
Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery

 , ,
, , 
Abstract
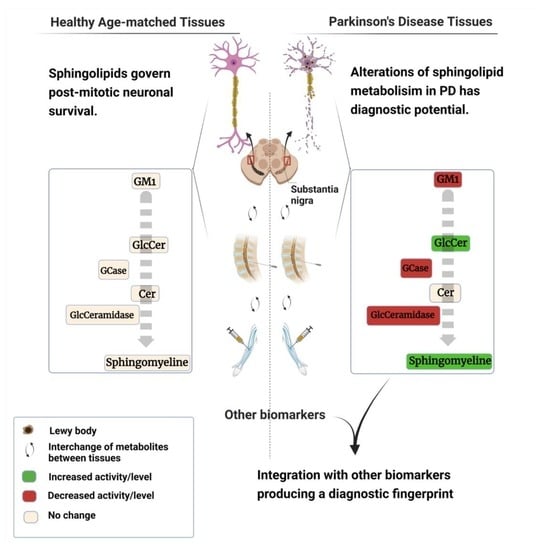
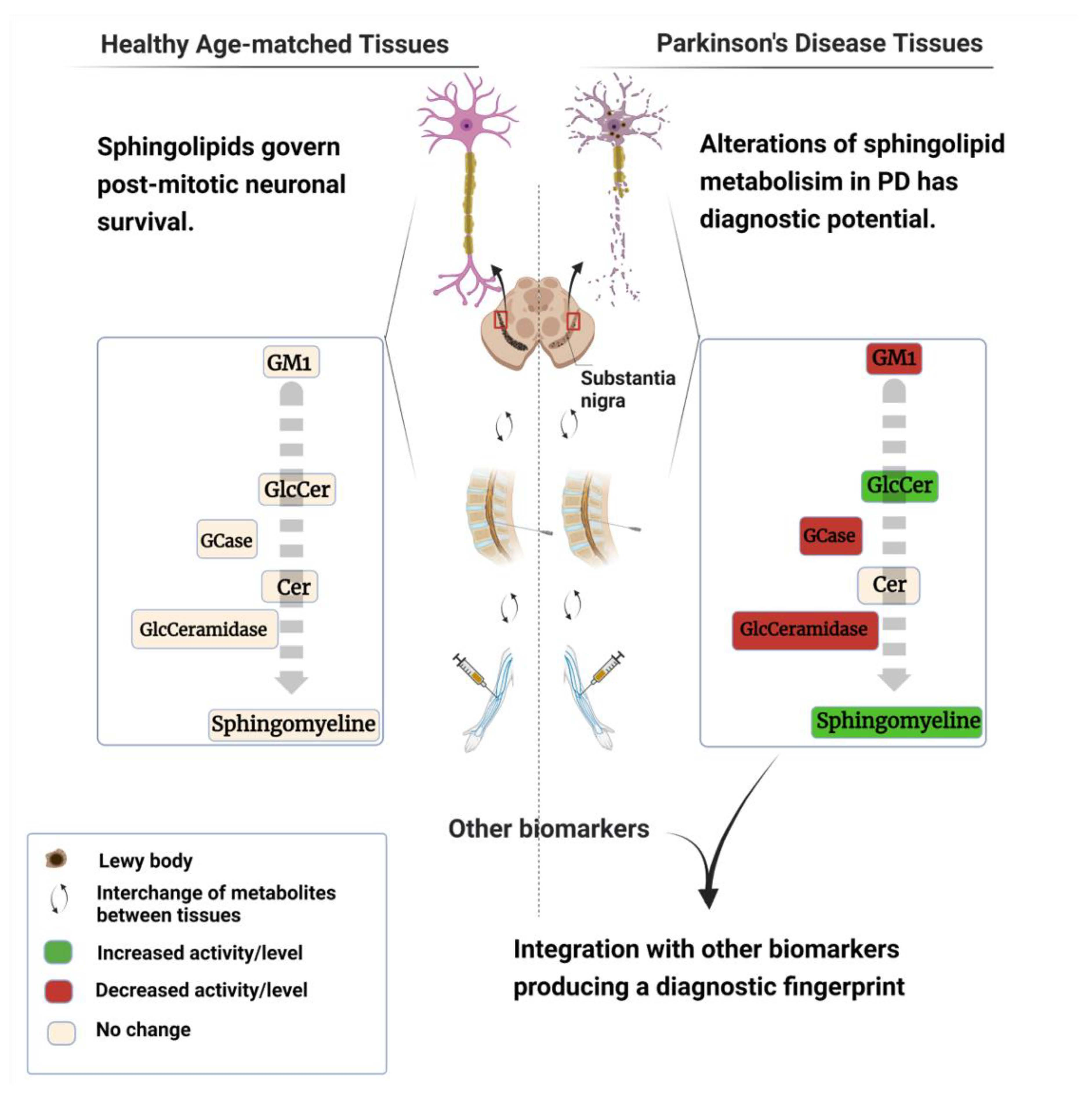
:
