
Neurodevelopmental Syndrome with Intellectual Disability, Speech Impairment, and Quadrupedia Is Associated with Glutamate Receptor Delta 2 Gene Defect

 , and
, and
Abstract
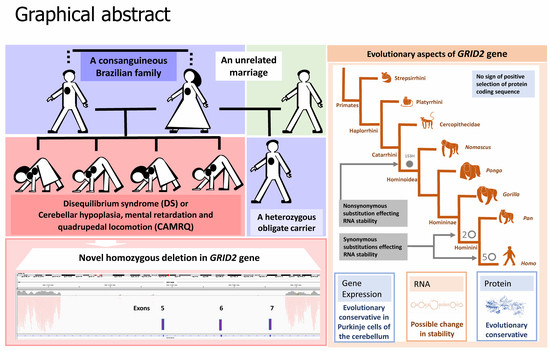
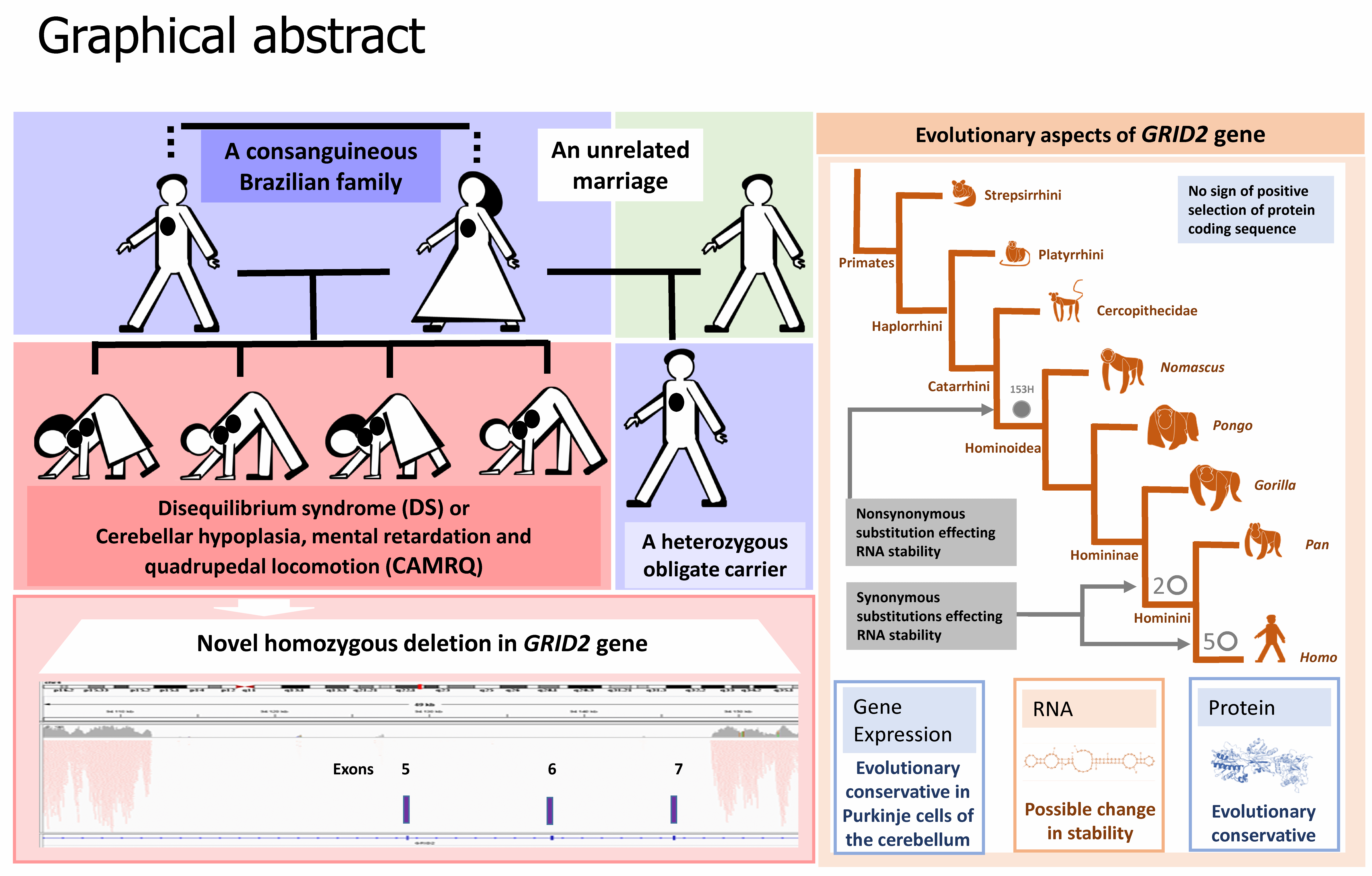
:
{kind=link}
{kind=link}
{kind=link}
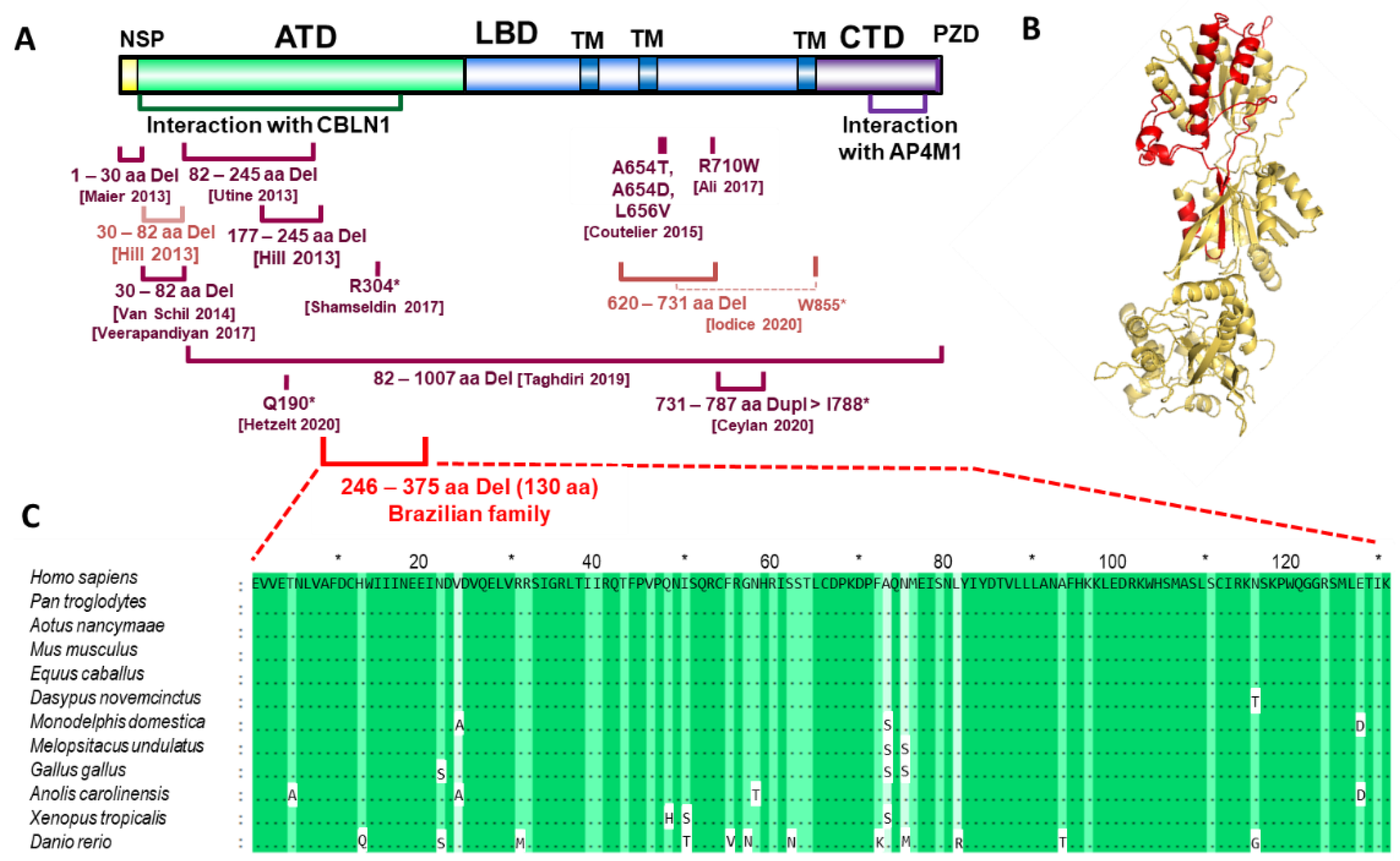{kind=link}
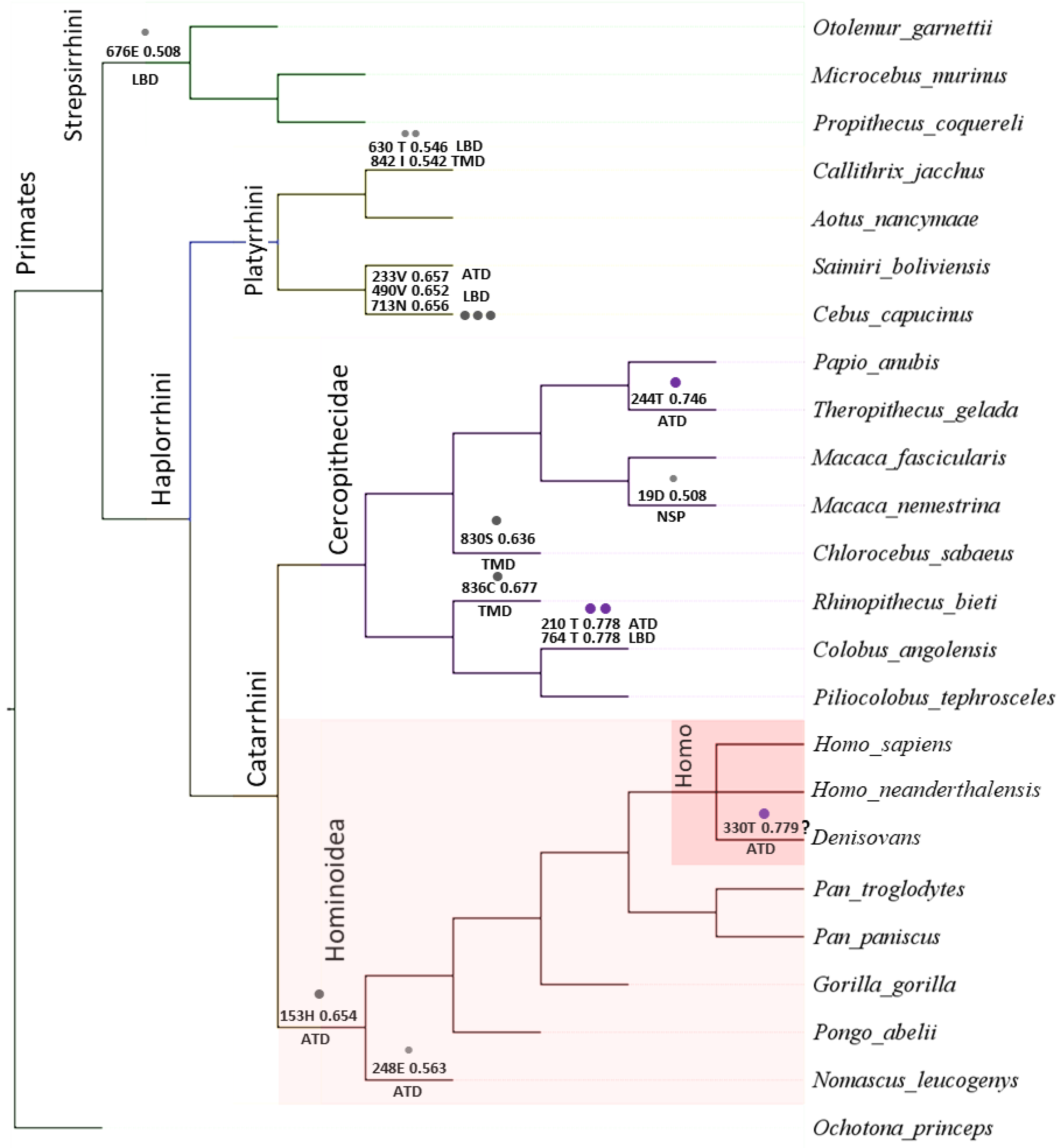{kind=link}
1. Introduction
2. Materials and Methods
3. Results
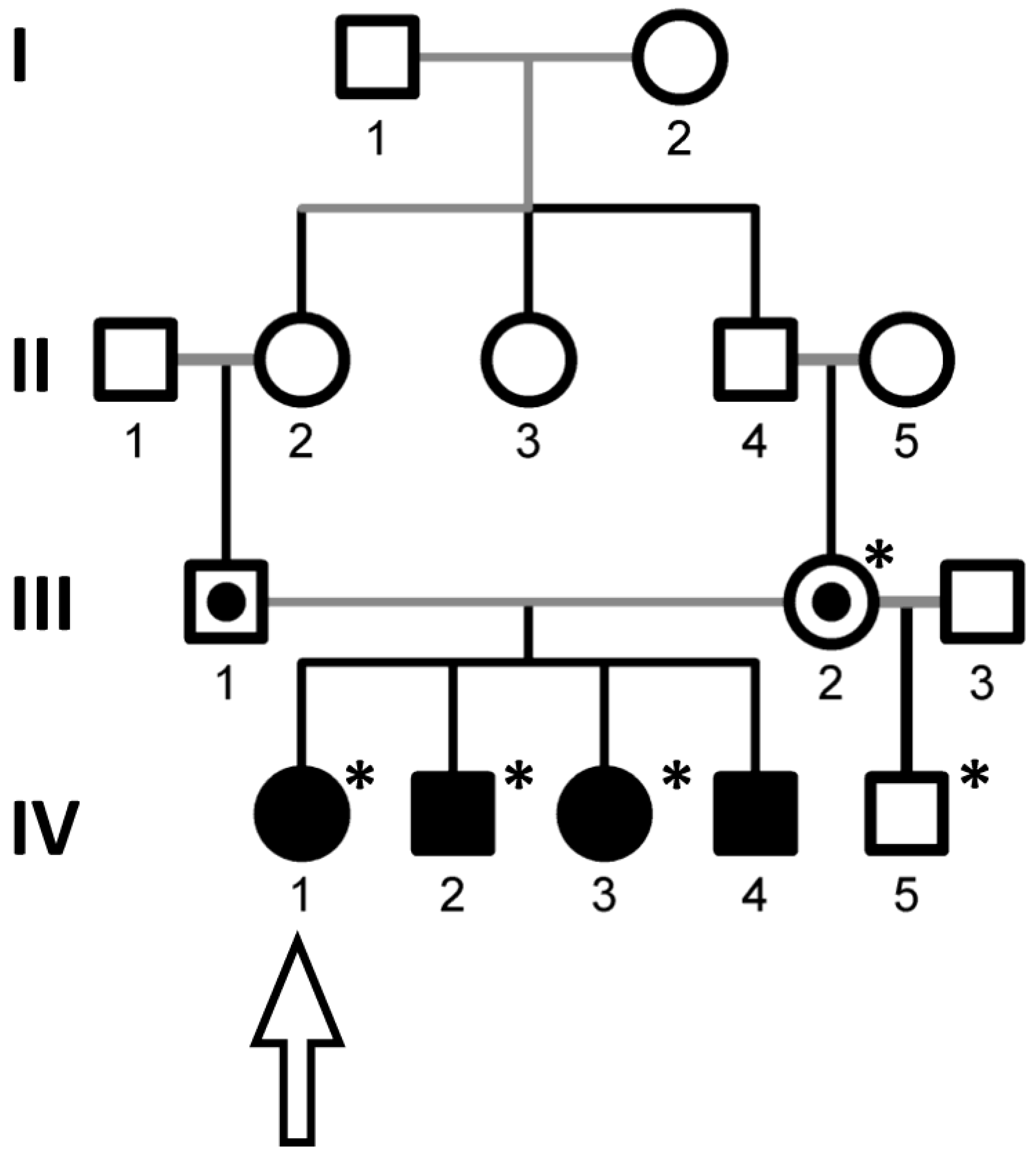
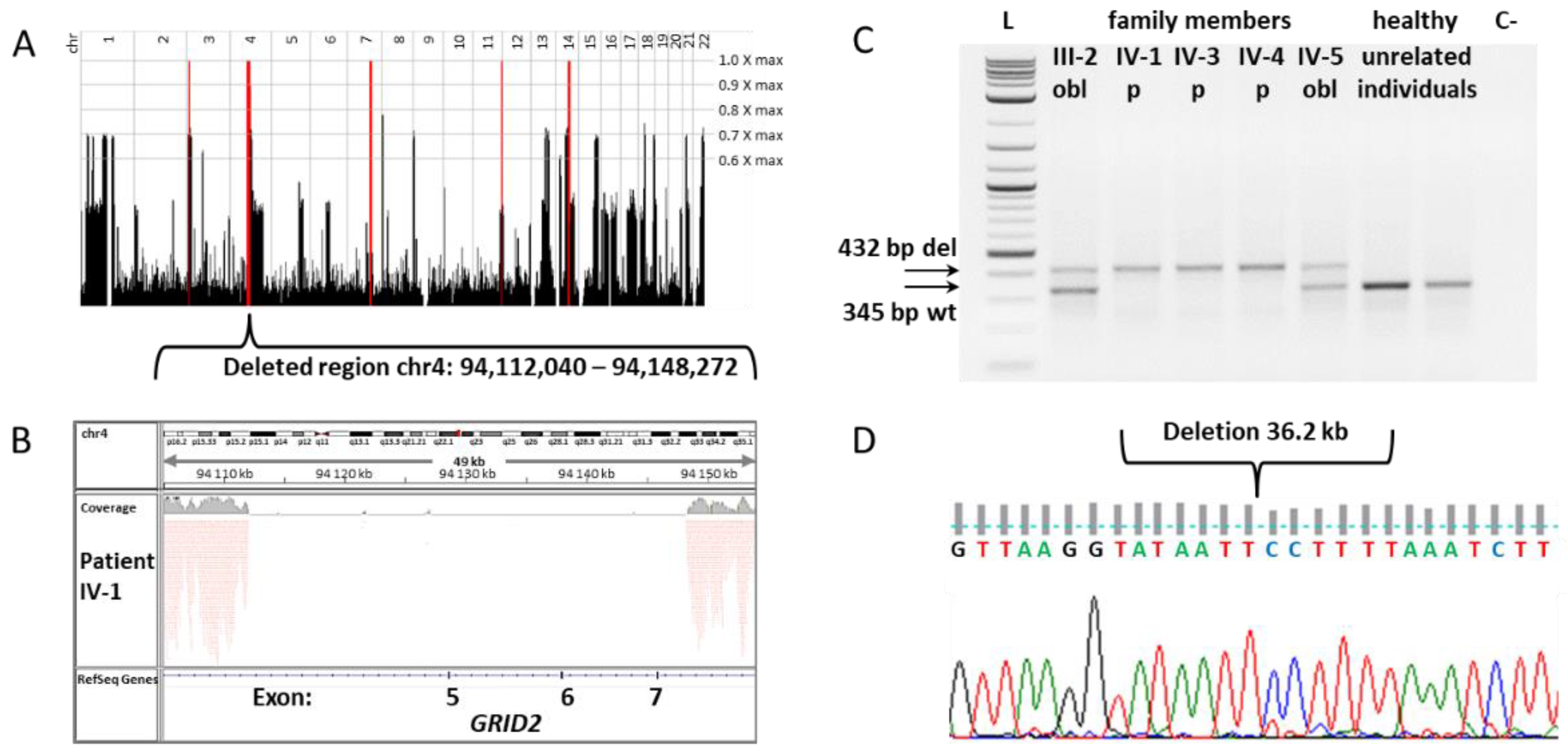
3.1. Genetic Study
3.2. Evolutionary Analysis of Protein-Coding Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Türkmen, S.; Demirhan, O.; Hoffmann, K.; Diers, A.; Zimmer, C.; Sperling, K.; Mundlos, S. Cerebellar hypoplasia and quadrupedal locomotion in humans as a recessive trait mapping to chromosome 17p. J. Med. Genet. 2006, 3, 461–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcelik, T.; Akarsu, N.; Uz, E.; Caglayan, S.; Gulsuner, S.; Onat, O.E.; Tan, M.; Tan, U. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 4232–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, U. A new syndrome with quadrupedal gait, primitive speech, and severe mental retardation as a live model for human evolution. Int. J. Neurosci. 2006, 116, 361–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, U.; Pençe, S.; Yilmaz, M.; Ozkur, A.; Karaca, S.; Tan, M.; Karataş, M. Unertan syndrome’ in two Turkish families in relation to devolution and emergence of Homo erectus: Neurological examination, MRI, and PET scans. Int. J. Neurosci. 2008, 118, 313–336. [Google Scholar] [CrossRef] [Green Version]
- Türkmen, S.; Guo, G.; Garshasbi, M.; Hoffmann, K.; Alshalah, A.J.; Mischung, C.; Kuss, A.; Humphrey, N.; Mundlos, S.; Robinson, P.N. CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet 2009, 5, e1000487. [Google Scholar] [CrossRef] [Green Version]
