
Multi-Target Effects of Novel Synthetic Coumarin Derivatives Protecting Aβ-GFP SH-SY5Y Cells against Aβ Toxicity

 , ,
, ,
Abstract
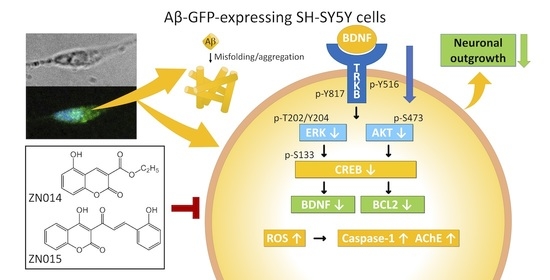
:
1. Introduction
2. Materials and Methods
2.1. Compounds and Cells
2.2. Bioavailability and Blood–Brain Barrier (BBB) Permeation Prediction
2.3. Thioflavin T Aggregation Assay
2.4. Aβ-GFP Fluorescence and Reactive Oxygen Species (ROS) Analyses
2.5. Neurite Outgrowth Analysis
2.6. Real-Time PCR Assay
2.7. Caspase-1 and AChE Assays
2.8. Western Blot Analysis
2.9. RNA Interference
- TRCN0000002243: 5’-CCGGCCAACTATCACATTTCTCGAActcgagTTCGAGAAATGTGATAGTTGGTTTTT-3’
- TRCN0000002245: 5’-CCGGGCACATCAAGCGACATAACATctcgagATGTT-ATGTCGCTTGATGTGCTTTTT-3’
- TRCN0000002246: 5’-CCGGCCTTGTTGTATTCCTGCCTTTctcgagAAAGGCAGGAATACAACAAGGTTTTT-3’
- TRC2.Void: 5’-CCGGAGTTCAGTTACGATATCATGTctcgagACATTCGCGAGTAACTGAACTTTTTT-3’
2.10. Parallel Artificial Membrane Permeability Assay (PAMPA)
2.11. Statistical Analysis
3. Results
3.1. Tested Coumarins and Amyloid Inhibition
3.2. Inhibition Aβ Aggregation and Oxidative Stress by Coumarin Derivatives
3.3. Inhibition of Caspase-1 and AChE and Promotion of Neurite Outgrowth by Coumarin Derivatives
3.4. Molecular Targets of New Coumarin Derivatives
3.5. TRKB Knockdown Attenuated the Neuroprotective Effects of New Coumarin Derivatives
3.6. The Potential of BBB Penetration of New Coumarin Derivatives
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, M.S.; Hemnani, T. Alzheimer’s disease pathogenesis and therapeutic interventions. J. Clin. Neurosci. 2004, 11, 456–467. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Akıncıoğlu, H.; Gülçin, İ. Potent acetylcholinesterase inhibitors: Potential drugs for Alzheimer’s disease. Mini Rev. Med. Chem. 2020, 20, 703–715. [Google Scholar] [CrossRef]
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702. [Google Scholar] [CrossRef]
- Holsinger, R.M.; Schnarr, J.; Henry, P.; Castelo, V.T.; Fahnestock, M. Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: Decreased levels in Alzheimer’s disease. Mol. Brain Res. 2000, 76, 347–354. [Google Scholar] [CrossRef]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Levine, E.S.; Dreyfus, C.F.; Black, I.B.; Plummer, M.R. Selective role for trkB neurotrophin receptors in rapid modulation of hippocampal synaptic transmission. Mol. Brain Res. 1996, 38, 300–303. [Google Scholar] [CrossRef]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonni, A.; Brunet, A.; West, A.E.; Datta, S.R.; Takasu, M.A.; Greenberg, M.E. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 1999, 286, 1358–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabuchi, A.; Sakaya, H.; Kisukeda, T.; Fushiki, H.; Tsuda, M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J. Biol. Chem. 2002, 277, 35920–35931. [Google Scholar] [CrossRef] [Green Version]
- Riccio, A.; Ahn, S.; Davenport, C.M.; Blendy, J.A.; Ginty, D.D. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 1999, 286, 2358–2361. [Google Scholar] [CrossRef] [PubMed]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef]
- Dlugosz, P.J.; Billen, L.P.; Annis, M.G.; Zhu, W.; Zhang, Z.; Lin, J.; Leber, B.; Andrews, D.W. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 2006, 25, 2287–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzon, D.J.; Fahnestock, M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J. Neurosci. 2007, 27, 2628–2635. [Google Scholar] [CrossRef] [Green Version]
- Poon, W.W.; Blurton-Jones, M.; Tu, C.H.; Feinberg, L.M.; Chabrier, M.A.; Harris, J.W.; Jeon, N.L.; Cotman, C.W. β-Amyloid impairs axonal BDNF retrograde trafficking. Neurobiol. Aging 2011, 32, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, A.H.; Merrill, D.A.; Coppola, G.; Tsukada, S.; Schroeder, B.E.; Shaked, G.M.; Wang, L.; Blesch, A.; Kim, A.; Conner, J.M.; et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat. Med. 2009, 15, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.M.; Damu, G.L.V.; Zhou, C.H. Current developments of coumarin compounds in medicinal chemistry. Curr. Pharm. Des. 2013, 19, 3884–3930. [Google Scholar] [CrossRef]