1. Introduction
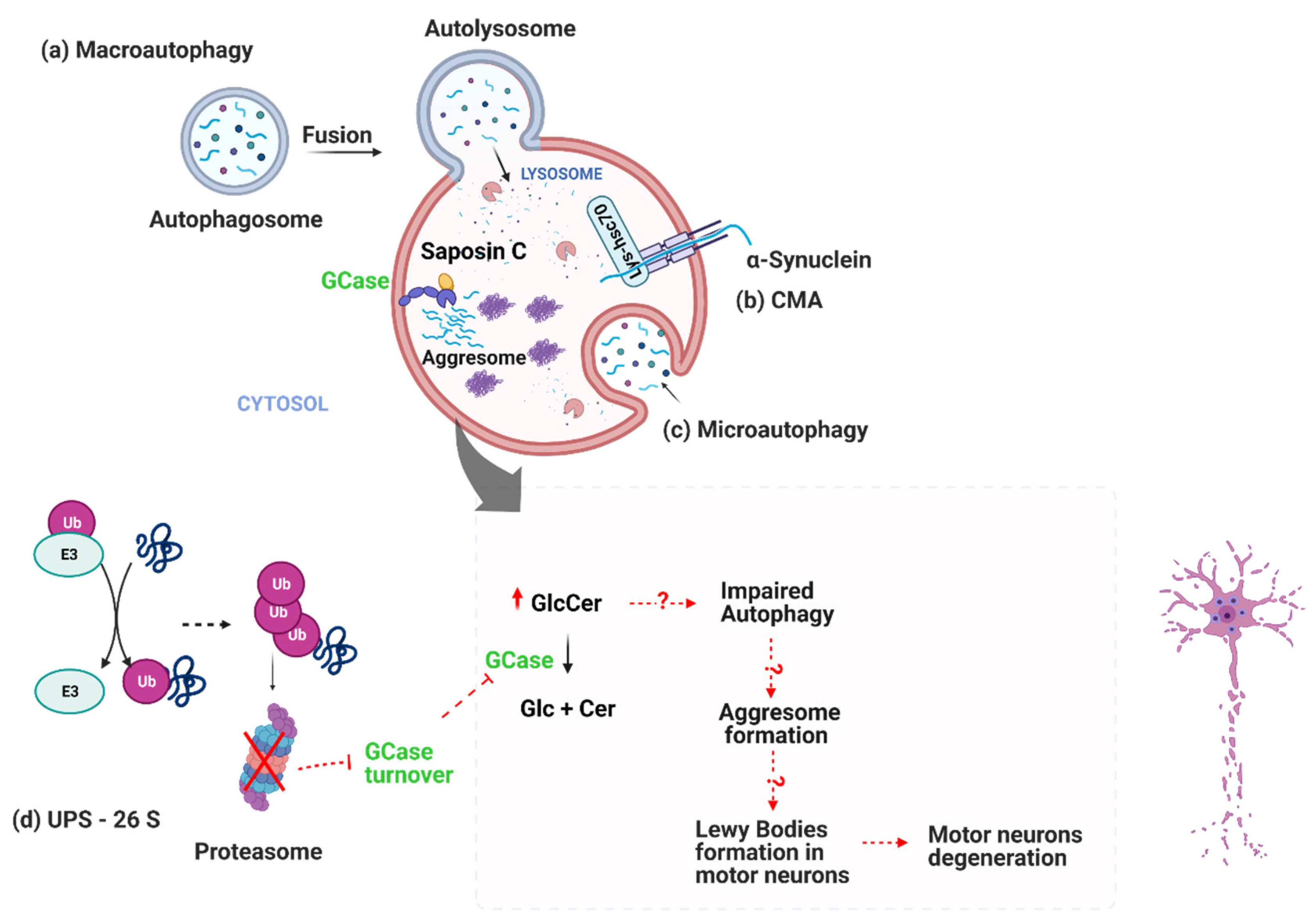
2. Protein Clearance System Impairment in PD-Ubiquitin-Proteasome System (UPS)
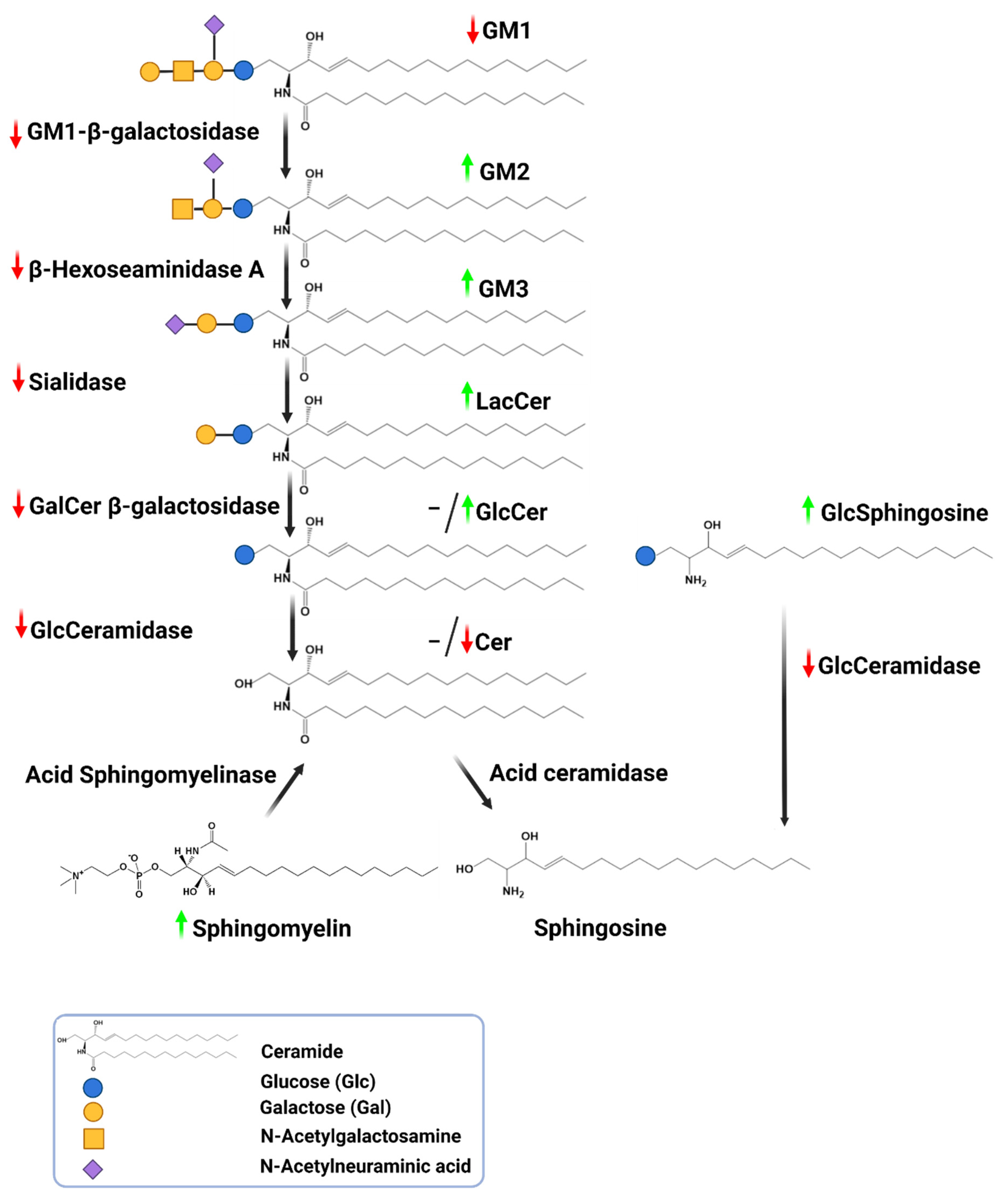
3. Protein Clearance System Impairment in PD-Autophagy-Lysosome Pathway Fails in Clearing Sphingolipids
3.1. Is GCase Activity a Potential Biomarker for iPD?
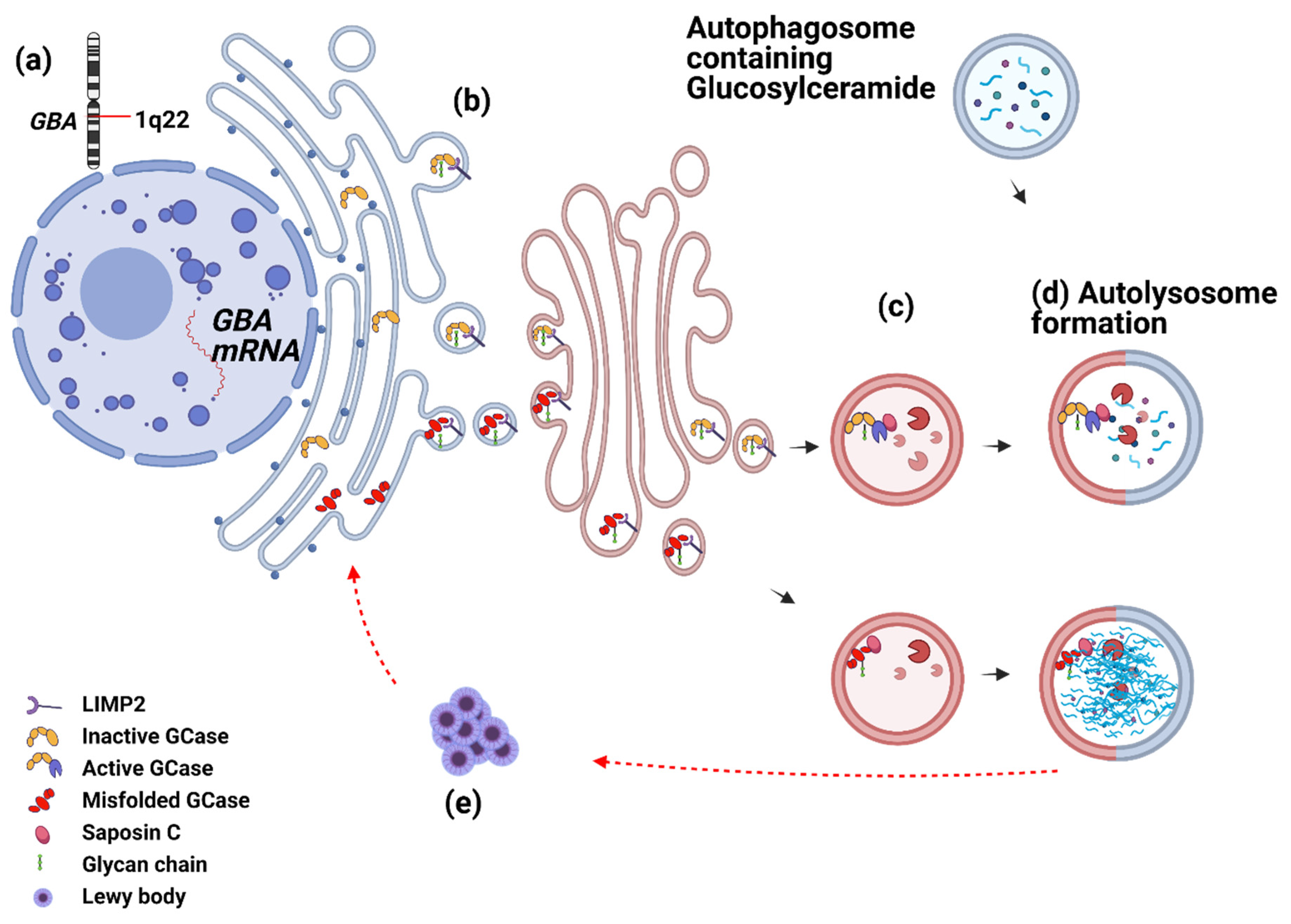
3.1.1. GCase from Birth to Death—What Can Go Wrong?
3.1.2. Saposin C and GCase
3.1.3. Do GCase Substrates Differentiate iPD?
4. Sphingolipids in PD Diagnosis
5. Would Integrating Biomarkers Make iPD Diagnosis Possible?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Poewe, W.; Litvan, I.; Lewis, S.; Lang, A.E.; Halliday, G.; Goetz, C.G.; Chan, P.; Slow, E.; Seppi, K.; et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2018, 33, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Logroscino, G.; Rizzo, G.; Copetti, M.; Arcuti, S.; Martino, D.; Fontana, A. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 2016, 86, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Progression of Parkinson Disease: Are we making progress in charting the course? Arch. Neurol. 2005, 62, 351–352. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef]
- Robbins, C.B.; Thompson, A.C.; Bhullar, P.K.; Koo, H.Y.; Agrawal, R.; Soundararajan, S.; Yoon, S.P.; Polascik, B.W.; Scott, B.L.; Grewal, D.S.; et al. Characterization of Retinal Microvascular and Choroidal Structural Changes in Parkinson Disease. JAMA Ophthalmol. 2021, 139, 182–188. [Google Scholar] [CrossRef]
- Rascunà, C.; Cicero, C.E.; Chisari, C.G.; Russo, A.; Giuliano, L.; Castellino, N.; Terravecchia, C.; Grillo, M.; Longo, A.; Avitabile, T. Retinal thickness and microvascular pathway in Idiopathic Rapid eye movement sleep behaviour disorder and Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 88, 40–45. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Zhu, C.; Li, G.; Kang, J.; Chen, F.; Yang, L.; Wang, X. The diagnostic value of SNpc using NM-MRI in Parkinson’s disease: Meta-analysis. Neurol. Sci. 2019, 40, 2479–2489. [Google Scholar] [CrossRef]
- Murakami, Y.; Kakeda, S.; Watanabe, K.; Ueda, I.; Ogasawara, A.; Moriya, J.; Ide, S.; Futatsuya, K.; Sato, T.; Okada, K.; et al. Usefulness of Quantitative Susceptibility Mapping for the Diagnosis of Parkinson Disease. Am. J. Neuroradiol. 2015, 36, 1102–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Watanabe, Y.; Tanaka, H.; Mihara, M.; Mochizuki, H.; Yamamoto, K.; Liu, T.; Wang, Y.; Tomiyama, N. Comprehensive MRI quantification of the substantia nigra pars compacta in Parkinson’s disease. Eur. J. Radiol. 2018, 109, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, P.; Krismer, F.; Poewe, W.; Seppi, K. Meta-analysis of dorsolateral nigral hyperintensity on magnetic resonance imaging as a marker for Parkinson’s disease. Mov. Disord. 2017, 32, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Matesan, M.; Gaddikeri, S.; Longfellow, K.; Miyaoka, R.; Elojeimy, S.; Elman, S.; Hu, S.-C.; Minoshima, S.; Lewis, D. I-123 DaTscan SPECT Brain Imaging in Parkinsonian Syndromes: Utility of the Putamen-to-Caudate Ratio. J. Neuroimaging 2018, 28, 629–634. [Google Scholar] [CrossRef]