- Valence, S.; Garel, C.; Barth, M.; Toutain, A.; Paris, C.; Amsallem, D.; Barthez, M.-A.; Mayer, M.; Rodriguez, D.; Burglen, L. RELN and VLDLR mutations underlie two distinguishable clinico-radiological phenotypes. Clin. Genet. 2016, 90, 545–549. [Google Scholar] [CrossRef]
- Gulsuner, S.; Tekinay, A.B.; Doerschner, K.; Boyaci, H.; Bilguvar, K.; Unal, H.; Ors, A.; Onat, O.E.; Atalar, E.; Basak, A.N.; et al. Homozygosity mapping and targeted genomic sequencing reveal the gene responsible for cerebellar hypoplasia and quadrupedal locomotion in a consanguineous kindred. Genome Res. 2011, 21, 1995–2003. [Google Scholar] [CrossRef] [Green Version]
- Onat, O.E.; Gulsuner, S.; Bilguvar, K.; Nazli Basak, A.; Topaloglu, H.; Tan, M.; Tan, U.; Gunel, M.; Ozcelik, T. Missense mutation in the ATPase, aminophospholipid transporter protein ATP8A2 is associated with cerebellar atrophy and quadrupedal locomotion. Eur. J. Hum. Genet. 2013, 21, 281–285. [Google Scholar] [CrossRef]
- Breuss, M.W.; Nguyen, T.; Srivatsan, A.; Leca, I.; Tian, G.; Fritz, T.; Hansen, A.H.; Musaev, D.; McEvoy-Venneri, J.; James, K.N.; et al. Uner Tan syndrome caused by a homozygous TUBB2B mutation affecting microtubule stability. Hum. Mol. Genet. 2017, 26, 258–269. [Google Scholar]
- Garcias, G.D.L.; Roth, M.D.G.M. A Brazilian family with quadrupedal gait, severe mental retardation, coarse facial characteristics, and hirsutism. Int. J. Neurosci. 2007, 117, 927–933. [Google Scholar] [CrossRef]