- Soto-Ortega, D.D.; Murphy, B.P.; Gonzalez-Velasquez, F.J.; Wilson, K.A.; Xie, F.; Wang, Q.; Moss, M.A. Inhibition of amyloid-β aggregation by coumarin analogs can be manipulated by functionalization of the aromatic center. Bioorgan. Med. Chem. 2011, 19, 2596–2602. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Chiu, Y.J.; Yang, S.M.; Chen, C.M.; Huang, C.C.; Lee-Chen, G.J.; Lin, W.; Chang, K.H. Novel synthetic chalcone-coumarin hybrid for Aβ aggregation reduction, antioxidation, and neuroprotection. CNS Neurosci. Ther. 2018, 24, 1286–1298. [Google Scholar] [CrossRef]
- Lin, T.H.; Chiu, Y.J.; Lin, C.H.; Lin, C.Y.; Chao, C.Y.; Chen, Y.C.; Yang, S.M.; Lin, W.; Hsieh-Li, H.M.; Wu, Y.R.; et al. Exploration of multi-target effects of 3-benzoyl-5-hydroxychromen-2-one in Alzheimer’s disease cell and mouse models. Aging Cell 2020, 19, e13169. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.X.; Zhang, K.Y.; Li, Y.C.; Chen, Y.W.; Yue, Y.S.; Xia, S.Z.; Li, Y.; Deng, H.H.; Jing, H.L.; Cao, Y.J. Imperatorin ameliorates learning and memory deficits through BDNF/TrkB and ERK/CaMKIIalpha/CREB signaling in prenatally-stressed female offspring. Phytother. Res. 2020, 34, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- Adu-Nti, F.; Gao, X.; Wu, J.M.; Li, J.; Iqbal, J.; Ahmad, R.; Ma, X.M. Osthole ameliorates estrogen deficiency-induced cognitive impairment in female mice. Front. Pharmacol. 2021, 12, 641909. [Google Scholar] [CrossRef]
- Lee, C.J.; Tsai, C.C.; Hong, S.H.; Chang, G.H.; Yang, M.C.; Möhlmann, L.; Lin, W. Preparation of furo[3,2-c]coumarins from 3-cinnamoyl-4-hydroxy-2H-chromen-2-ones and acyl chlorides: A Bu3P-mediated C-acylation/cyclization sequence. Angew. Chem. Int. Ed. 2015, 54, 8502–8505. [Google Scholar] [CrossRef]
- Jabbari, A.; Mousavian, M.; Seyedi, S.M.; Bakavoli, M.; Sadeghian, H. O-prenylated 3-carboxycoumarins as a novel class of 15-LOX-1 inhibitors. PLoS ONE 2017, 12, e0171789. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Chiu, Y.J.; Chen, S.L.; Huang, C.H.; Lin, C.H.; Lin, T.H.; Lee, C.M.; Ramesh, C.; Wu, C.H.; Huang, C.C.; et al. The potential of synthetic indolylquinoline derivatives for Aβ aggregation reduction by chemical chaperone activity. Neuropharmacology 2016, 101, 309–319. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Ibrahim, A.; Fish, P.V.; Cole, S.; Lewis, M.L.; de Groot, M.J.; Reynolds, D.P. Predicting penetration across the blood-brain barrier from simple descriptors and fragmentation schemes. J. Chem. Inf. Model. 2007, 47, 170–175. [Google Scholar] [CrossRef]
- Ottaviani, G.; Martel, S.; Escarala, C.; Nicolle, E.; Carrupt, P.A. The PAMPA technique as a HTS tool for partition coefficients determination in different solvent/water systems. Eur. J. Pharm. Sci. 2008, 35, 68–75. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hitchcock, S.A.; Pennington, L.D. Structure—Brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.Q. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H., III. [18] Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Kim, Y.; Min, J.; Kim, D.J.; Chang, Y.T.; Hecht, M.H. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem. Biol. 2006, 1, 461–469. [Google Scholar] [CrossRef]
- Påhlman, S.; Ruusala, A.I.; Abrahamsson, L.; Mattsson, M.E.; Esscher, T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: A comparison with phorbolester-induced differentiation. Cell Differ. 1984, 14, 135–144. [Google Scholar] [CrossRef]