- Beauchamp, L.C.; Villemagne, V.L.; Finkelstein, D.I.; Doré, V.; Bush, A.I.; Barnham, K.J.; Rowe, C.C. Reduced striatal vesicular monoamine transporter 2 in REM sleep behavior disorder: Imaging prodromal parkinsonism. Sci. Rep. 2020, 10, 17631. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of α-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.; Isacson, O. Selective loss of 20S proteasome α-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H.V. Chaperone-Mediated Autophagy Markers in Parkinson Disease Brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Lerche, S.; Sjödin, S.; Brinkmalm, A.; Blennow, K.; Wurster, I.; Roeben, B.; Zimmermann, M.; Hauser, A.; Liepelt-Scarfone, I.; Waniek, K.; et al. CSF Protein Level of Neurotransmitter Secretion, Synaptic Plasticity, and Autophagy in PD and DLB. Mov. Disord. 2021, 36, 2595–2604. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Alteration of autophagosomal proteins (LC3, GABARAP and GATE-16) in Lewy body disease. Neurobiol. Dis. 2011, 43, 690–697. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Gordon, S.E.; Shambrook, M.; Nasiri Kenari, A.; Coleman, B.M.; Perugini, M.A.; Lawson, V.A.; Finkelstein, D.I.; Hill, A.F. An intact membrane is essential for small extracellular vesicle-induced modulation of α-synuclein fibrillization. J. Extracell. Vesicles 2020, 10, e12034. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 2021, 110, 436–451.e11. [Google Scholar] [CrossRef]
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Reza, S.M.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef]
- Klemann, C.J.H.M.; Martens, G.J.M.; Sharma, M.; Martens, M.B.; Isacson, O.; Gasser, T.; Visser, J.E.; Poelmans, G. Integrated molecular landscape of Parkinson’s disease. NPJ Park. Dis. 2017, 3, 14. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Zhang, R.; Pan, C.; Xu, J.; Sun, H.; Hua, P.; Zhang, L.; Zhang, W.; Xu, P.; Ma, C.; et al. Prevalence and genotype-phenotype correlations of GBA -related Parkinson disease in a large Chinese cohort. Eur. J. Neurol. 2021, 29, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Custodia, A.; Aramburu-Núñez, M.; Correa-Paz, C.; Posado-Fernández, A.; Gómez-Larrauri, A.; Castillo, J.; Gómez-Muñoz, A.; Sobrino, T.; Ouro, A. Ceramide Metabolism and Parkinson’s Disease—Therapeutic Targets. Biomolecules 2021, 11, 945. [Google Scholar] [CrossRef] [PubMed]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Lerche, S.; Schulte, C.; Wurster, I.; Machetanz, G.; Roeben, B.; Zimmermann, M.; Deuschle, C.; Hauser, A.; Böhringer, J.; Krägeloh-Mann, I.; et al. The Mutation Matters: CSF Profiles of GCase, Sphingolipids, α-Synuclein in PDGBA. Mov. Disord. 2021, 36, 1216–1228. [Google Scholar] [CrossRef]
- Alselehdar, S.K.; Chakraborty, M.; Chowdhury, S.; Alcalay, R.N.; Surface, M.; Ledeen, R. Subnormal GM1 in PBMCs: Promise for Early Diagnosis of Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 11522. [Google Scholar] [CrossRef]
