
STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact

 , , ,
, , ,  and
and
Abstract
:1. Introduction
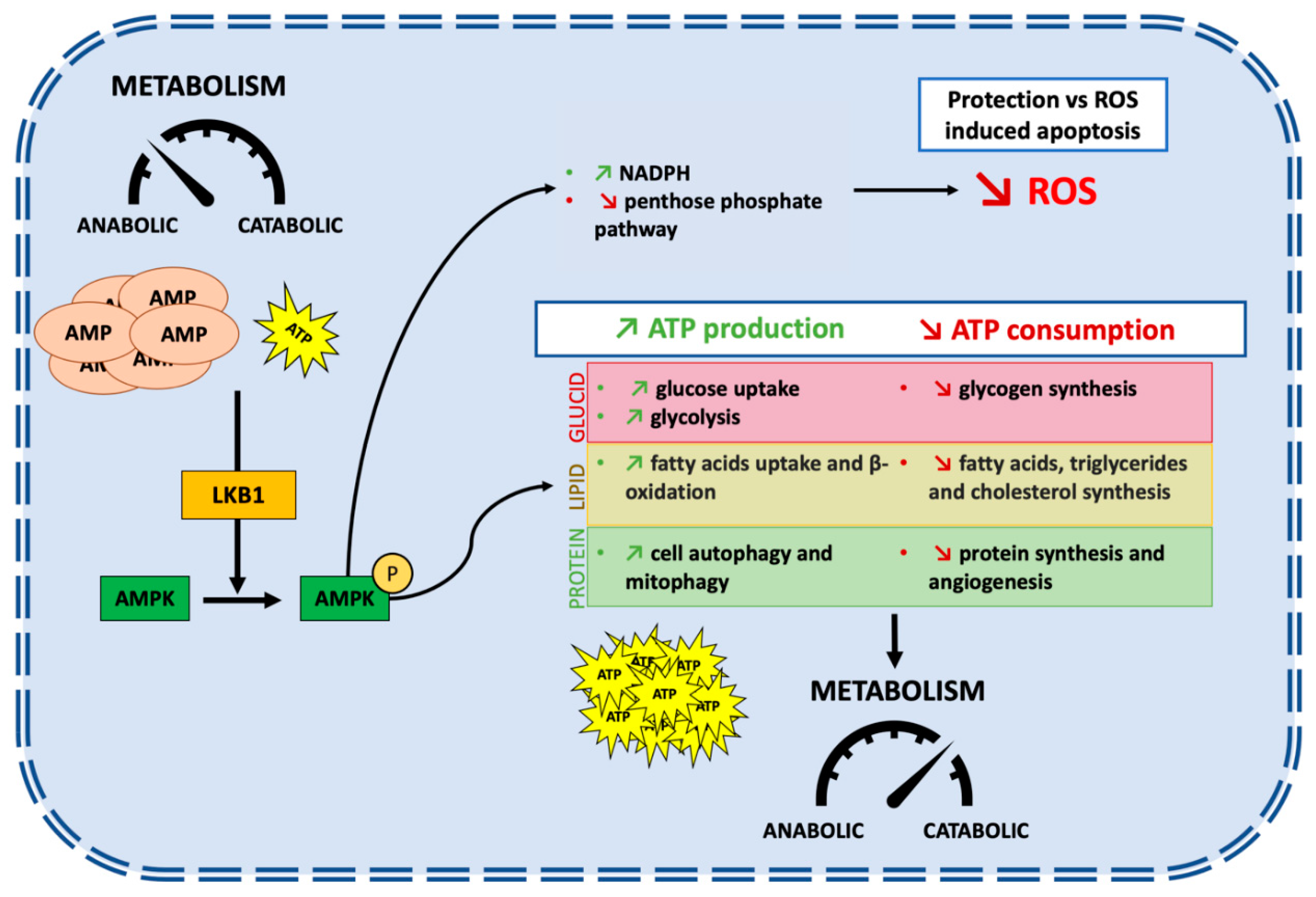
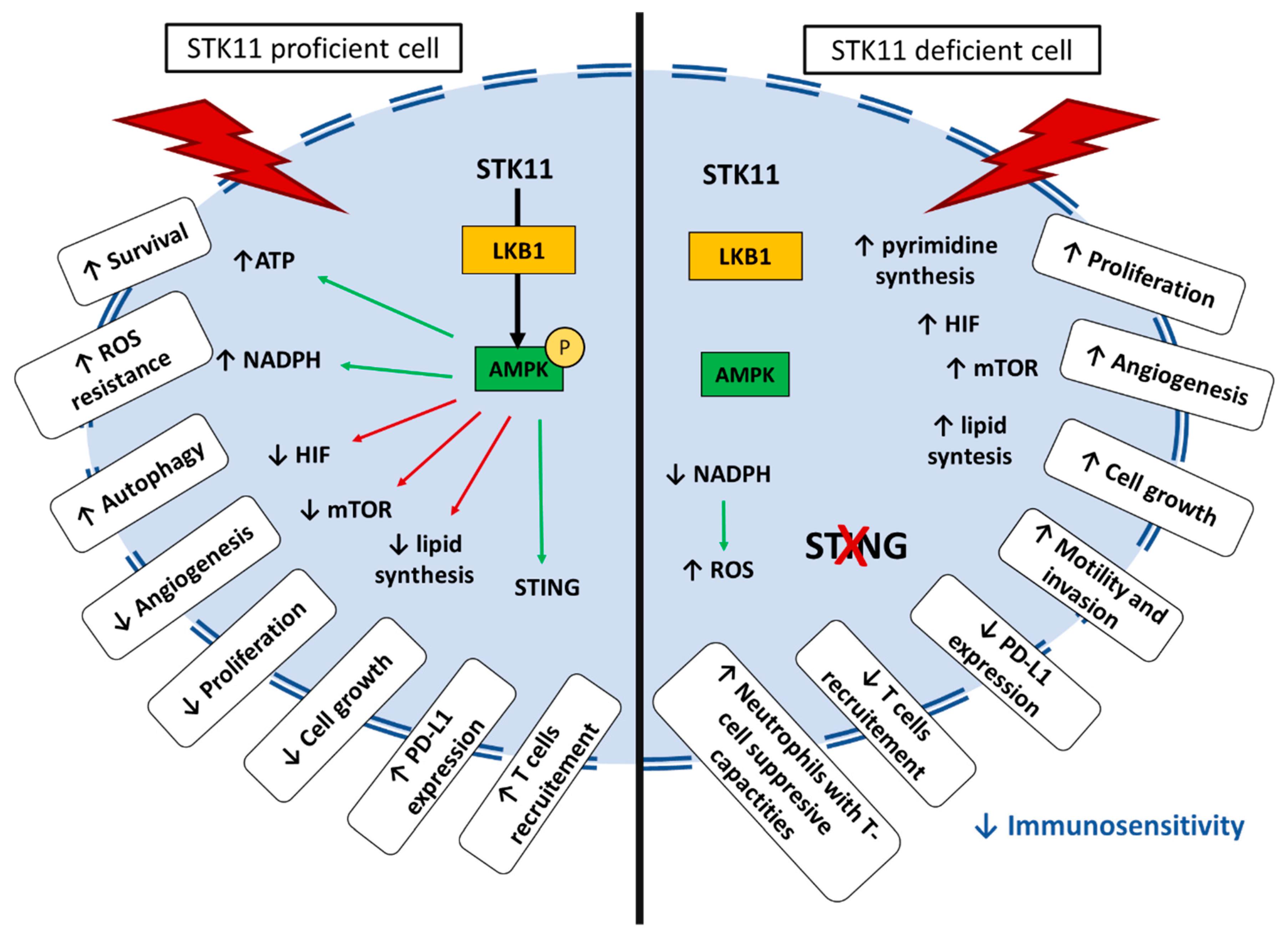
2. STK11/LKB1: An Overview of Biological Functions
3. STK11/LKB1: A Tumor Suppressor
3.1. Molecular Mechanisms of STK11/LKB1 Inactivation in Cancers
3.2. STK11/LKB1 Alterations Promote Lung Cancer Cell Survival and Invasion
4. STK11/LKB1 Alterations and Co-Occurring Mutations: Prognostic Impact in Lung Cancer Patients
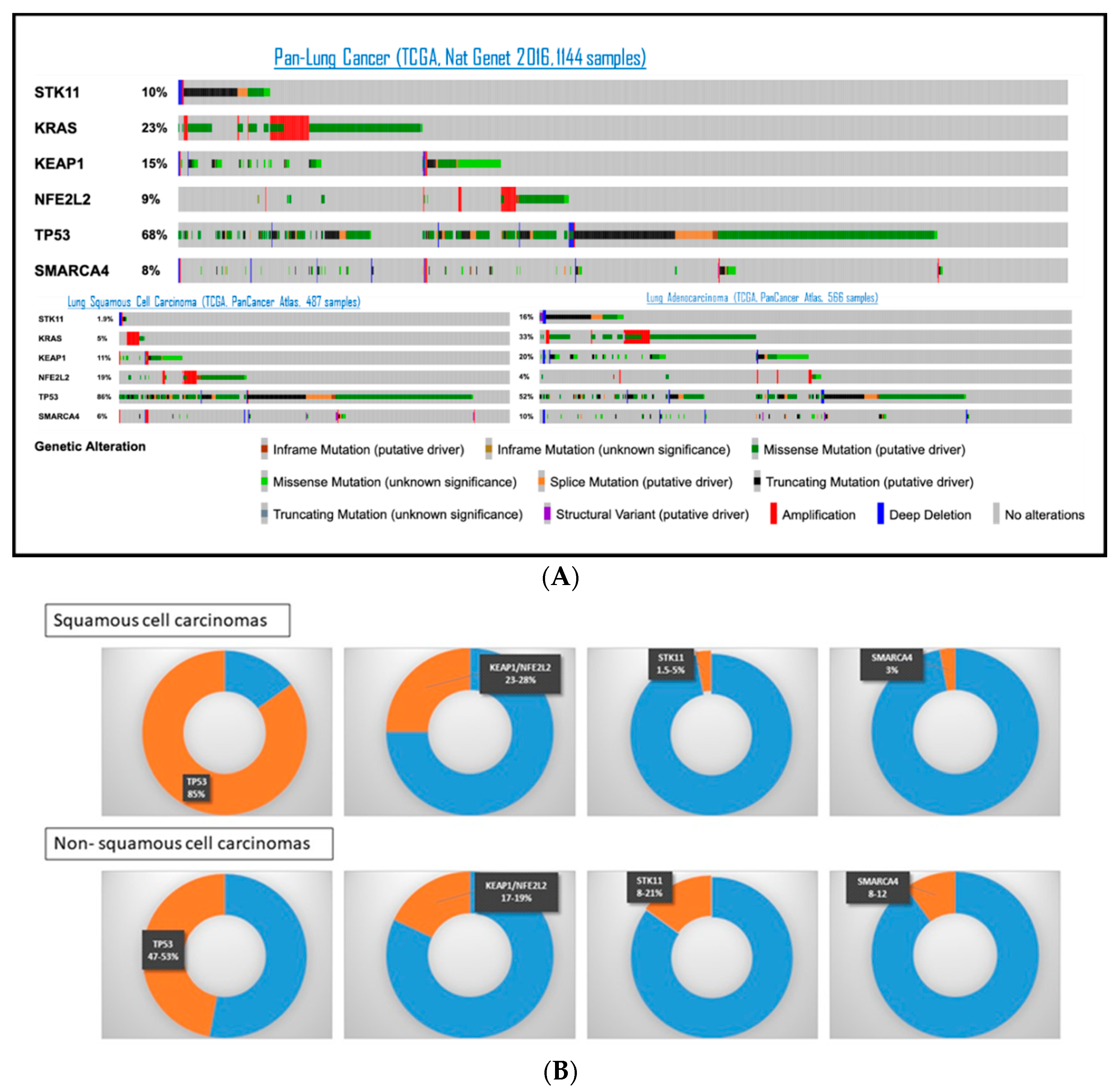
4.1. STK11/LKB1 Co-Occurring Genomic Alterations in Lung Cancer
4.2. Prognostic Impact of STK11/LKB1 Alterations in Lung Cancer before the Era of Immunotherapy
5. Immune Impact of STK11/LKB1 Alterations in Lung Cancer Patients
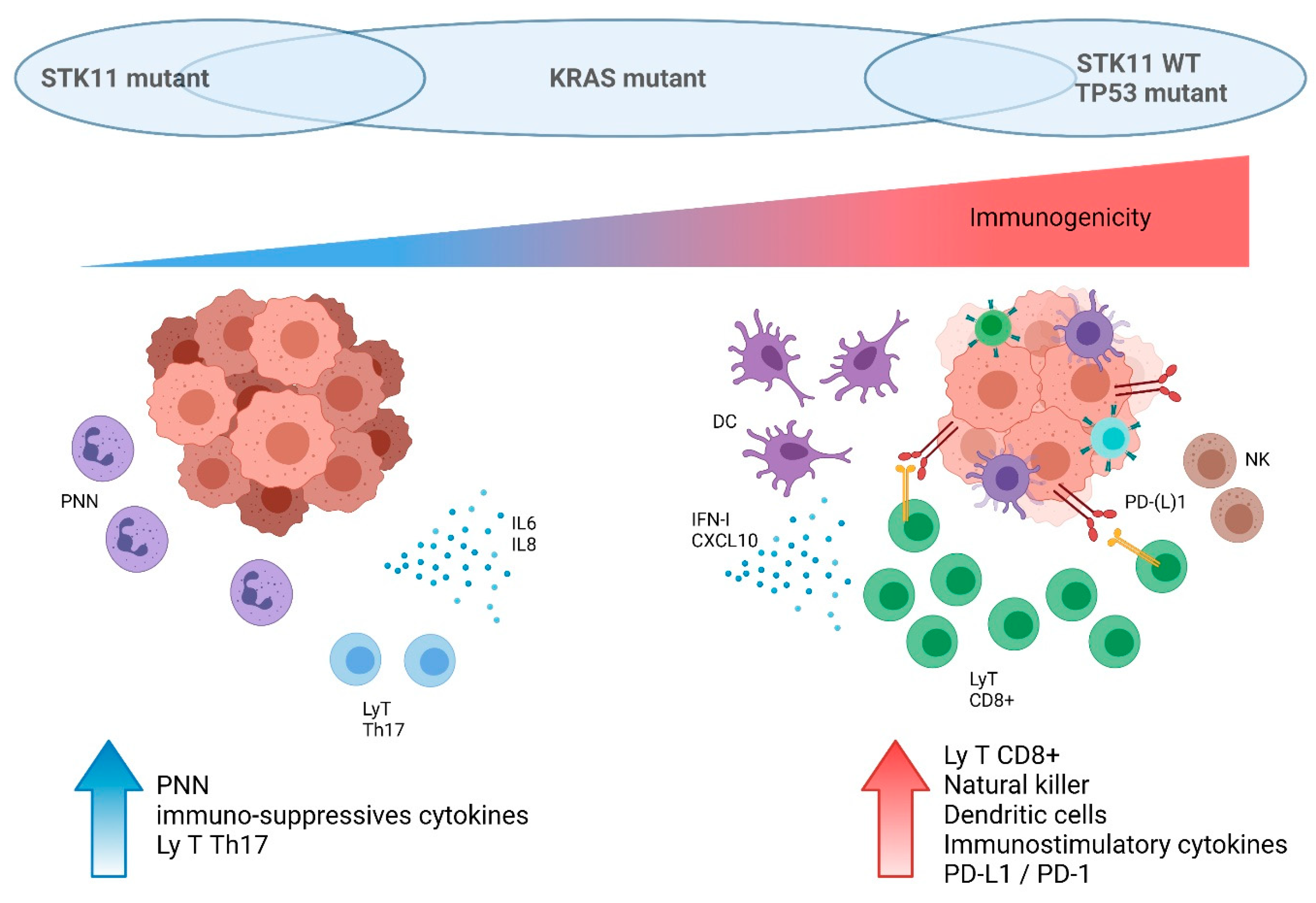
5.1. Tumor-Extrinsic Impact of STK11/LKB1 Alteration: Interaction with Immune System
5.2. STK11/LKB1 Alterations Negatively Impact Immune Surveillance in NSCLC Patients
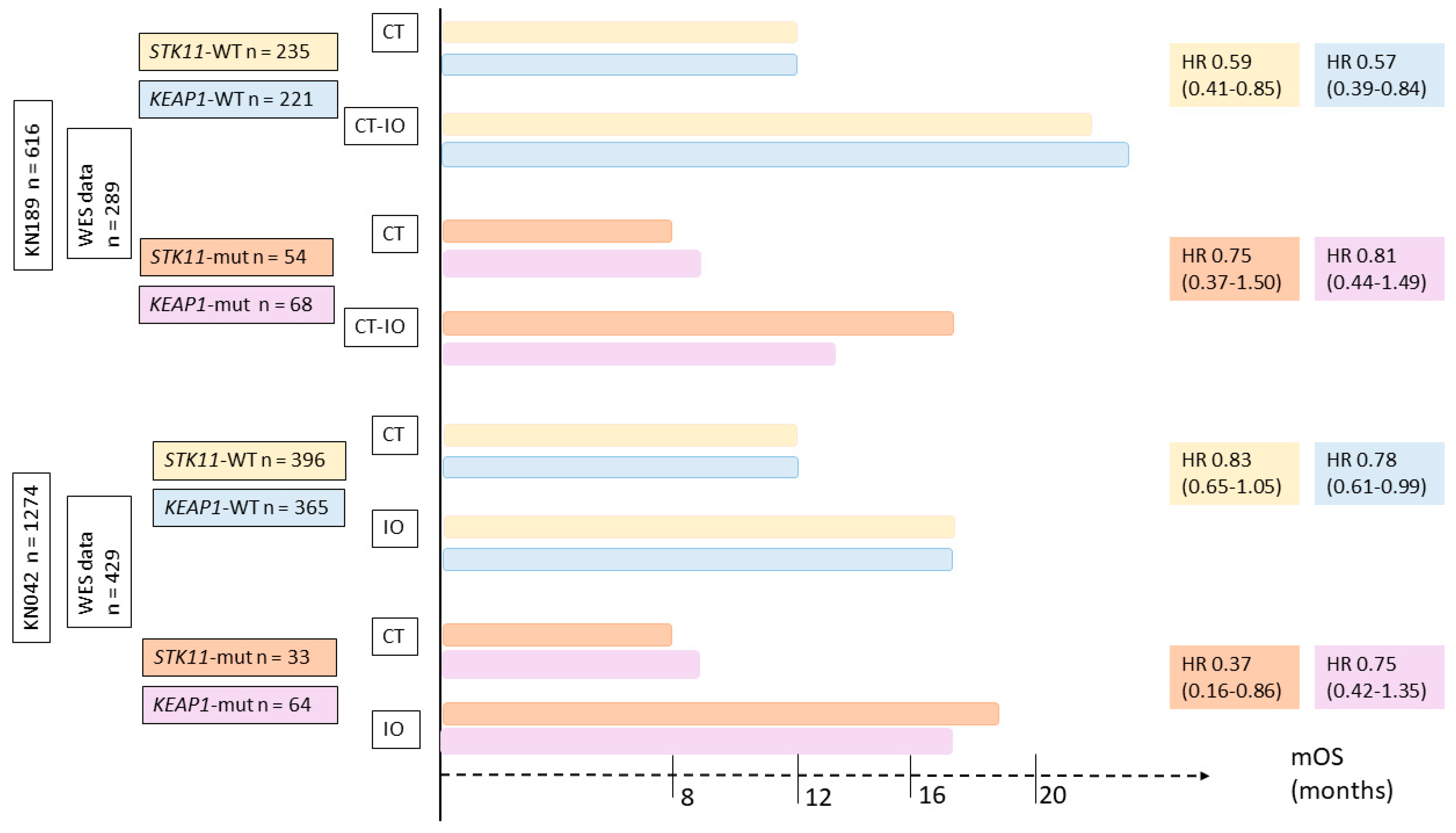
5.3. Impact of SKT11/LKB1 Alterations on ICI Efficacy in NSCLC Patients
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beggs, A.D.; Latchford, A.R.; Vasen, H.F.A.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; et al. Peutz-Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumor suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Tomlinson, I.; Markie, D.; Järvinen, H.; Sistonen, P.; Björkqvist, A.M.; Knuutila, S.; Salovaara, R.; Bodmer, W.; Shibata, D.; et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat. Genet. 1997, 15, 87–90. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Launonen, V. Mutations in the human LKB1/STK11 gene. Hum. Mutat. 2005, 26, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [Green Version]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Munday, M.R. Regulation of mammalian acetyl-CoA carboxylase. Biochem. Soc. Trans. 2002, 30, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Ferré, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsin, A.-S.; Bouzin, C.; Bertrand, L.; Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 2002, 277, 30778–30783. [Google Scholar] [CrossRef] [Green Version]