- Lin, C.C.; Lee, I.T.; Wu, W.L.; Lin, W.N.; Yang, C.M. Adenosine triphosphate regulates NADPH oxidase activity leading to hydrogen peroxide production and COX-2/PGE2 expression in A549 cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L401–L412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, J.; Noel, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, P.; Ding, Q.; Wu, C.; Zhang, W.; Tang, B. Observation of acetylcholinesterase in stress-induced depression phenotypes by two-photon fluorescence imaging in the mouse brain. J. Am. Chem. Soc. 2019, 141, 2061–2068. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Alvarez, A.; Perez, C.A.; Moreno, R.D.; Vicente, M.; Linker, C.; Casanueva, O.I.; Soto, C.; Garrido, J. Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer’s fibrils: Possible role of the peripheral site of the enzyme. Neuron 1996, 16, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The β-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008, 131, 90–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, N.A.; Facci, L.; Owen, D.E.; Soden, P.E.; Burbidge, S.A.; Prinjha, R.K.; Richardson, J.C.; Skaper, S.D. Aβ1-42 reduces synapse number and inhibits neurite outgrowth in primary cortical and hippocampal neurons: A quantitative analysis. J. Neurosci. Methods 2008, 175, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Bezar, I.F.; Petusky, S.L.; Huang, Y. Comparison of blood-brain barrier permeability assays: In situ brain perfusion, MDR1-MDCKII and PAMPA-BBB. J. Pharm. Sci. 2009, 98, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Mensch, J.; Melis, A.; Mackie, C.; Verreck, G.; Brewster, M.E.; Augustijns, P. Evaluation of various PAMPA models to identify the most discriminating method for the prediction of BBB permeability. Eur. J. Pharm. Biopharm. 2010, 74, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, D.; Godugu, C.; Dubey, P.K. A review on pharmacological properties of coumarins. Mini Rev. Med. Chem. 2018, 18, 113–141. [Google Scholar] [CrossRef]
- Anand, P.; Singh, B.; Singh, N. A review on coumarins as acetylcholinesterase inhibitors for Alzheimer’s disease. Bioorgan. Med. Chem. 2012, 20, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Piazzi, L.; Cavalli, A.; Colizzi, F.; Belluti, F.; Bartolini, M.; Mancinni, F.; Recanatini, M.; Andrisana, V.; Rampa, A. Multi-target-directed coumarin derivatives: hAChE and BACE1 inhibitors as potential anti-Alzheimer compounds. Bioorgan. Med. Chem. Lett. 2008, 18, 423–426. [Google Scholar] [CrossRef]
- Schober, A.; Minichiello, L.; Keller, M.; Huber, K.; Layer, P.G.; Roig-López, J.L.; García-Arrarás, J.E.; Klein, R.; Unsicker, K. Reduced acetylcholinesterase (AChE) activity in adrenal medulla and loss of sympathetic preganglionic neurons in TrkA-deficient, but not TrkB-deficient, mice. J. Neurosci. 1997, 17, 891–903. [Google Scholar] [CrossRef]
- Tonnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.H.; Dong, F.X. The relevant targets of anti-oxidative stress: A review. J. Drug Target 2021, 29, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, E.H.M.; Sayed, A.M.; Hussein, O.E.; Mahmoud, A.M. Coumarins as modulators of the Keap1/Nrf2/ARE signaling pathway. Oxid. Med. Cell. Longev. 2020, 2020, 1675957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, M.; Roghani, M.; Sarfallah, A.; Riazi, G.H. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int. Immunopharmacol. 2018, 54, 78–85. [Google Scholar] [CrossRef]
- Howley, B.; Fearnhead, H.O. Caspases as therapeutic targets. J. Cell. Mol. Med. 2008, 12, 1502–1516. [Google Scholar] [CrossRef] [Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.Y.; Chung, K.S.; Jin, J.S.; Lee, Y.C.; An, H.J. The inhibitory effect of nodakenin on mast-cell-mediated allergic inflammation via downregulation of NF-κB and caspase-1 activation. J. Cell. Biochem. 2017, 118, 3993–4001. [Google Scholar] [CrossRef]