- Alcalay, R.; Wolf, P.; Levy, O.; Kang, U.J.; Waters, C.; Fahn, S.; Ford, B.; Kuo, S.; Vanegas, N.; Shah, H. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol. Dis. 2018, 112, 85–90. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.; Van Der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Sakharkar, M.K.; Singh, S.K.K.; Rajamanickam, K.; Essa, M.M.; Yang, J.; Chidambaram, S.B. A systems biology approach towards the identification of candidate therapeutic genes and potential biomarkers for Parkinson’s disease. PLoS ONE 2019, 14, e0220995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.E.; Samant, R.S. Alternative systems for misfolded protein clearance: Life beyond the proteasome. FEBS J. 2021, 288, 4464–4487. [Google Scholar] [CrossRef]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between Early-Onset Parkinson’s Disease and Mutations in the Parkin Gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.A.; Kim, D.; Hwang, H.; Kim, M.S.; Ma, S.-X.; Kwon, S.-H.; Kweon, S.H.; Yoo, J.M.; Choi, S.; Kwon, S.H.; et al. TRIP12 ubiquitination of glucocerebrosidase contributes to neurodegeneration in Parkinson’s disease. Neuron 2021, 109, 3758–3774.e11. [Google Scholar] [CrossRef]
- McNaught, K.S.P.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Cerri, S.; Blandini, F. Role of Autophagy in Parkinson’s Disease. Curr. Med. Chem. 2019, 26, 3702–3718. [Google Scholar] [CrossRef]
- Lu, J.; Wu, M.; Yue, Z. Autophagy and Parkinson’s Disease. In Autophagy: Biology and Diseases: Clinical Science; Le, W., Ed.; Springer: Singapore, 2020; pp. 21–51. [Google Scholar]
- Shen, W.; Jiang, L.; Zhao, J.; Wang, H.; Hu, M.; Chen, L.; Chen, Y. Bioactive lipids and their metabolism: New therapeutic opportunities for Parkinson’s disease. Eur. J. Neurosci. 2022, 55, 846–872. [Google Scholar] [CrossRef]
- Mielke, M.M.; Maetzler, W.; Haughey, N.J.; Bandaru, V.V.R.; Savica, R.; Deuschle, C.; Gasser, T.; Hauser, A.-K.; Gräber-Sultan, S.; Schleicher, E.; et al. Plasma Ceramide and Glucosylceramide Metabolism Is Altered in Sporadic Parkinson’s Disease and Associated with Cognitive Impairment: A Pilot Study. PLoS ONE 2013, 8, e73094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, R.B.; Perotte, A.; Zhou, B.; Liong, C.; Shorr, E.J.; Marder, K.S.; Kang, U.J.; Waters, C.H.; Levy, O.A.; Xu, Y.; et al. Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: A lipidomic analysis. PLoS ONE 2017, 12, e0172348. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.K.; Li, H.; Muñoz, S.S.; Knoch, B.; Batterham, M.; Murphy, K.E.; Halliday, G.M.; Garner, B. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov. Disord. 2014, 29, 518–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, Y.E.; Park, H.; Chiang, M.S.R.; Tuncali, I.; Liu, G.; Locascio, J.J.; Shirvan, J.; Hutten, S.J.; Rotunno, M.S.; Viel, C.; et al. Glucosylceramide in cerebrospinal fluid of patients with GBA-associated and idiopathic Parkinson’s disease enrolled in PPMI. NPJ Park. Dis. 2021, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Irigoyen, J.; Cartas-Cejudo, P.; Iruarrizaga-Lejarreta, M.; Santamaría, E. Alteration in the Cerebrospinal Fluid Lipidome in Parkinson’s Disease: A Post-Mortem Pilot Study. Biomedicines 2021, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Hadaczek, P.; Wu, G.; Sharma, N.; Ciesielska, A.; Bankiewicz, K.; Davidow, A.; Lu, Z.-H.; Forsayeth, J.; Ledeen, R.W. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp. Neurol. 2015, 263, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lu, Z.H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; Van De Berg, W.D.J. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. 2013, 28, 747–754. [Google Scholar] [CrossRef]