- Seelow, D.; Schuelke, M.; Hildebrandt, F.; Nürnberg, P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic. Acids. Res. 2009, 37, W593–W599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows—Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- 1000 Genomes Project Consortium; Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Pruitt, K.D.; Brown, G.R.; Hiatt, S.M.; Thibaud-Nissen, F.; Astashyn, A.; Ermolaeva, O.; Farrell, C.M.; Hart, J.; Landrum, M.J.; McGarvey, K.M.; et al. RefSeq: An update on mammalian reference sequences. Nucleic Acids Res. 2014, 42, D756–D763. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database.Oxf. 2016, 2016, baw093. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuánez, H.N.; Horvath, J.E.; Moreira, M.A.M.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A molecular phylogeny of living primates. PLoS Genet 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evol. 2008, 25, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Zuker, M.; Jacobson, A.B. Using reliability information to annotate RNA secondary structures. RNA 1998, 4, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Waugh, A.; Gendron, P.; Altman, R.; Brown, J.W.; Case, D.; Gautheret, D.; Harvey, S.C.; Leontis, N.; Westbrook, J.; Westhof, E.; et al. RNAML: A standard syntax for exchanging RNA information. RNA 2002, 8, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. Gene expression atlas at the European bioinformatics institute. Nucleic Acids Res. 2010, 38, D690–D698. [Google Scholar]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; Dimauro, S.; Hirano, M. A mutation in para-hydroxybenzoate-polyprenyl transferase.COQ2) causes primary coenzyme Q10 deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Jakobs, B.S.; van den Heuvel, L.P.; Smeets, R.J.P.; de Vries, M.C.; Hien, S.; Schaible, T.; Smeitink, J.A.M.; Wevers, R.A.; Wortmann, S.B.; Rodenburg, R.J.T. A novel mutation in COQ2 leading to fatal infantile multisystem disease. J. Neurol. Sci. 2013, 326, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N. Engl. J. Med. 2013, 369, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Ogaki, K.; Fujioka, S.; Heckman, M.G.; Rayaprolu, S.; Soto-Ortolaza, A.I.; Labbé, C.; Walton, R.L.; Lorenzo-Betancor, O.; Wang, X.; Asmann, Y.; et al. Analysis of COQ2 gene in multiple system atrophy. Mol. Neurodegener 2014, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, R.; Pineda, M.; Aracil, A.; Vilaseca, M.-A.; Briones, P.; Sánchez-Alcázar, J.-A.; Navas, P.; Artuch, R. Clinical, biochemical and molecular aspects of cerebellar ataxia and Coenzyme Q10 deficiency. Cerebellum 2007, 6, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.B.; Porta, S.; Michael Baer, G.; Xu, Y.; Suh, E.; Kwong, L.K.; Elman, L.; Grossman, M.; Lee, V.M.-Y.; Irwin, D.J.; et al. Expansion of the classification of FTLD-TDP: Distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017, 134, 65–78. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.E.; Cummings, B.B.; et al. The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [Green Version]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-specific Expression by Genome-wide Integration of Transcriptomics and Antibody-based Proteomics. Mol. Cell Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Ding, S.-L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta2, a bidirectional synapse organizer. Science 2010, 328, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Lee, S.-J.; Yasumura, M.; Takeuchi, T.; Yoshida, T.; Ra, M.; Taguchi, R.; Sakimura, K.; Mishina, M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell 2010, 141, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Yuzaki, M. Mutation in hotfoot-4J mice results in retention of delta2 glutamate receptors in ER. Eur. J. Neurosci. 2002, 16, 1507–1516. [Google Scholar] [CrossRef]