- Wojtaszewski, J.F.P.; Nielsen, J.N.; Jørgensen, S.B.; Frøsig, C.; Birk, J.B.; Richter, E.A. Transgenic models--a scientific tool to understand exercise-induced metabolism: The regulatory role of AMPK (5′-AMP-activated protein kinase) in glucose transport and glycogen synthase activity in skeletal muscle. Biochem. Soc. Trans. 2003, 31, 1290–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ji, J.; Yan, X.-H. Cross-Talk between AMPK and mTOR in Regulating Energy Balance. Crit. Rev. Food Sci. Nutr. 2012, 52, 373–381. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Onorati, A.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species (Apex, NC) 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whang, Y.M.; Park, S.I.; Trenary, I.A.; Egnatchik, R.A.; Fessel, J.P.; Kaufman, J.M.; Carbone, D.P.; Young, J.D. LKB1 deficiency enhances sensitivity to energetic stress induced by erlotinib treatment in non-small-cell lung cancer (NSCLC) cells. Oncogene 2016, 35, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Bonanno, L.; Zulato, E.; Pavan, A.; Attili, I.; Pasello, G.; Conte, P.; Indraccolo, S. LKB1 and Tumor Metabolism: The Interplay of Immune and Angiogenic Microenvironment in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Cespedes, M.; Parrella, P.; Esteller, M.; Nomoto, S.; Trink, B.; Engles, J.M.; Westra, W.H.; Herman, J.G.; Sidransky, D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62, 3659–3662. [Google Scholar] [PubMed]
- Hezel, A.F.; Gurumurthy, S.; Granot, Z.; Swisa, A.; Chu, G.C.; Bailey, G.; Dor, Y.; Bardeesy, N.; Depinho, R.A. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol. Cell. Biol. 2008, 28, 2414–2425. [Google Scholar] [CrossRef] [Green Version]
- Wingo, S.N.; Gallardo, T.D.; Akbay, E.A.; Liang, M.-C.; Contreras, C.M.; Boren, T.; Shimamura, T.; Miller, D.S.; Sharpless, N.E.; Bardeesy, N.; et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 2009, 4, e5137. [Google Scholar] [CrossRef]
- Gill, R.K.; Yang, S.-H.; Meerzaman, D.; Mechanic, L.E.; Bowman, E.D.; Jeon, H.-S.; Roy Chowdhuri, S.; Shakoori, A.; Dracheva, T.; Hong, K.-M.; et al. Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene 2011, 30, 3784–3791. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Choi, J.E.; Na, Y.K.; Lee, E.J.; Lee, W.K.; Choi, Y.Y.; Yoon, G.S.; Jeon, H.-S.; Kim, D.S.; Park, J.Y. Genetic and epigenetic alterations of the LKB1 gene and their associations with mutations in TP53 and EGFR pathway genes in Korean non-small cell lung cancers. Lung Cancer 2013, 81, 194–199. [Google Scholar] [CrossRef]