- Parnetti, L.; Paciotti, S.; Eusebi, P.; Dardis, A.; Zampieri, S.; Chiasserini, D.; Tasegian, A.; Tambasco, N.; Bembi, B.; Calabresi, P.; et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in parkinson’s disease patients. Mov. Disord. 2017, 32, 1423–1431. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.; Moloney, E.B.; Macbain, Z.K.; Hallett, P.J.; Isacson, O. Fibroblasts from idiopathic Parkinson’s disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2. Mol. Brain 2021, 14, 16. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O.; Kanfer, J.N.; Bradley, R.M.; Shapiro, D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J. Clin. Investig. 1966, 45, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Caminiti, S.P.; Carli, G.; Avenali, M.; Blandini, F.; Perani, D. Clinical and Dopamine Transporter Imaging Trajectories in a Cohort of Parkinson’s Disease Patients with GBA Mutations. Mov. Disord. 2021, 37, 106–118. [Google Scholar] [CrossRef]
- O’Regan, G.; Desouza, R.-M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Park. Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Mallett, V.; Ross, J.P.; Alcalay, R.N.; Ambalavanan, A.; Sidransky, E.; Dion, P.A.; Rouleau, G.A.; Gan-Or, Z. GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurol. Genet. 2016, 2, e104. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2012, 28, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson Risk in Ashkenazi Jewish Patients With Gaucher Disease and GBA Heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Castrioto, A.; Chiasserini, D.; Persichetti, E.; Tambasco, N.; El-Agnaf, O.; Calabresi, P. Cerebrospinal fluid biomarkers in Parkinson disease. Nat. Rev. Neurol. 2013, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Omer, N.; Giladi, N.; Gurevich, T.; Bar-Shira, A.; Gana-Weisz, M.; Glinka, T.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; Mabrouk, O.S.; et al. Glucocerebrosidase Activity is not Associated with Parkinson’s Disease Risk or Severity. Mov. Disord. 2021, 37, 190–195. [Google Scholar] [CrossRef]
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. 2021, 36, 2719–2730. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvagnion, C.; Marlet, F.R.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain 2022. [Google Scholar] [CrossRef] [PubMed]
- Pol-Fachin, L.; Siebert, M.; Verli, H.; Saraiva-Pereira, M.L. Glycosylation is crucial for a proper catalytic site organization in human glucocerebrosidase. Glycoconj. J. 2016, 33, 237–244. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Kishimoto, Y. Saposin proteins: Structure, function, and role in human lysosomal storage disorders. FASEB J. 1991, 5, 301–308. [Google Scholar] [CrossRef]
- Fabbro, D.; Grabowski, G.A. Human acid beta-glucosidase. Use of inhibitory and activating monoclonal antibodies to investigate the enzyme’s catalytic mechanism and saposin A and C binding sites. J. Biol. Chem. 1991, 266, 15021–15027. [Google Scholar] [CrossRef]
