamodière, E.L.; Stankowski, J.N.; Bauer, P.; Betancor, O.L.; et al. (Patho-)physiological relevance of PINK 1-dependent ubiquitin phosphorylation. EMBO Rep. 2015, 16, 1114–1130. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Greene, J.C.; Whitworth, A.; Kuo, I.; Andrews, L.A.; Feany, M.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [Green Version]
- Pesah, Y.; Pham, T.; Burgess, H.; Middlebrooks, B.; Verstreken, P.; Zhou, Y.; Harding, M.; Bellen, H.; Mardon, G. Drosophila parkinmutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 2004, 131, 2183–2194. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nat. Cell Biol. 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nat. Cell Biol. 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Yang, Y.; Gehrke, S.; Imai, Y.; Huang, Z.; Ouyang, Y.; Wang, J.; Yang, L.; Beal, M.F.; Vogel, O.H.; Lu, B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA 2006, 103, 10793–10798. [Google Scholar] [CrossRef] [Green Version]
- Todd, A.M.; Staveley, B.E. Pink1 suppresses α-synuclein-induced phenotypes in a Drosophila model of Parkinson’s disease. Genome 2008, 51, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- De Castro, I.P.; Costa, A.C.; Lam, D.; Tufi, R.; Fedele, V.; Moisoi, N.; Dinsdale, D.; Deas, E.; Loh, S.H.Y.; Martins, L.M. Genetic analysis of mitochondrial protein misfolding in Drosophila melanogaster. Cell Death Differ. 2012, 19, 1308–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincow, E.S.; Merrihew, G.; Thomas, R.E.; Shulman, N.J.; Beyer, R.P.; MacCoss, M.J.; Pallanck, L.J. The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6400–6405. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-Associated Parkinson’s Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufi, R.; Gandhi, S.; de Castro, I.P.; Lehmann, S.; Angelova, P.R.; Dinsdale, D.; Deas, E.; Plun-Favreau, H.; Nicotera, P.; Abramov, A.; et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat. Cell Biol. 2014, 16, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, S.; Loh, S.H.Y.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, 8844–8853. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with High Ca2+ Close to IP 3-Sensitive Channels that Are Sensed by Neighboring Mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.-S.; Huh, S.; Liu, S.; Lee, D.-Y.; Hong, S.H.; Yu, K.; Lu, B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca2+ Homeostasis in Neural Stem Cell Development. Dev. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.; Schwarz, T.L. PINK1 and Parkin Target Miro for Phosphorylation and Degradation to Arrest Mitochondrial Motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s Disease–Associated Kinase PINK1 Regulates Miro Protein Level and Axonal Transport of Mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Stevens, D.A.; Kang, S.-U.; Jiang, H.; Lee, Y.-I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.-H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.-I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) Repression of PGC-1α Contributes to Neurodegeneration in Parkinson’s Disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirooznia, S.K.; Yuan, C.; Khan, M.R.; Karuppagounder, S.S.; Wang, L.; Xiong, Y.; Kang, S.U.; Lee, Y.; Dawson, V.L.; Dawson, T.M. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol. Neurodegener. 2020, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nat. Cell Biol. 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Andreazza, S.; Whitworth, A.J. The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci. Rep. 2020, 10, 2693. [Google Scholar] [CrossRef]
- Julienne, H.; Buhl, E.; Leslie, D.S.; Hodge, J.J. Drosophila PINK1 and parkin loss-of-function mutants display a range of non-motor Parkinson’s disease phenotypes. Neurobiol. Dis. 2017, 104, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Vilain, S.; Vints, K.; Gounko, N.; Verstreken, P.; Vandenberghe, W. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. eLife 2018, 7, 35878. [Google Scholar] [CrossRef]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 phosphorylates Drp1 S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, 48686. [Google Scholar] [CrossRef]