- Tanwar, P.S.; Mohapatra, G.; Chiang, S.; Engler, D.A.; Zhang, L.; Kaneko-Tarui, T.; Ohguchi, Y.; Birrer, M.J.; Teixeira, J.M. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 2014, 35, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, J.; Li, P.; Mao, X.; Li, W.; Yang, J.; Liu, P. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J. Exp. Clin. Cancer Res. 2014, 33, 70. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Y.; Jiang, S.-H.; Liu, D.-J.; Yang, X.-M.; Huo, Y.-M.; Li, J.; Hua, R.; Zhang, Z.-G.; Sun, Y.-W. Decreased LKB1 predicts poor prognosis in Pancreatic Ductal Adenocarcinoma. Sci. Rep. 2015, 5, 10575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yin, L.; Song, G.; Han, X.; Yin, Z.; Luo, D. LKB1 loss cooperating with BRAF V600E promotes melanoma cell invasion and migration by up-regulation MMP-2 via PI3K/Akt/mTOR pathway. Oncotarget 2017, 8, 113847–113857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, N.; Goodman, A.M.; Barkauskas, D.A.; Kurzrock, R. STK11 alterations in the pan-cancer setting: Prognostic and therapeutic implications. Eur. J. Cancer 2021, 148, 215–229. [Google Scholar] [CrossRef]
- Granado-Martínez, P.; Garcia-Ortega, S.; González-Sánchez, E.; McGrail, K.; Selgas, R.; Grueso, J.; Gil, R.; Naldaiz-Gastesi, N.; Rhodes, A.C.; Hernandez-Losa, J.; et al. STK11 (LKB1) missense somatic mutant isoforms promote tumor growth, motility and inflammation. Commun. Biol. 2020, 3, 366. [Google Scholar] [CrossRef]
- Borzi, C.; Galli, G.; Ganzinelli, M.; Signorelli, D.; Vernieri, C.; Garassino, M.C.; Sozzi, G.; Moro, M. Beyond LKB1 Mutations in Non-Small Cell Lung Cancer: Defining LKB1less Phenotype to Optimize Patient Selection and Treatment. Pharmaceuticals 2020, 13, 385. [Google Scholar] [CrossRef]
- Zheng, F.; Yuan, X.; Chen, E.; Ye, Y.; Li, X.; Dai, Y. Methylation of STK11 promoter is a risk factor for tumor stage and survival in clear cell renal cell carcinoma. Oncol. Lett. 2017, 14, 3065–3070. [Google Scholar] [CrossRef] [Green Version]
- Trojan, J.; Brieger, A.; Raedle, J.; Esteller, M.; Zeuzem, S. 5′-CpG island methylation of the LKB1/STK11 promoter and allelic loss at chromosome 19p13.3 in sporadic colorectal cancer. Gut 2000, 47, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, X.; Song, G.; Luo, D. Prognostic significance of LKB1 promoter methylation in cutaneous malignant melanoma. Oncol. Lett. 2017, 14, 2075–2080. [Google Scholar] [CrossRef] [Green Version]