- Sun, Y.; Qi, X.; Grabowski, G.A. Saposin C Is Required for Normal Resistance of Acid beta-Glucosidase to Proteolytic Degradation. J. Biol. Chem. 2003, 278, 31918–31923. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Ramanathan, A.; Yuen, T.; Bhowmik, D.; Mathew, M.; Munshi, L.B.; Javaid, S.; Bloch, M.; Lizneva, D.; Rahimova, A.; et al. Mechanism of glucocerebrosidase activation and dysfunction in Gaucher disease unraveled by molecular dynamics and deep learning. Proc. Natl. Acad. Sci. USA 2019, 116, 5086–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Msc, L.K.; Hons, R.Y.J.W.B.; Heilbron, K.; Msc, J.A.R.; Bts, S.B.L.; Blauwendraat, C.; Alam Bsc, A.; Arnulf, I.; Mbbs, F.M.T.M.H.; Dauvilliers, Y.; et al. Fine-Mapping of SNCA in Rapid Eye Movement Sleep Behavior Disorder and Overt Synucleinopathies. Ann. Neurol. 2020, 87, 584–598. [Google Scholar] [CrossRef]
- Bencheikh, B.O.A.; Leveille, E.; Ruskey, J.A.; Spiegelman, D.; Liong, C.; Fon, E.A.; Rouleau, G.; Dauvilliers, Y.; Dupre, N.; Alcalay, R.N.; et al. Sequencing of the GBA coactivator, Saposin C, in Parkinson disease. Neurobiol. Aging 2018, 72, 187.e1–187.e3. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, I.; Blumenreich, S.; Futerman, A.H. GBA mutations, glucosylceramide and Parkinson’s disease. Curr. Opin. Neurobiol. 2021, 72, 148–154. [Google Scholar] [CrossRef]
- Van Echten-Deckert, G.; Alam, S. Sphingolipid metabolism—An ambiguous regulator of autophagy in the brain. Biol. Chem. 2018, 399, 837–850. [Google Scholar] [CrossRef]
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362. [Google Scholar] [CrossRef] [Green Version]
- Martinez, Z.; Zhu, M.; Han, A.S.; Fink, A.L. GM1 Specifically Interacts with α-Synuclein and Inhibits Fibrillation. Biochemistry 2007, 46, 1868–1877. [Google Scholar] [CrossRef]
- Miyagi, T.; Wada, T.; Iwamatsu, A.; Hata, K.; Yoshikawa, Y.; Tokuyama, S.; Sawada, M. Molecular Cloning and Characterization of a Plasma Membrane-associated Sialidase Specific for Gangliosides. J. Biol. Chem. 1999, 274, 5004–5011. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Tang, Y.; Zhao, L.; Wang, Q.; Qin, W.; Ji, X.; Zhang, J.; Jia, J. Associations between plasma ceramides and cognitive and neuropsychiatric manifestations in Parkinson’s disease dementia. J. Neurol. Sci. 2016, 370, 82–87. [Google Scholar] [CrossRef]
- Surface, M.; Balwani, M.; Waters, C.; Haimovich, A.; Gan-Or, Z.; Marder, K.S.; Hsieh, T.; Song, L.; Padmanabhan, S.; Hsieh, F.; et al. Plasma Glucosylsphingosine in GBA1 Mutation Carriers with and without Parkinson’s Disease. Mov. Disord. 2021, 37, 416–421. [Google Scholar] [CrossRef]
- Heinzel, S.; Berg, D.; Gasser, T.; Chen, H.; Yao, C.; Postuma, R.B.; Disease, M.T.F. Update of the MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2019, 34, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L.; Arizona Parkinson’s Disease Consortium. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef]
- Chung, C.-C.; Chan, L.; Chen, J.-H.; Bamodu, O.A.; Hong, C.-T. Neurofilament light chain level in plasma extracellular vesicles and Parkinson’s disease. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420975917. [Google Scholar] [CrossRef]
