
LSD1: Expanding Functions in Stem Cells and Differentiation




Abstract
:1. Introduction
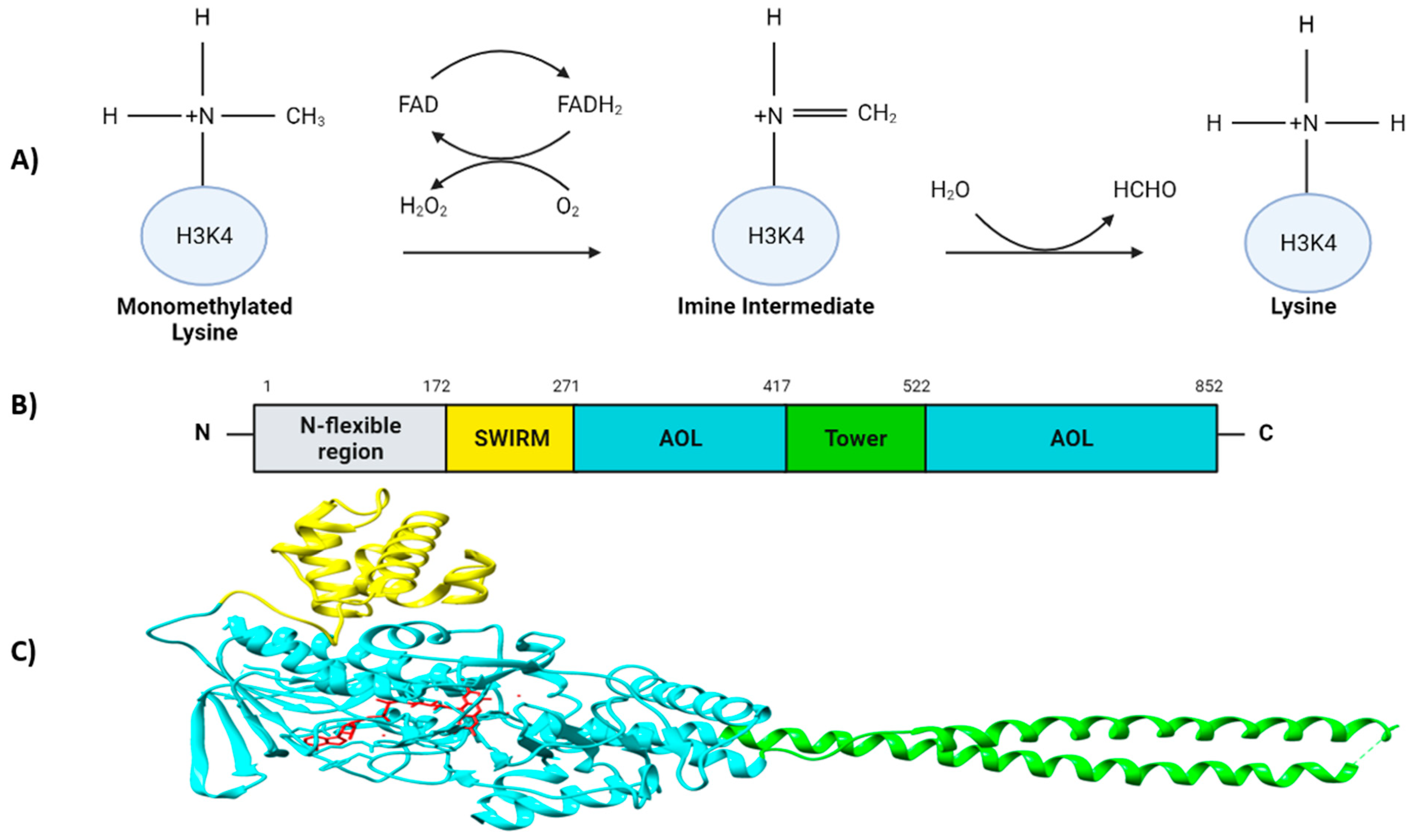
2. Structure and Enzymatic Activity of LSD1
3. Regulation of Gene Expression Mediated by LSD1: Transcriptional Repression and Activation
4. Non-Canonical Targets of LSD1 beyond Demethylation of Histone Lysine Residues
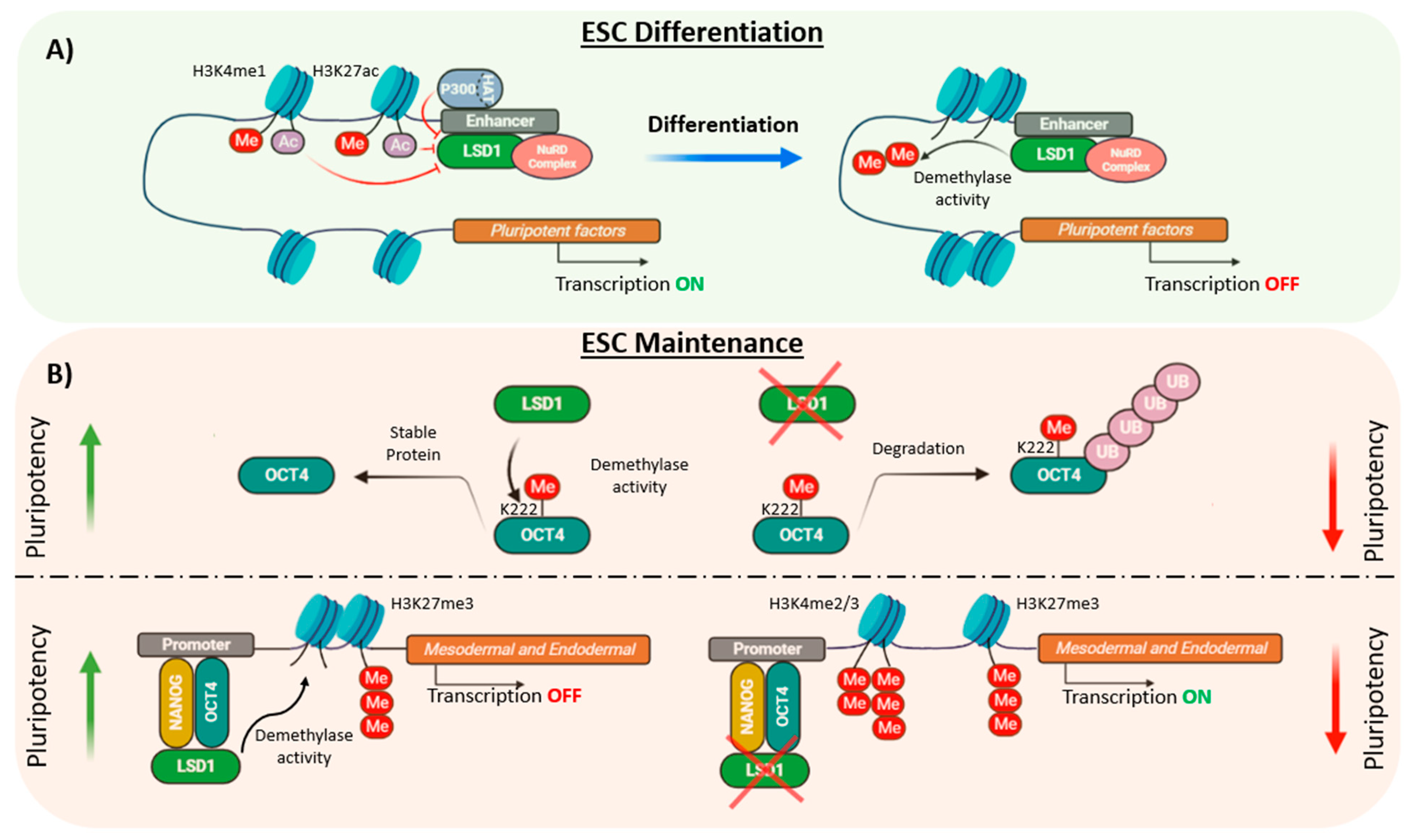
5. LSD1: Self-Renewal or Pluripotency?
6. LSD1 in Somatic Cell Reprogramming
7. Crosstalk with DNA Methylation
8. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Black, J.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Telese, F.; Tan, Y.; Li, W.; Jin, C.; He, X.; Basnet, H.; Ma, Q.; Merkurjev, D.; Zhu, X.; et al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 2015, 18, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.A.; Ziller, M.J.; Gu, H.; Trapnell, C.; Donaghey, J.; Tsankov, A.; Shalek, A.K.; Kelley, D.R.; Shishkin, A.A.; Issner, R.; et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 2013, 153, 1149–1163. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Bilodeau, S.; Orlando, D.A.; Hoke, H.A.; Frampton, G.; Foster, C.T.; Cowley, S.; Young, R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Amente, S.; Lania, L.; Majello, B. The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Acta Bioenerg. 2013, 1829, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulos, P.; Blobel, G.; Hoelz, A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat. Struct. Mol. Biol. 2006, 13, 626–632. [Google Scholar] [CrossRef]
- Huang, J.; Sengupta, R.; Espejo, A.; Lee, M.G.; Dorsey, J.A.; Richter, M.; Opravil, S.; Shiekhattar, R.; Bedford, M.T.; Jenuwein, T.; et al. p53 is regulated by the lysine demethylase LSD1. Nat. Cell Biol. 2007, 449, 105–108. [Google Scholar] [CrossRef]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2008, 41, 125–129. [Google Scholar] [CrossRef]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Battaglioli, E.; Mattevi, A. Human Histone Demethylase LSD1 Reads the Histone Code. J. Biol. Chem. 2005, 280, 41360–41365. [Google Scholar] [CrossRef] [Green Version]