- Lao, G.; Liu, P.; Wu, Q.; Zhang, W.; Liu, Y.; Yang, L.; Ma, C. Mir-155 promotes cervical cancer cell proliferation through suppression of its target gene LKB1. Tumour Biol. 2014, 35, 11933–11938. [Google Scholar] [CrossRef]
- Figueroa-González, G.; Carrillo-Hernández, J.F.; Perez-Rodriguez, I.; Cantú de León, D.; Campos-Parra, A.D.; Martínez-Gutiérrez, A.D.; Coronel-Hernández, J.; García-Castillo, V.; López-Camarillo, C.; Peralta-Zaragoza, O.; et al. Negative Regulation of Serine Threonine Kinase 11 (STK11) through miR-100 in Head and Neck Cancer. Genes 2020, 11, 1058. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, L.; Falk-Sörqvist, E.; Smeds, P.; Berglund, A.; Sundström, M.; Mattsson, J.S.; Brandén, E.; Koyi, H.; Isaksson, J.; Brunnström, H.; et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer 2019, 130, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; König, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Ding, B.; Xu, J.; Mao, K.; Zhang, P.; Xue, Q. Relevance of STK11 Mutations Regarding Immune Cell Infiltration, Drug Sensitivity, and Cellular Processes in Lung Adenocarcinoma. Front. Oncol. 2020, 10, 580027. [Google Scholar] [CrossRef]
- Schabath, M.B.; Welsh, E.A.; Fulp, W.J.; Chen, L.; Teer, J.K.; Thompson, Z.J.; Engel, B.E.; Xie, M.; Berglund, A.E.; Creelan, B.C.; et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016, 35, 3209–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchinetti, F.; Bluthgen, M.V.; Tergemina-Clain, G.; Faivre, L.; Pignon, J.-P.; Planchard, D.; Remon, J.; Soria, J.-C.; Lacroix, L.; Besse, B. LKB1/STK11 mutations in non-small cell lung cancer patients: Descriptive analysis and prognostic value. Lung Cancer Amst. Neth. 2017, 112, 62–68. [Google Scholar] [CrossRef]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 876–889. [Google Scholar] [CrossRef]
- Shire, N.J.; Klein, A.B.; Golozar, A.; Collins, J.M.; Fraeman, K.H.; Nordstrom, B.L.; McEwen, R.; Hembrough, T.; Rizvi, N.A. STK11 (LKB1) mutations in metastatic NSCLC: Prognostic value in the real world. PLoS ONE 2020, 15, e0238358. [Google Scholar] [CrossRef]
- Shang, X.; Li, Z.; Sun, J.; Zhao, C.; Lin, J.; Wang, H. Survival analysis for non-squamous NSCLC patients harbored STK11 or KEAP1 mutation receiving atezolizumab. Lung Cancer 2021, 154, 105–112. [Google Scholar] [CrossRef]