- Rotunno, M.S.; Lane, M.; Zhang, W.; Wolf, P.; Oliva, P.; Viel, C.; Wills, A.-M.; Alcalay, R.N.; Scherzer, C.R.; Shihabuddin, L.S.; et al. Cerebrospinal fluid proteomics implicates the granin family in Parkinson’s disease. Sci. Rep. 2020, 10, 2479. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-Y.; Chen, W.; Xu, W.; Li, J.-Q.; Hou, X.-H.; Ou, Y.-N.; Yu, J.-T.; Tan, L. Neurofilament Light Chain in Cerebrospinal Fluid and Blood as a Biomarker for Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019, 72, 1353–1361. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Biomarker ID | Brain | CSF | Blood | References, Patient Population, Comments | |||
|---|---|---|---|---|---|---|---|---|
| Other Parts | SN | |||||||
| Sphingolipid pathway | Sphingolipids | Ceramide | −53% (ACC) (***), −26% (OC) (NS) | NS| +393% (***) | +5.48% (C16:0) (***), +3.34 (C18:0) (NS), +5.25% (C20:0) (**), 6.35% (C22:0) (**), +3.1% (C24:0) (NS), +4.2% (C26:0) (NS), +7.21% (C24:1) (**), +6.9% (C26:1) (*) [52] | −20.4% (C22:0) (***), −18.4% (C24:1) (***), −26.2% (C26:0) (***), −19.2% (C20:1) (**), −22.4 (C22:1) (***) [53] | Brain: Total ceramide in anterior cingulate cortex (ACC) and OC of iPD vs. HC [54] CSF: NS in PD-GBA vs. PD-GBA wild-type [37,55] and PD-GBA wild-type vs. HC [55], increased in postmortem CSF of PD vs. HC [56] Plasma: PD-GBA wild-type vs. HC [52,53] | ||
| Sphingomyelin | −0.4% (****)|+0.98% (*)|+200% (***) | +6.9% (C20:1) (**) | CSF: −0.4% in PD-GBA vs. HC [55], +0.98% in PD-GBA wild-type vs. HC [37], +200% in in postmortem CSF of PD vs. HC [56] Plasma: PD-GBA wild-type vs. HC [53] | |||||
| Glycosphingolipids | Monosialoganglioside GM1a | −27% (**) | −22% in 70s (*), NS in 80s (1st cohort), −26% in 80s (*) (2nd cohort)| −73% (***) | −17% NS | −23% (**)| −32% (****), −57% (****), −37% (***) | Brain (OC): in iPD vs. HC [57] SN: in iPD vs. HC [40], −73% of GM1 positive dopaminergic neurons in iPD vs. HC [58] CSF: iPD vs. HC [40] Serum: −23% iPD vs. HC [40] PBMC: −32% in PD-GBA wild-type vs. HC, −57% in PD-GBA vs. HC, −37% in PD-GBA vs. PD-GBA wild-type [38] | ||
| Ganglioside GD1a | −28% (**) | −39% in 70s (*), NS in 80s (1st cohort), NS in 80s (2nd cohort) | −38% (**) | −20% (**) | Brain (OC): in iPD vs. HC [57] SN & CSF: in iPD vs. HC [40] Serum: in iPD vs. HC [40] | |||
| Ganglioside GD3 | −33% (*) | Brain (OC): in iPD vs. HC [57] SN & CSF: in iPD vs. HC [40] | ||||||
| Ganglioside GD1b | −12% (*) | −16% in 70s (*) −21% in 80s (*) (1st cohort), −31% in 80s (*) (2nd cohort) | −42% (***) | |||||
| Ganglioside GT1b | NS | −23% NS in 70s −27% in 80s (*) (1st cohort), −34% in 80s (*) (2nd cohort) | −51% (***) | |||||
| Ganglioside GM2 | +23% (*) | −15% NS | CSF & Serum: in iPD vs. HC [40] | |||||
| Ganglioside GM3 | +40% (*) | −8% NS|+14.5% (***) | CSF: in iPD vs. HC [40] Serum: NS in iPD vs. HC [40] Plasma: +14.5% in PD-GBA wild-type vs. HC [52] | |||||
| Gangliosides (sum of GM1a, GD1a, GD1b and GT1b) | −71% in 70s (**) NS in 80s (1st cohort), −67% in 80s (**) (2nd cohort) | −61% (**) | SN & CSF: in iPD vs. HC [40] | |||||
| Lactosylceramide (LacCer) | NS (1st cohort), NS (2nd cohort) | +22% (***) | NS| +2% (C16:0) (**), +4.8% (C18:0) (**), +4.5% (C22:0) (***), +4.4% (C24:0) (**), +4.7% (C24:1) (**) | SN: in NS in iPD vs. HC [40] CSF: in iPD vs. HC [40] Serum: NS in iPD vs. HC [40] Plasma: in PD-GBA wild-type vs. HC [52] | ||||