- Maier, A.; Klopocki, E.; Horn, D.; Tzschach, A.; Holm, T.; Meyer, R.; Meyer, T. De novo partial deletion in GRID2 presenting with complicated spastic paraplegia. Muscle Nerve 2014, 49, 289–292. [Google Scholar] [CrossRef]
- Utine, G.E.; Haliloğlu, G.; Salanci, B.; Çetinkaya, A.; Kiper, P.Ö.; Alanay, Y.; Aktas, D.; Boduroğlu, K.; Alikaşifoğlu, M. A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J. Child Neurol. 2013, 28, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Hills, L.B.; Masri, A.; Konno, K.; Kakegawa, W.; Lam, A.-T.N.; Lim-Melia, E.; Chandy, N.; Hill, R.S.; Partlow, J.N.; Al-Saffar, M.; et al. Deletions in GRID2 lead to a recessive syndrome of cerebellar ataxia and tonic upgaze in humans. Neurology 2013, 81, 1378–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Schil, K.; Meire, F.; Karlstetter, M.; Bauwens, M.; Verdin, H.; Coppieters, F.; Scheiffert, E.; Van Nechel, C.; Langmann, T.; Deconinck, N.; et al. Early onset autosomal recessive cerebellar ataxia associated with retinal dystrophy: New human hotfoot phenotype caused by homozygous GRID2 deletion. Genet. Med. 2015, 17, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutelier, M.; Burglen, L.; Mundwiller, E.; Abada-Bendib, M.; Rodriguez, D.; Chantot-Bastaraud, S.; Rougeot, C.; Cournelle, M.-A.; Milh, M.; Toutain, A.; et al. GRID2 mutations span from congenital to mild adult-onset cerebellar ataxia. Neurology 2015, 84, 1751–1759. [Google Scholar] [CrossRef]
- Veerapandiyan, A.; Enner, S.; Thulasi, V.; Ming, X. A Rare Syndrome of GRID2 Deletion in 2 Siblings. Child Neurol. Open 2017, 4, 2329048X17726168. [Google Scholar] [CrossRef] [Green Version]