- Armon, S.; Hofman, P.; Ilié, M. Perspectives and Issues in the Assessment of SMARCA4 Deficiency in the Management of Lung Cancer Patients. Cells 2021, 10, 1920. [Google Scholar] [CrossRef]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.C.; Kohno, T.; Iwakawa, R.; Moriguchi, T.; Kiyono, T.; Morishita, K.; Sanchez-Cespedes, M.; Akiyama, T.; Yokota, J. Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of human lung cancer cells. Lung Cancer 2010, 70, 136–145. [Google Scholar] [CrossRef]
- Yao, Y.-H.; Cui, Y.; Qiu, X.-N.; Zhang, L.-Z.; Zhang, W.; Li, H.; Yu, J.-M. Attenuated LKB1-SIK1 signaling promotes epithelial-mesenchymal transition and radioresistance of non-small cell lung cancer cells. Chin. J. Cancer 2016, 35, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giatromanolaki, A.; Koukourakis, M.I.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.C.; Harris, A.L. Relation of hypoxia inducible factor 1 alpha and 2 alpha in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881–890. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Shackelford, D.B. Targeting LKB1 in cancer—Exposing and exploiting vulnerabilities. Br. J. Cancer 2015, 113, 574–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Le, A. Glutamine Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Charlier, D.; Nguyen Le Minh, P.; Roovers, M. Regulation of carbamoylphosphate synthesis in Escherichia coli: An amazing metabolite at the crossroad of arginine and pyrimidine biosynthesis. Amino Acids 2018, 50, 1647–1661. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.-F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.C. Regulation of Glutathione Synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Caso, R.; Tan, K.S.; Mastrogiacomo, B.; Sanchez-Vega, F.; Liu, Y.; Connolly, J.G.; Murciano-Goroff, Y.R.; Bott, M.J.; Adusumilli, P.S.; et al. KRASG12C Mutation Is Associated with Increased Risk of Recurrence in Surgically Resected Lung Adenocarcinoma. Clin. Cancer Res. 2021, 27, 2604–2612. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.-H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Izumchenko, E.; Chang, X.; Brait, M.; Fertig, E.; Kagohara, L.T.; Bedi, A.; Marchionni, L.; Agrawal, N.; Ravi, R.; Jones, S.; et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat. Commun. 2015, 6, 8258. [Google Scholar] [CrossRef] [PubMed]
- Arbour, K.C.; Rizvi, H.; Plodkowski, A.J.; Hellmann, M.D.; Knezevic, A.; Heller, G.; Yu, H.A.; Ladanyi, M.; Kris, M.G.; Arcila, M.E.; et al. Treatment Outcomes and Clinical Characteristics of Patients with KRAS-G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2209-2015. [Google Scholar] [CrossRef]