- Forneris, F.; Binda, C.; Battaglioli, E.; Mattevi, A. LSD1: Oxidative chemistry for multifaceted functions in chromatin regulation. Trends Biochem. Sci. 2008, 33, 181–189. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Y.; Wang, F.; Wan, K.; Yamane, K.; Zhang, Y.; Lei, M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1). Proc. Natl. Acad. Sci. USA 2006, 103, 13956–13961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Kim, T.Y.; Kim, M.S.; Park, S.H.; Jang, Y.K. Nuclear import of human histone lysine-specific demethylase LSD1. J. Biochem. 2014, 156, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Da, G.; Lenkart, J.; Zhao, K.; Shiekhattar, R.; Cairns, B.R.; Marmorstein, R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. USA 2006, 103, 2057–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, A.; Takezawa, S.; Schule, R.; Kitagawa, H.; Kato, S. Transrepressive Function of TLX Requires the Histone Demethylase LSD1. Mol. Cell. Biol. 2008, 28, 3995–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tochio, N.; Umehara, T.; Koshiba, S.; Inoue, M.; Yabuki, T.; Aoki, M.; Seki, E.; Watanabe, S.; Tomo, Y.; Hanada, M.; et al. Solution Structure of the SWIRM Domain of Human Histone Demethylase LSD1. Structure 2006, 14, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forneris, F.; Binda, C.; Adamo, A.; Battaglioli, E.; Mattevi, A. Structural Basis of LSD1-CoREST Selectivity in Histone H3 Recognition. J. Biol. Chem. 2007, 282, 20070–20074. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef]
- Kim, S.-A.; Chatterjee, N.; Jennings, M.J.; Bartholomew, B.; Tan, S. Extranucleosomal DNA enhances the activity of the LSD1/CoREST histone demethylase complex. Nucleic Acids Res. 2015, 43, 4868–4880. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Zhu, J.; Yennawar, N.; Eek, P.; Tan, S. Crystal structure of the LSD1/CoREST histone demethylase bound to its nucleosome substrate. Mol. Cell 2020, 78, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Karytinos, A.; Forneris, F.; Profumo, A.; Ciossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A Novel Mammalian Flavin-dependent Histone Demethylase. J. Biol. Chem. 2009, 284, 17775–17782. [Google Scholar] [CrossRef] [Green Version]
- van Essen, D.; Zhu, Y.; Saccani, S. A feed-forward circuit controlling inducible NF-κB target gene activation by promoter histone demethylation. Mol. Cell 2010, 39, 750–760. [Google Scholar] [CrossRef]
- Yang, Z.; Jiang, J.; Stewart, D.M.; Qi, S.; Yamane, K.; Li, J.; Zhang, Y.; Wong, J. AOF1 is a histone H3K4 demethylase possessing demethylase activity-independent repression function. Cell Res. 2010, 20, 276–287. [Google Scholar] [CrossRef]