- Taghdiri, M.; Kashef, A.; Abbassi, G.; Moshtagh, A.; Sadatian, N.; Fardaei, M.; Najafi, K.; Kariminejad, R. Further delineation of the phenotype caused by a novel large homozygous deletion of GRID2 gene in an adult patient. Clin. Case Rep. 2019, 7, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceylan, A.C.; Acar Arslan, E.; Erdem, H.B.; Kavus, H.; Arslan, M.; Topaloğlu, H. Autosomal recessive spinocerebellar ataxia 18 caused by homozygous exon 14 duplication in GRID2 and review of the literature. Acta Neurol. Belg. 2020, 121, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Hetzelt, K.L.M.L.; Kraus, C.; Kusnik, S.; Thiel, C.T.; Uebe, S.; Ekici, A.B.; Trollmann, R.; Reis, A.; Zweier, C. A case of severe autosomal recessive spinocerebellar ataxia type 18 with a novel nonsense variant in GRID2. Eur. J. Med. Genet. 2020, 63, 103998. [Google Scholar] [CrossRef] [PubMed]
- Lalouette, A.; Guénet, J.-L.; Vriz, S. Hotfoot Mouse Mutations Affect the δ2 Glutamate Receptor Gene and Are Allelic to Lurcher. Genomics 1998, 50, 9–13. [Google Scholar] [CrossRef]
- Burada, A.P.; Vinnakota, R.; Kumar, J. The architecture of GluD2 ionotropic delta glutamate receptor elucidated by cryo-EM. J. Struct. Biol. 2020, 211, 107546. [Google Scholar] [CrossRef]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, K.; Nicholas, H.; Deerfield, D. GeneDoc: Analysis and visualization of genetic variation. EMBNEW News 1997, 4, 14. [Google Scholar]
- Edwards, N.C.; Hing, Z.A.; Perry, A.; Blaisdell, A.; Kopelman, D.B.; Fathke, R.; Plum, W.; Newell, J.; Allen, C.E.; S., G.; et al. Characterization of Coding Synonymous and Non-Synonymous Variants in ADAMTS13 Using Ex Vivo and In Silico Approaches. PLoS ONE 2012, 7, e38864. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Zulfiqar, S.; Klar, J.; Wikström, J.; Ullah, F.; Khan, A.; Abdullah, U.; Baig, S.; Dahl, N. Homozygous GRID2 missense mutation predicts a shift in the D-serine binding domain of GluD2 in a case with generalized brain atrophy and unusual clinical features. BMC Med. Genet. 2017, 18, 144. [Google Scholar] [CrossRef] [Green Version]