| Total glycosphingolipids (GlcCer, LacCer and gangliosides) | +31% NS in 70s +65% in 80s (***) (1st cohort), +39% in 80s (*) (2nd cohort) | SN: in iPD vs. HC [40] | ||||||
| C18-Sphingosine | +86% in 70s (*), NS in 80s | |||||||
| Sphinganine | +87% in 70s (*), NS in 80s | |||||||
| Glucosylsphingosine | NS (Putamen) | +16% NS in 70s +116% (**) in 80s [40]| +77% in 60s (*), NS in 70s and 80s [59] | Brain: in PD-GBA wild-type vs. HC [59] SN: C18-glucosylsphingosine in iPD vs. HC [40], in PD-GBA wild-type vs. HC [59] | |||||
| Glucosylceramide | +37% NS in 70s +74% (***) in 80s, +45% in 80s (*) (2nd cohort) | NS, +18% (****) | NS [40]| +7.5% (***) (C16:0), +6% (*) (C18:0), +4.3 (*) (C20:0), +5.5 (***) (C22:0), +5.7% (**) (C24:0), NS (C26:0, C16:1, C22:1, C24:1) | SN: in iPD vs. HC [40] CSF: NS in PD-GBA vs. HC, +18% in PD-GBA vs. HC [55] Serum: NS in iPD vs. HC [40] Plasma: Monohexosylceramides in PD-GBA wild-type vs. HC [52] | ||||
| Enzyme Activity | GCase | −49% (*) in 60s, −44% (*) in 70s (Putamen) [59]| −20% (*) (Caudate) [35] | −34% in 70s (****), −26% in 80s (**) (1st cohort), −79% in 80s (*) (2nd cohort) [40]| −47% in 60e, NS in 70s and 80s [59]| −14% (*) [35] | NS| −28% (***) | −28% (*), −29% (*) | Brain: in PD vs. HC [35], in PD-GBA wild-type vs. HC [59]. SN: in PD-GBA and iPD [36], in iPD at their 60s not in 70s or 80s [59], in PD vs. HC [35] CSF: NS in PD vs. HC [37,60], −28% in PD vs. HC [61] Blood (Monocytes): in PD vs. HC (−28%), in PD-GBA wild-type vs. HC wild-type (−29%). NS in lymphocytes [62] | ||
| β-galactosidase | −0.3% NS | −71% in 70s (*), −77% in 80s (2nd cohort)| −11% NS | +37% (**) | Brain (Caudate): in PD vs. HC [35] SN: in iPD vs. HC [40], NS in PD vs. HC [35] CSF: in PD vs. HC [60] | ||||
| α-galactosidase | −59% in 70s (****), −65% in 80s (***) (1st cohort), −28% in 80s (*) (2nd cohort) | −7% (*), −9% (*) | SN: in iPD vs. HC [40] Blood: Dried blood spots from PD vs. HC [39]. −9% in iPD-GBA wild-type over 40 age of onset vs. HC [39]. | |||||
| β-hexosaminidase | −4.1% NS (Caudate) | −31% in 80s (**) (1st cohort), −23% (**) (2nd cohort)| +4.5% NS | −9% NS| −8% NS | Brain tissue: NS in PD vs. HC [35] SN: in iPD vs. HC [40], NS in PD vs. HC [35] CSF: −9% in PD vs. HC [61], −8% in PD vs. HC [60] | ||||
| Neuraminidase/Sialidase | −42% NS in 70s, −52% in 80s (*) (1st cohort), −54% in 80s (*) (2nd cohort) | SN: in iPD vs. HC [40] | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esfandiary, A.; Finkelstein, D.I.; Voelcker, N.H.; Rudd, D. Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery. Cells 2022, 11, 1353. https://doi.org/10.3390/cells11081353
Esfandiary A, Finkelstein DI, Voelcker NH, Rudd D. Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery. Cells. 2022; 11(8):1353. https://doi.org/10.3390/cells11081353
Chicago/Turabian StyleEsfandiary, Ali, David Isaac Finkelstein, Nicolas Hans Voelcker, and David Rudd. 2022. "Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery" Cells 11, no. 8: 1353. https://doi.org/10.3390/cells11081353
APA StyleEsfandiary, A., Finkelstein, D. I., Voelcker, N. H., & Rudd, D. (2022). Clinical Sphingolipids Pathway in Parkinson’s Disease: From GCase to Integrated-Biomarker Discovery. Cells, 11(8), 1353. https://doi.org/10.3390/cells11081353