
Conserved Mitotic Phosphorylation of a Proteasome Subunit Regulates Cell Proliferation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Transfection, and Infection
2.2. Plasmids
2.3. Antibodies and Reagents
2.4. Cell Proliferation Assay and Flow Cytometry
2.5. CRISPR/Cas9-Mediated Genome Editing
2.6. Immunoblot, Immunoprecipitation, and Streptavidin Pulldown
2.7. Fluorescence Microscopy
2.8. Yeast Genetics
2.9. Sucrose Gradient Ultracentrifugation
2.10. Mass Spectrometry
2.11. Data Analysis and Presentation
3. Results
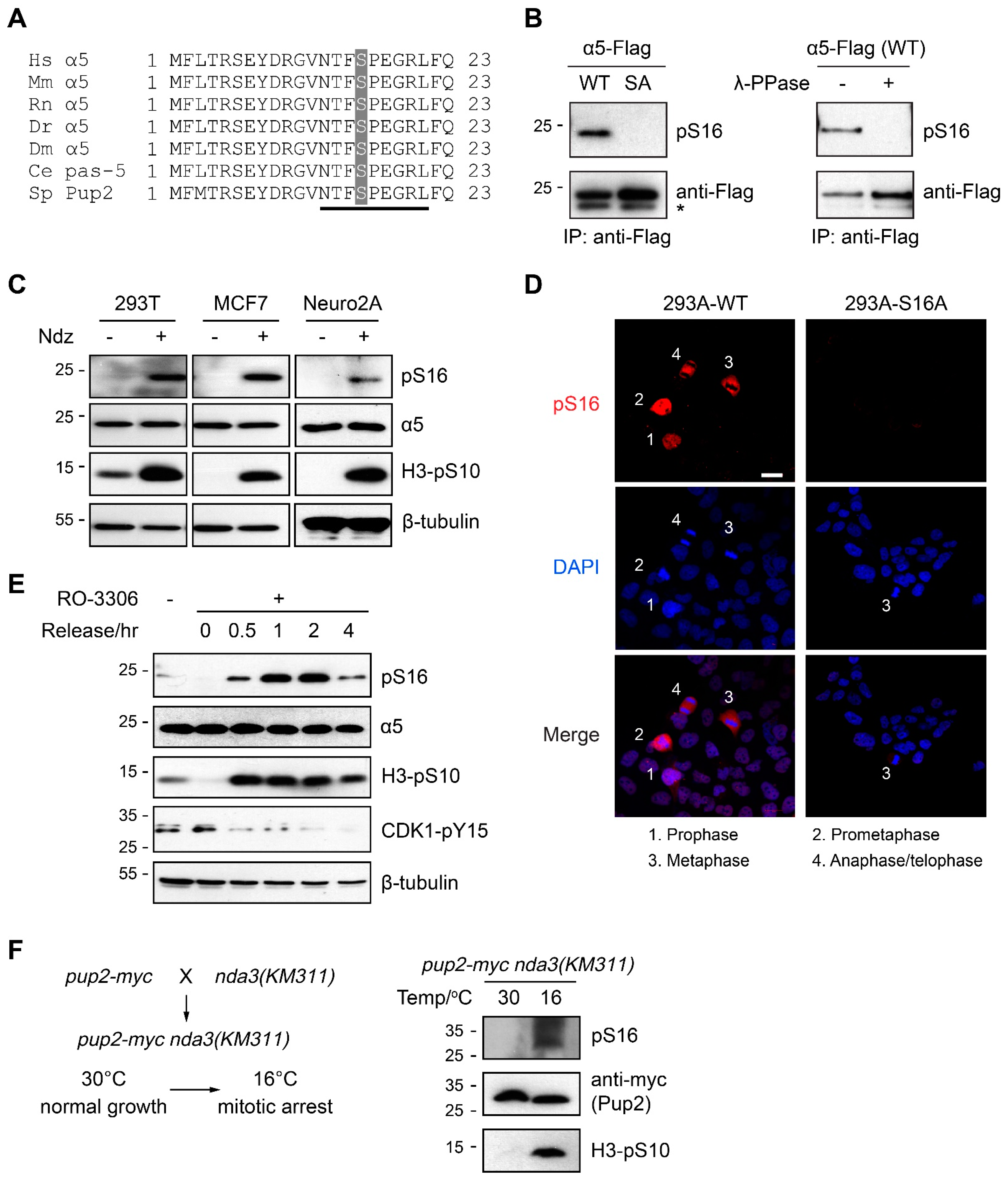
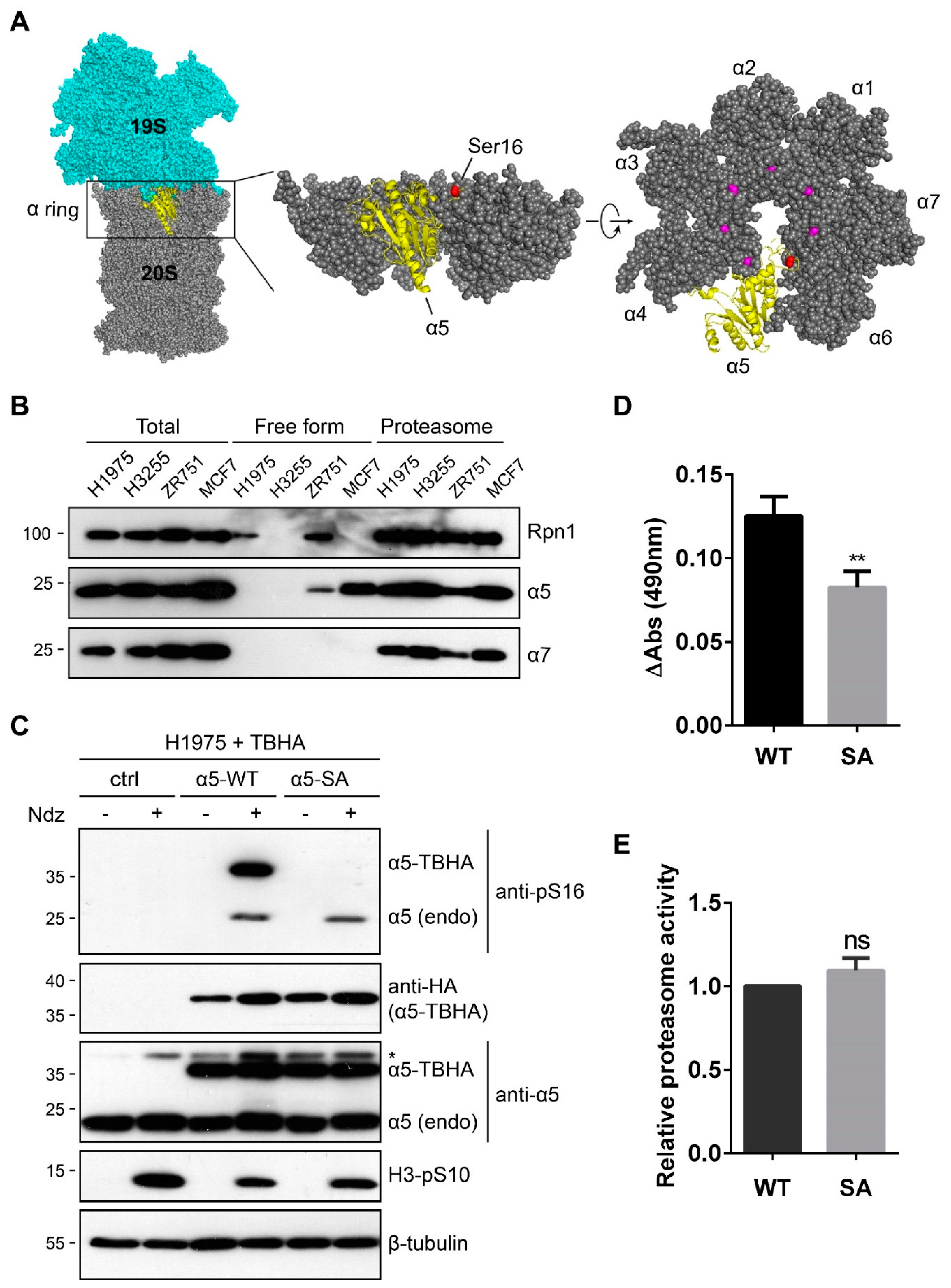
3.1. α5-S16 Is a Conserved Phosphosite Associated with Mitosis
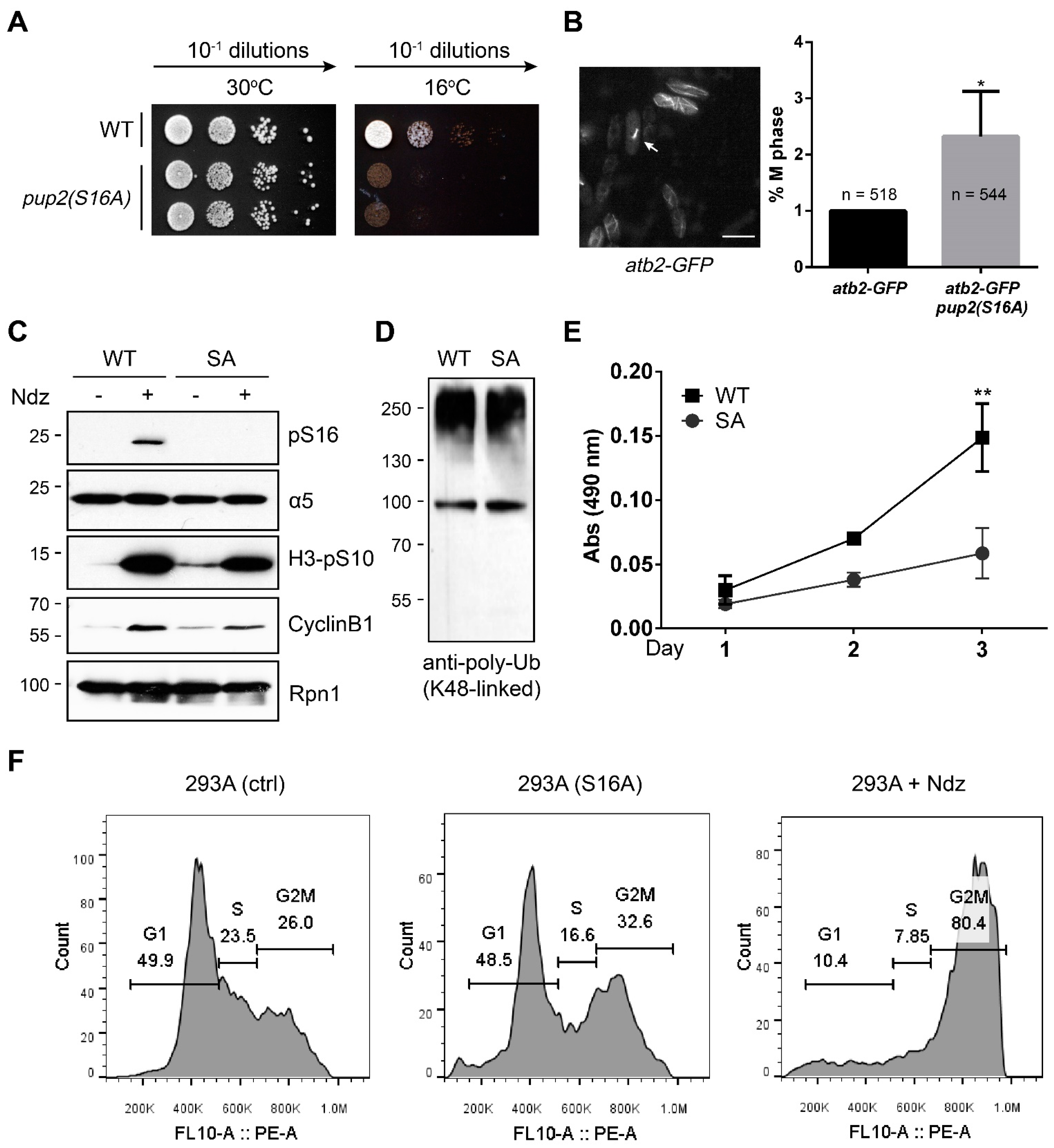
3.2. The α5-S16A Mutation Impaired Cell Proliferation
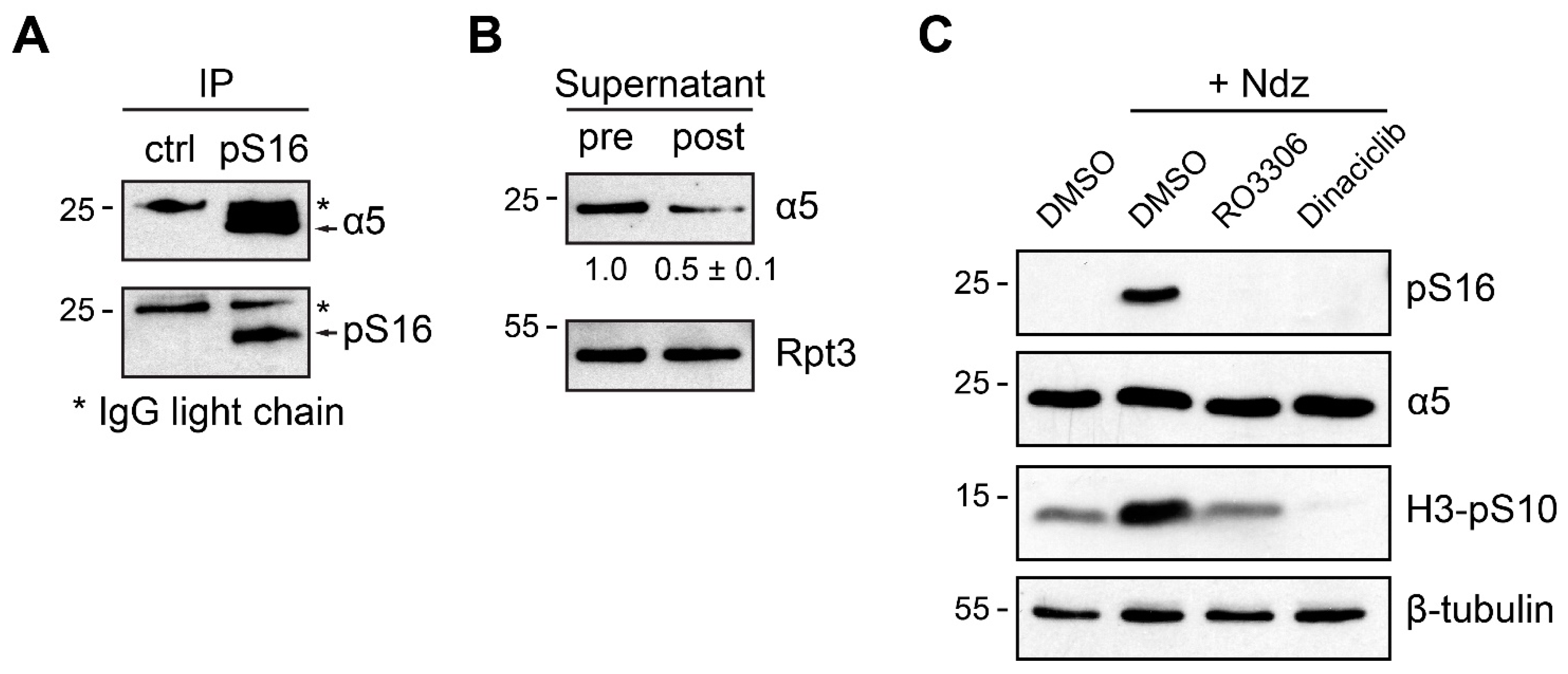
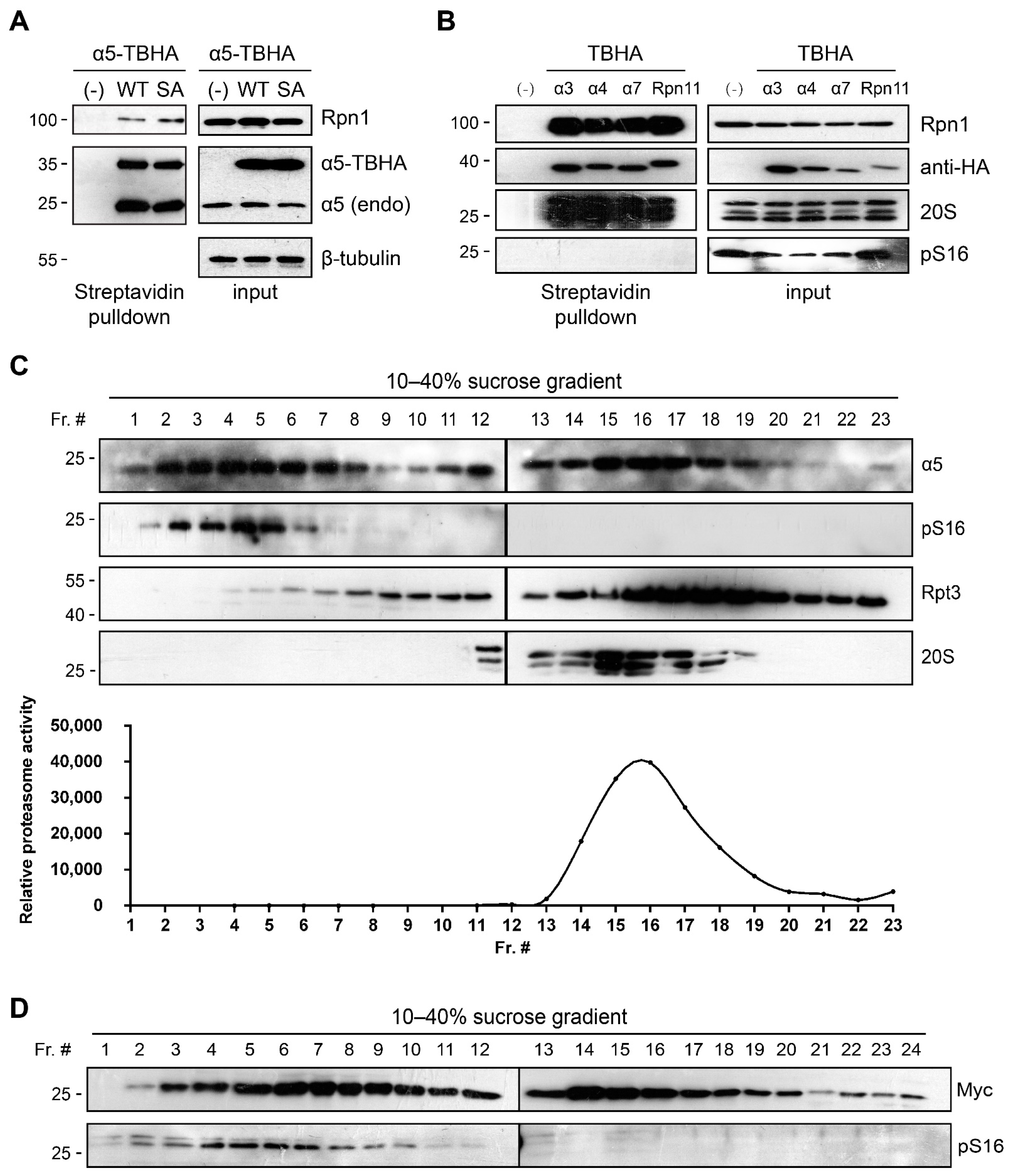
3.3. α5-S16 Phosphorylation Did Not Occur on the Assembled Proteasome Complexes
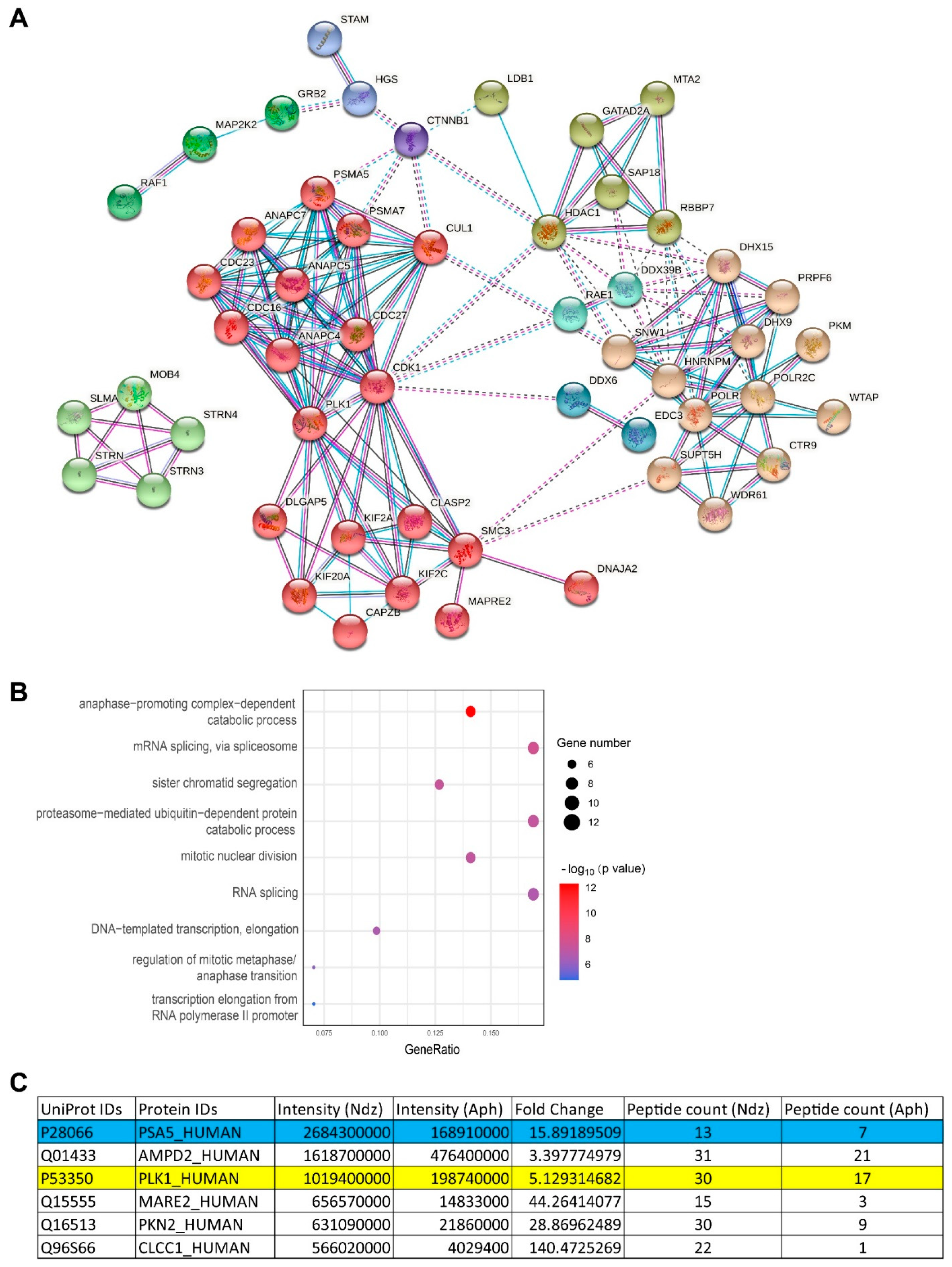
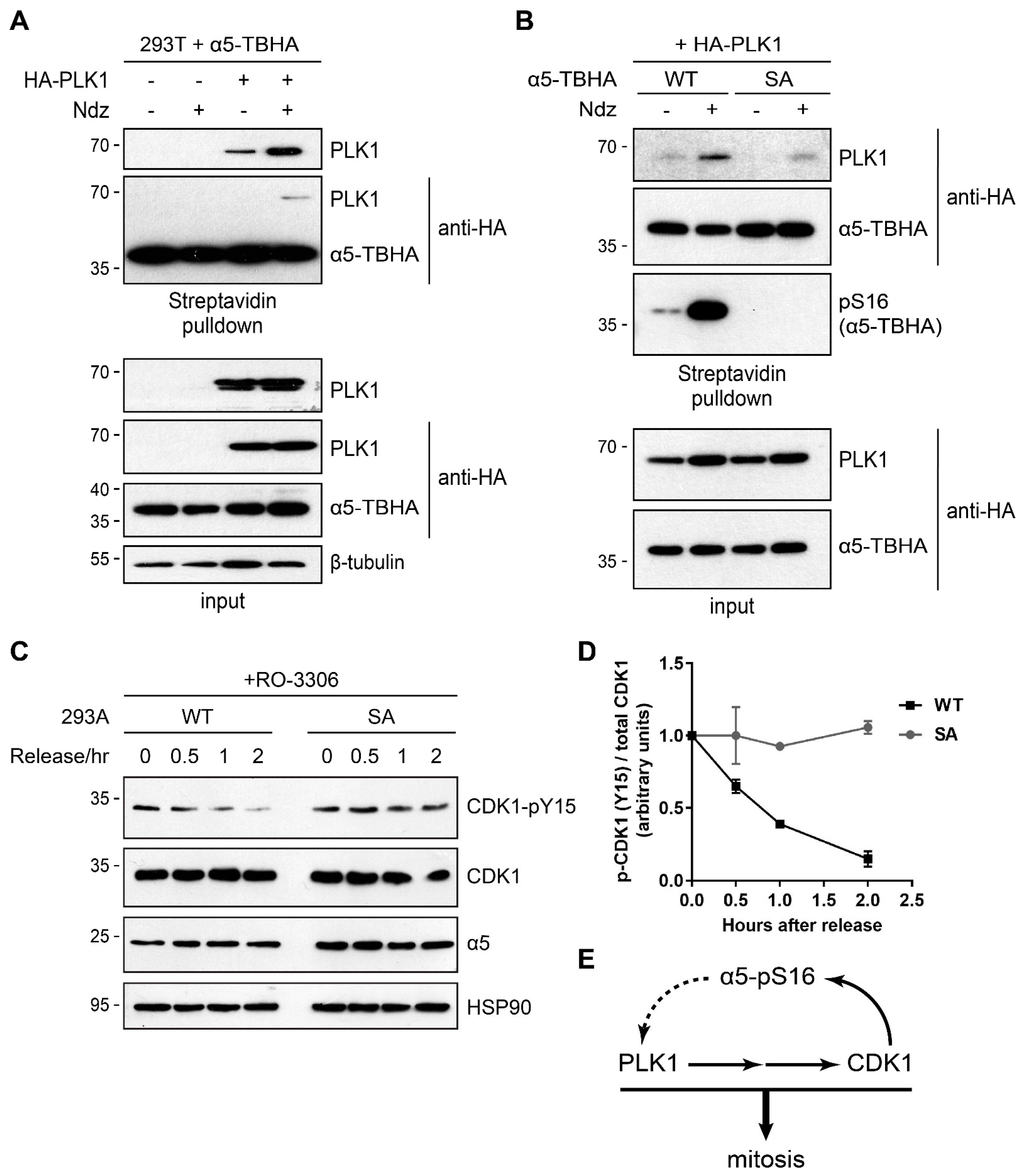