- Reck, M.; Carbone, D.P.; Garassino, M.; Barlesi, F. Targeting KRAS in non-small-cell lung cancer: Recent progress and new approaches. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, S0923-753402045-7. [Google Scholar] [CrossRef]
- Arbour, K.C.; Jordan, E.; Kim, H.R.; Dienstag, J.; Yu, H.A.; Sanchez-Vega, F.; Lito, P.; Berger, M.; Solit, D.B.; Hellmann, M.; et al. Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Sima, C.S.; Shen, R.; Kass, S.; Gainor, J.; Shaw, A.; Hames, M.; Iams, W.; Aston, J.; Lovly, C.M.; et al. Prognostic impact of KRAS mutation subtypes in 677 patients with metastatic lung adenocarcinomas. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, J.V.; Ricciuti, B.; Spurr, L.F.; Gupta, H.; Li, Y.Y.; Glass, C.; Nishino, M.; Cherniack, A.D.; Lindsay, J.; Sharma, B.; et al. SMARCA4 and Other SWItch/Sucrose NonFermentable Family Genomic Alterations in NSCLC: Clinicopathologic Characteristics and Outcomes to Immune Checkpoint Inhibition. J. Thorac. Oncol. 2021, 16, 1176–1187. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; Doshi, P.; Dobrin, R.; Szustakowski, J.; Walsh, A.M. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2020, 5, e000706. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pore, N.; Wu, S.; Standifer, N.; Jure-Kunkel, M.; de Los Reyes, M.; Shrestha, Y.; Halpin, R.; Rothstein, R.; Mulgrew, K.; Blackmore, S.; et al. Resistance to durvalumab and durvalumab plus tremelimumab is associated with functional STK11 mutations in non-small-cell lung cancer patients and is reversed by STAT3 knockdown. Cancer Discov. 2021, candisc.1543.2020. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Biton, J.; Mansuet-Lupo, A.; Pécuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Rizvi, H.; Bandlamudi, C.; Sauter, J.L.; Travis, W.D.; Rekhtman, N.; Plodkowski, A.J.; Perez-Johnston, R.; Sawan, P.; Beras, A.; et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and Molecular Characteristics Associated with Survival among Patients Treated with Checkpoint Inhibitors for Advanced Non-Small Cell Lung Carcinoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- Dudnik, E.; Moskovitz, M.; Daher, S.; Shamai, S.; Hanovich, E.; Grubstein, A.; Shochat, T.; Wollner, M.; Bar, J.; Merimsky, O.; et al. Effectiveness and safety of nivolumab in advanced non-small cell lung cancer: The real-life data. Lung Cancer 2018, 126, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Morita, R.; Okishio, K.; Shimizu, J.; Saito, H.; Sakai, H.; Kim, Y.H.; Hataji, O.; Yomota, M.; Nishio, M.; Aoe, K.; et al. Real-world effectiveness and safety of nivolumab in patients with non-small cell lung cancer: A multicenter retrospective observational study in Japan. Lung Cancer 2020, 140, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salehi-Rad, R.; Li, R.; Tran, L.M.; Lim, R.J.; Abascal, J.; Momcilovic, M.; Park, S.J.; Ong, S.L.; Shabihkhani, M.; Huang, Z.L.; et al. Novel Kras-mutant murine models of non-small cell lung cancer possessing co-occurring oncogenic mutations and increased tumor mutational burden. Cancer Immunol. Immunother. 2021, 70, 2389–2400. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti–Programmed Cell Death (PD)-1 and Anti–Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non–Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Gadgeel, S.M.; Rodriguez-Abreu, D.; Felip, E.; Esteban, E.; Speranza, G.; Reck, M.; Hui, R.; Boyer, M.; Garon, E.B.; Horinouchi, H.; et al. Abstract LB-397: Pembrolizumab plus pemetrexed and platinum vs. placebo plus pemetrexed and platinum as first-line therapy for metastatic nonsquamous NSCLC: Analysis of KEYNOTE-189 by STK11 and KEAP1 status. Cancer Res. 2020, 80, LB-397. [Google Scholar] [CrossRef]
- Cho, B.C.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; Castro, G.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; Loboda, A.; et al. Abstract CT084: Relationship between STK11 and KEAP1 mutational status and efficacy in KEYNOTE-042: Pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. Cancer Res. 2020, 80, CT084. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutational Status | Total Number of Mutated Patients | Number of Patients with Co-Mutation Gene/STK11 | % Co-Mutation |
|---|---|---|---|
| KRAS | 259 | 63 | 24.32 |
| NTRK1 | 96 | 18 | 18.75 |
| NRAS | 30 | 4 | 13.33 |
| SMARCA4 | 94 | 11 | 11.70 |
| BRAF | 80 | 9 | 11.25 |
| NKX2-1 | 99 | 11 | 11.11 |
| ALK | 57 | 5 | 8.77 |
| ROS1 | 71 | 6 | 8.45 |
| CDKN2A (HD) | 342 | 25 | 7.29 |
| TP53 | 776 | 50 | 6.44 |
| RET | 48 | 3 | 6.25 |
| MAP2K1 | 19 | 1 | 5.26 |
| EGFR | 164 | 7 | 4.27 |
| ERBB2 | 50 | 2 | 4.00 |
| MET | 57 | 2 | 3.51 |
| PIK3CA | 276 | 9 | 3.26 |
| Publication | Population | Number of Patients | Impact of STK11 Mutation on OS | Conclusion |
|---|---|---|---|---|