- Terriente-Felix, A.; Wilson, E.L.; Whitworth, A.J. Drosophila phosphatidylinositol-4 kinase fwd promotes mitochondrial fission and can suppress Pink1/parkin phenotypes. PLoS Genet. 2020, 16, e1008844. [Google Scholar] [CrossRef]
- Han, Y.; Zhuang, N.; Wang, T. Roles of PINK1 in regulation of systemic growth inhibition induced by mutations of PTEN in Drosophila. Cell Rep. 2021, 34, 108875. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-L.; Chou, A.-H.; Wu, A.-S.; Chen, S.-Y.; Weng, Y.-H.; Kao, Y.-C.; Yeh, T.-H.; Chu, P.-J.; Lu, C.-S. PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-L.; Chou, A.-H.; Yeh, T.-H.; Li, A.H.; Chen, Y.-L.; Kuo, Y.-L.; Tsai, S.-R.; Yu, S.-T. PINK1 mutants associated with recessive Parkinson’s disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol. Dis. 2007, 28, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, T.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.-I.; Bower, A.; Jiang, H.; Kang, S.-U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nat. Cell Biol. 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Hang, L.; Thundyil, J.; Goh, G.W.Y.; Lim, K.-L. AMP Kinase Activation is Selectively Disrupted in the Ventral Midbrain of Mice Deficient in Parkin or PINK1 Expression. Neuromol. Med. 2019, 21, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rüb, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2014, 24, 1061–1076. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Barini, E.; Pohjolan-Pirhonen, R.; Brooks, S.P.; Singh, F.; Burel, S.; Balk, K.; Kumar, A.; Garriga, L.M.; Prescott, A.R.; et al. Phosphorylation of Parkin at serine 65 is essential for its activation in vivo. Open Biol. 2018, 8, 180108. [Google Scholar] [CrossRef] [Green Version]
- Zhi, L.; Qin, Q.; Muqeem, T.; Seifert, E.L.; Liu, W.; Zheng, S.; Li, C.; Zhang, H. Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol. Aging 2019, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sardi, S.P.; Cedarbaum, J.M.; Brundin, P. Targeted Therapies for Parkinson’s Disease: From Genetics to the Clinic. Mov. Disord. 2018, 33, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Wells, G.; Campanella, N.D.G.M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moors, T.E.; Hoozemans, J.J.M.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.-C.; Van De Berg, W.D.J. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A ΔΨm Independent Pharmacological Regulator of Mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, srep44373. [Google Scholar] [CrossRef]
- Abudu, Y.P.; Pankiv, S.; Mathai, B.J.; Lamark, T.; Johansen, T.; Simonsen, A. NIPSNAP1 and NIPSNAP2 act as “eat me” signals to allow sustained recruitment of autophagy receptors during mitophagy. Autophagy 2019, 15, 1845–1847. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.; Phu, L.; Reichelt, M.; Bakalarski, C.; Song, Q.; Foreman, O.; Kirkpatrick, D.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nat. Cell Biol. 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Wang, Y.; Serricchio, M.; Jauregui, M.; Shanbhag, R.; Stoltz, T.; Di Paolo, C.T.; Kim, P.K.; McQuibban, G.A. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 2015, 11, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.-L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP 8 regulates mitophagy by removing K 6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Muqit, M.M. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vizziello, M.; Borellini, L.; Franco, G.; Ardolino, G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells 2021, 10, 3022. https://doi.org/10.3390/cells10113022
Vizziello M, Borellini L, Franco G, Ardolino G. Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells. 2021; 10(11):3022. https://doi.org/10.3390/cells10113022
Chicago/Turabian StyleVizziello, Maria, Linda Borellini, Giulia Franco, and Gianluca Ardolino. 2021. "Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease" Cells 10, no. 11: 3022. https://doi.org/10.3390/cells10113022
APA StyleVizziello, M., Borellini, L., Franco, G., & Ardolino, G. (2021). Disruption of Mitochondrial Homeostasis: The Role of PINK1 in Parkinson’s Disease. Cells, 10(11), 3022. https://doi.org/10.3390/cells10113022