3.4. Ser16-Phosphorylated α5 Bound Mitotic Regulators Including PLK1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kaldis, P. Cell Cycle Regulation; Springer: New York, NY, USA, 2006. [Google Scholar]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.; Vuong, J.; Rogers, S.; Malumbres, M.; O’Donoghue, S.I. SnapShot: Phosphoregulation of Mitosis. Cell 2017, 169, 1358. [Google Scholar] [CrossRef]
- Holt, L.J.; Tuch, B.B.; Villén, J.; Johnson, A.D.; Gygi, S.P.; Morgan, D.O. Global Analysis of Cdk1 Substrate Phosphorylation Sites Provides Insights into Evolution. Science 2009, 325, 1682–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta Bioenerg. 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Bard, J.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Murata, S.; Yashiroda, H.; Tanaka, K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10, 104–115. [Google Scholar] [CrossRef]
- Howell, L.A.; Peterson, A.K.; Tomko, R.J., Jr. Proteasome subunit α1 overexpression preferentially drives canonical proteasome biogenesis and enhances stress tolerance in yeast. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Peak-Chew, S.-Y.; Juszkiewicz, S.; Narvaez, A.J.; Hegde, R.S. Identification of a quality-control factor that monitors failures during proteasome assembly. Science 2021, 373, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Schmidt, M.; Kloetzel, P.M. Maturation of mammalian 20 S proteasome: Purification and characterization of 13 S and 16 S proteasome precursor complexes. J. Mol. Biol. 1997, 268, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, L.; Hendil, K.B. Proteasome subunit zeta, a putative ribonuclease, is also found as a free monomer. Mol. Biol. Rep. 1999, 26, 119–123. [Google Scholar] [CrossRef]
- Guo, X.; Engel, J.L.; Xiao, J.; Tagliabracci, V.S.; Wang, X.; Huang, L.; Dixon, J.E. UBLCP1 is a 26S proteasome phosphatase that regulates nuclear proteasome activity. Proc. Natl. Acad. Sci. USA 2011, 108, 18649–18654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Huang, X.; Chen, M.J. Reversible phosphorylation of the 26S proteasome. Protein Cell 2017, 8, 255–272. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Wang, X.; Wang, Z.; Banerjee, S.; Yang, J.; Huang, L.; Dixon, J.E. Site-specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat. Cell Biol. 2016, 18, 202–212. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, W.; Zhang, Y.; Wiley, S.E.; Zuo, T.; Zheng, Y.; Chen, N.; Chen, L.; Wang, X.; Zheng, Y.; et al. Reversible phosphorylation of Rpn1 regulates 26S proteasome assembly and function. Proc. Natl. Acad. Sci. USA 2020, 117, 328–336. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.; Shu, X.; Chen, Q.; Wei, T.; Wang, H.; Wang, X.; Wu, Q.; Zhang, X.; Liu, X.; et al. Proteasome regulation by reversible tyrosine phosphorylation at the membrane. Oncogene 2021, 40, 1942–1956. [Google Scholar] [CrossRef]
- Guo, X.; Dixon, J.E. The 26S proteasome: A cell cycle regulator regulated by cell cycle. Cell Cycle 2016, 15, 875–876. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Wei, T.; Wang, J.; Lee, J.J.; Gutierrez, H.L.; Chapman, O.; Wiley, S.E.; Mayfield, J.E.; Tandon, V.; Juarez, E.F.; et al. Inhibition of dual-specificity tyrosine phosphorylation-regulated kinase 2 perturbs 26S proteasome-addicted neoplastic progression. Proc. Natl. Acad. Sci. USA 2019, 116, 24881–24891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Chen, C.-F.; Baker, P.R.; Chen, P.-L.; Kaiser, P.; Huang, L. Mass Spectrometric Characterization of the Affinity-Purified Human 26S Proteasome Complex. Biochemistry 2007, 46, 3553–3565. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Goldberg, A.L. Regulating protein breakdown through proteasome phosphorylation. Biochem. J. 2017, 474, 3355–3371. [Google Scholar] [CrossRef] [Green Version]
- Swaffer, M.P.; Jones, A.; Flynn, H.R.; Snijders, B.; Nurse, P. Quantitative Phosphoproteomics Reveals the Signaling Dynamics of Cell-Cycle Kinases in the Fission Yeast