- Alvarez, A.; Opazo, C.; Alarcón, R.; Garrido, J.; Inestrosa, N.C. Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol. 1997, 272, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Colović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racchi, M.; Mazzucchelli, M.; Porrello, E.; Lanni, C.; Govoni, S. Acetylcholinesterase inhibitors: Novel activities of old molecules. Pharm. Res. 2004, 50, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.R.; Dragunow, M. Is CREB a key to neuronal survival? Trends Neurosci. 2000, 23, 48–53. [Google Scholar] [CrossRef]
- Deutsch, P.J.; Hoeffler, J.P.; Jameson, J.L.; Lin, J.C.; Habener, J.F. Structural determinants for transcriptional activation by cAMP-responsive DNA elements. J. Biol. Chem. 1988, 263, 18466–18472. [Google Scholar] [CrossRef]
- Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007, 274, 3210–3217. [Google Scholar] [CrossRef]
- Jonas, E.A. Molecular participants in mitochondrial cell death channel formation during neuronal ischemia. Exp. Neurol. 2009, 218, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid β peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J. Neurosci. 1996, 16, 7533–7539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siracusa, R.; Scuto, M.; Fusco, R.; Trovato, A.; Ontario, M.L.; Crea, R.; Di Paola, R.; Cuzzocrea, S.; Calabrese, V. Anti-inflammatory and anti-oxidant activity of Hidrox® in rotenone-induced Parkinson’s disease in mice. Antioxidants 2020, 9, 824. [Google Scholar] [CrossRef]
- Miquel, S.; Champ, C.; Day, J.; Aarts, E.; Bahr, B.A.; Bakker, M.; Bánáti, D.; Calabrese, V.; Cederholm, T.; Cryan, J.; et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res. Rev. 2018, 42, 40–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunetti, G.; Di Rosa, G.; Scuto, M.; Leri, M.; Stefani, M.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Healthspan maintenance and prevention of Parkinson’s-like phenotypes with hydroxytyrosol and oleuropein aglycone in C. elegans. Int. J. Mol. Sci. 2020, 21, 2588. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | MW | HBD | HBA | cLogP | PSA (Å2) | BBB Score (Threshold: 0.02) |
|---|---|---|---|---|---|---|
| ZN009 | 338.31 | 1 | 6 | 2.44 | 89.9 | 0.68 |
| ZN010 | 300.69 | 1 | 4 | 4.23 | 63.6 | 0.15 |
| ZN011 | 296.28 | 1 | 5 | 2.74 | 72.8 | 0.04 |
| ZN014 | 234.21 | 1 | 5 | 2.17 | 72.8 | 0.19 |
| ZN015 | 308.29 | 2 | 5 | 3.28 | 83.8 | 0.14 |
| Compound Name | Measured Pe (10−6 cm/s) or % Transport | BBB Permeability Classification a |
|---|---|---|
| ZN014 | 0.56 ± 0.01 | Low |
| ZN015 | 5.16 ± 0.11 | High |
| 7,8-DHF | 6.32 ± 1.35 | High |
| Carbamazepine | 9.85 ± 0.60 | High marker |
| Theophylline | 0.13 ± 0.00 | Low marker |
| Lucifer yellow | 0.00 (% Transport) | Integrity marker (well-accepted membrane integrity) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.-C.; Chang, K.-H.; Chiu, Y.-J.; Chen, Y.-R.; Lung, T.-H.; Hsieh-Li, H.M.; Su, M.-T.; Sun, Y.-C.; Chen, C.-M.; Lin, W.; et al. Multi-Target Effects of Novel Synthetic Coumarin Derivatives Protecting Aβ-GFP SH-SY5Y Cells against Aβ Toxicity. Cells 2021, 10, 3095. https://doi.org/10.3390/cells10113095
Huang C-C, Chang K-H, Chiu Y-J, Chen Y-R, Lung T-H, Hsieh-Li HM, Su M-T, Sun Y-C, Chen C-M, Lin W, et al. Multi-Target Effects of Novel Synthetic Coumarin Derivatives Protecting Aβ-GFP SH-SY5Y Cells against Aβ Toxicity. Cells. 2021; 10(11):3095. https://doi.org/10.3390/cells10113095
Chicago/Turabian StyleHuang, Ching-Chia, Kuo-Hsuan Chang, Ya-Jen Chiu, Yi-Ru Chen, Tsai-Hui Lung, Hsiu Mei Hsieh-Li, Ming-Tsan Su, Ying-Chieh Sun, Chiung-Mei Chen, Wenwei Lin, and et al. 2021. "Multi-Target Effects of Novel Synthetic Coumarin Derivatives Protecting Aβ-GFP SH-SY5Y Cells against Aβ Toxicity" Cells 10, no. 11: 3095. https://doi.org/10.3390/cells10113095
APA StyleHuang, C. -C., Chang, K. -H., Chiu, Y. -J., Chen, Y. -R., Lung, T. -H., Hsieh-Li, H. M., Su, M. -T., Sun, Y. -C., Chen, C. -M., Lin, W., & Lee-Chen, G. -J. (2021). Multi-Target Effects of Novel Synthetic Coumarin Derivatives Protecting Aβ-GFP SH-SY5Y Cells against Aβ Toxicity. Cells, 10(11), 3095. https://doi.org/10.3390/cells10113095