- Iodice, A.; Spagnoli, C.; Cangini, M.; Soliani, L.; Rizzi, S.; Salerno, G.G.; Frattini, D.; Pisani, F.; Fusco, C. Long-term follow-up in infantile-onset SCAR18: A case report. J. Clin. Neurosci. 2020, 77, 232–234. [Google Scholar] [CrossRef]
- Xu, Y.; Mak, H.Y.; Lukmantara, I.; Li, Y.E.; Hoehn, K.L.; Huang, X.; Du, X.; Yang, H. CDP-DAG synthase 1 and 2 regulate lipid droplet growth through distinct mechanisms. J. Biol. Chem. 2019, 294, 1674016755. [Google Scholar] [CrossRef] [PubMed]
- Webb, E.A.; AlMutair, A.; Kelberman, D.; Bacchelli, C.; Chanudet, E.; Lescai, F.; Andoniadou, C.L.; Banyan, A.; Alsawaid, A.; Alrifai, M.T.; et al. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 2013, 134, 3096–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latimer, B.; Lovejoy, C.O. Metatarsophalangeal joints of Australopithecus afarensis. Am. J. Phys. Anthropol. 1990, 83, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Fernández, P.J.; Holowka, N.B.; Demes, B.; Jungers, W.L. Form and function of the human and chimpanzee forefoot: Implications for early hominin bipedalism. Sci. Rep. 2016, 6, 30532. [Google Scholar] [CrossRef] [Green Version]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Adaptation of mRNA structure to control protein folding. RNA Biol. 2017, 14, 1649–1654. [Google Scholar] [CrossRef]
- Dhindsa, R.S.; Copeland, B.R.; Mustoe, A.M.; Goldstein, D.B. Natural Selection Shapes Codon Usage in the Human Genome. Am. J. Hum. Genet. 2020, 107, 83–95. [Google Scholar] [CrossRef]