| Rivizi J Clin Oncol 2018 | MSK-IMPACT Cohort
| n = 240 received ICI Comparison to a non-ICI group (n = 608) | STK11-mutant tumors were significantly enriched in the no durable benefit group (progressive disease or stable disease shorter than 6 moths), compared to the non-ICI group | Alterations in STK11 were associated with lack of benefit from ICI |
| Skoulidis Cancer Discov 2018 | MDACC cohort
| n = 66 All patients received ICI (no other groups) | STK11 mutations were associated with shorter OS
| Alterations in STK11 were associated with lack of benefit from ICI |
| Papillon-Cavanagh ESMO Open 2020 | Retrospective real-world cohort
| n= 2276 574 (25%) received first-line ICI-based therapies | STK11 mutations were associated with shorter OS
| STK11 confers a poor prognosis, regardless of treatment class with ICI or chemotherapy or targeted therapies |
| Shire Plos One 2020 | Retrospective real-word cohort
| n = 2407 patients 270 (11%) received first-line ICI-based therapies | Median OS was shorter for patients with STK11-mut compared with patients with STK11 WT:
| STK11 confers a poor prognosis, regardless of treatment class with ICI or chemotherapy |
| Shang Lung Cancer 2021 | Selected population from the POPLAR and OAK studies
| n = 598 304 received atezolizumab 294 received docetaxel | Median OS was shorter for patients with STK11-mut compared with patients with STK11 WT:
| STK11 confers a poor prognosis, regardless of treatment class with ICI (atezolizumab) or chemotherapy (docetaxel) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pons-Tostivint, E.; Lugat, A.; Fontenau, J.-F.; Denis, M.G.; Bennouna, J. STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells 2021, 10, 3129. https://doi.org/10.3390/cells10113129
Pons-Tostivint E, Lugat A, Fontenau J-F, Denis MG, Bennouna J. STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells. 2021; 10(11):3129. https://doi.org/10.3390/cells10113129
Chicago/Turabian StylePons-Tostivint, Elvire, Alexandre Lugat, Jean-François Fontenau, Marc Guillaume Denis, and Jaafar Bennouna. 2021. "STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact" Cells 10, no. 11: 3129. https://doi.org/10.3390/cells10113129
APA StylePons-Tostivint, E., Lugat, A., Fontenau, J. -F., Denis, M. G., & Bennouna, J. (2021). STK11/LKB1 Modulation of the Immune Response in Lung Cancer: From Biology to Therapeutic Impact. Cells, 10(11), 3129. https://doi.org/10.3390/cells10113129