Schizosaccharomyces pombe. Cell Rep. 2018, 24, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, Y.; Toda, T.; Yanagida, M. The NDA3 gene of fission yeast encodes beta-tubulin: A cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell 1984, 39, 349–358. [Google Scholar] [CrossRef]
- Al-Fageeh, M.B.; Smales, C.M. Control and regulation of the cellular responses to cold shock: The responses in yeast and mammalian systems. Biochem. J. 2006, 397, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Moore, J.K. Microtubule dynamics at low temperature: Evidence that tubulin recycling limits assembly. Mol. Biol. Cell 2020, 31, 1154–1166. [Google Scholar] [CrossRef]
- Bordas, J.; Mandelkow, E.M.; Mandelkow, E. Stages of tubulin assembly and disassembly studied by time-resolved synchrotron X-ray scattering. J. Mol. Biol. 1983, 164, 89–135. [Google Scholar] [CrossRef]
- Nijhawan, D.; Zack, T.I.; Ren, Y.; Strickland, M.R.; Lamothe, R.; Schumacher, S.E.; Tsherniak, A.; Besche, H.C.; Rosenbluh, J.; Shehata, S.; et al. Cancer Vulnerabilities Unveiled by Genomic Loss. Cell 2012, 150, 842–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerdt, B.C.V.D.; Medema, R.H. Polo-like kinases: A team in control of the division. Cell Cycle 2006, 5, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Nabais, C.; Jana, S.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Feng, Y.; Longo, D.L.; Ferris, D.K. Polo-like kinase interacts with proteasomes and regulates their activity. Cell Growth Differ. 2001, 12, 29–37. [Google Scholar] [PubMed]
- Elia, A.E.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.; Smerdon, S.J.; Yaffe, M.B. The Molecular Basis for Phosphodependent Substrate Targeting and Regulation of Plks by the Polo-Box Domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Nakajima, H.; Toyoshima-Morimoto, F.; Taniguchi, E.; Nishida, E. Identification of a Consensus Motif for Plk (Polo-like Kinase) Phosphorylation Reveals Myt1 as a Plk1 Substrate. J. Biol. Chem. 2003, 278, 25277–25280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rêgo, A.T.; da Fonseca, P.C. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duan, J.; Li, W.; Shu, X.; Yang, B.; He, X.; Guo, X. Conserved Mitotic Phosphorylation of a Proteasome Subunit Regulates Cell Proliferation. Cells 2021, 10, 3075. https://doi.org/10.3390/cells10113075
Duan J, Li W, Shu X, Yang B, He X, Guo X. Conserved Mitotic Phosphorylation of a Proteasome Subunit Regulates Cell Proliferation. Cells. 2021; 10(11):3075. https://doi.org/10.3390/cells10113075
Chicago/Turabian StyleDuan, Jinyuan, Wenzhu Li, Xin Shu, Bing Yang, Xiangwei He, and Xing Guo. 2021. "Conserved Mitotic Phosphorylation of a Proteasome Subunit Regulates Cell Proliferation" Cells 10, no. 11: 3075. https://doi.org/10.3390/cells10113075
APA StyleDuan, J., Li, W., Shu, X., Yang, B., He, X., & Guo, X. (2021). Conserved Mitotic Phosphorylation of a Proteasome Subunit Regulates Cell Proliferation. Cells, 10(11), 3075. https://doi.org/10.3390/cells10113075