- Elegheert, J.; Kakegawa, W.; Clay, J.E.; Shanks, N.F.; Behiels, E.; Matsuda, K.; Kohda, K.; Miura, E.; Rossmann, M.; Mitakidis, N.; et al. Structural basis for integration of GluD receptors within synaptic organizer complexes. Science 2016, 353, 295–299. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigorenko, A.P.; Protasova, M.S.; Lisenkova, A.A.; Reshetov, D.A.; Andreeva, T.V.; Garcias, G.D.L.; Martino Roth, M.D.G.; Papassotiropoulos, A.; Rogaev, E.I. Neurodevelopmental Syndrome with Intellectual Disability, Speech Impairment, and Quadrupedia Is Associated with Glutamate Receptor Delta 2 Gene Defect. Cells 2022, 11, 400. https://doi.org/10.3390/cells11030400
Grigorenko AP, Protasova MS, Lisenkova AA, Reshetov DA, Andreeva TV, Garcias GDL, Martino Roth MDG, Papassotiropoulos A, Rogaev EI. Neurodevelopmental Syndrome with Intellectual Disability, Speech Impairment, and Quadrupedia Is Associated with Glutamate Receptor Delta 2 Gene Defect. Cells. 2022; 11(3):400. https://doi.org/10.3390/cells11030400
Chicago/Turabian StyleGrigorenko, Anastasia P., Maria S. Protasova, Alexandra A. Lisenkova, Denis A. Reshetov, Tatiana V. Andreeva, Gilberto De Lima Garcias, Maria Da Graça Martino Roth, Andreas Papassotiropoulos, and Evgeny I. Rogaev. 2022. "Neurodevelopmental Syndrome with Intellectual Disability, Speech Impairment, and Quadrupedia Is Associated with Glutamate Receptor Delta 2 Gene Defect" Cells 11, no. 3: 400. https://doi.org/10.3390/cells11030400
APA StyleGrigorenko, A. P., Protasova, M. S., Lisenkova, A. A., Reshetov, D. A., Andreeva, T. V., Garcias, G. D. L., Martino Roth, M. D. G., Papassotiropoulos, A., & Rogaev, E. I. (2022). Neurodevelopmental Syndrome with Intellectual Disability, Speech Impairment, and Quadrupedia Is Associated with Glutamate Receptor Delta 2 Gene Defect. Cells, 11(3), 400. https://doi.org/10.3390/cells11030400