- Zhang, Q.; Qi, S.; Xu, M.; Yu, L.; Tao, Y.; Deng, Z.; Wu, W.; Li, J.; Chen, Z.; Wong, J. Structure-function analysis reveals a novel mechanism for regulation of histone demethylase LSD2/AOF1/KDM1b. Cell Res. 2013, 23, 225–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nat. Cell Biol. 2007, 446, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Barbera, A.J.; Xu, Y.; Rutenberg, M.; Leonor, T.; Bi, Q.; Lan, F.; Mei, P.; Yuan, G.C.; Lian, C.; et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 2010, 39, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, G.W.; Wang, Y.; Russanova, V.R.; Hirai, T.; Qin, J.; Nakatani, Y.; Howard, B.H. Stable Histone Deacetylase Complexes Distinguished by the Presence of SANT Domain Proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem. 2001, 276, 6817–6824. [Google Scholar] [CrossRef] [Green Version]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, M.-A.; Dong, Y.; Lane, W.S.; Speicher, D.W.; Shiekhattar, R. A Candidate X-linked Mental Retardation Gene Is a Component of a New Family of Histone Deacetylase-containing Complexes. J. Biol. Chem. 2003, 278, 7234–7239. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.K.; Hassig, C.A.; Schnitzler, G.R.; Kingston, R.E.; Schreiber, S.L. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 1998, 395, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Collins, R.; De Cegli, R.; Alpatov, R.; Horton, J.; Shi, X.; Gozani, O.; Cheng, X.; Shi, Y. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 2007, 448, 718–722. [Google Scholar] [CrossRef]
- Reynolds, N.; Latos, P.; Hynes-Allen, A.; Loos, R.; Leaford, D.; O’Shaughnessy, A.; Mosaku, O.; Signolet, J.; Brennecke, P.; Kalkan, T.; et al. NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 2012, 10, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Garcia-Bassets, I.; Kwon, Y.-S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.-G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; et al. Histone Methylation-Dependent Mechanisms Impose Ligand Dependency for Gene Activation by Nuclear Receptors. Cell 2007, 128, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Zibetti, C.; Adamo, A.; Binda, C.; Forneris, F.; Toffolo, E.; Verpelli, C.; Ginelli, E.; Mattevi, A.; Sala, C.; Battaglioli, E. Alternative Splicing of the Histone Demethylase LSD1/KDM1 Contributes to the Modulation of Neurite Morphogenesis in the Mammalian Nervous System. J. Neurosci. 2010, 30, 2521–2532. [Google Scholar] [CrossRef]
- Laurent, B.; Ruitu, L.; Murn, J.; Hempel, K.; Ferrao, R.; Xiang, Y.; Liu, S.; Garcia, B.A.; Wu, H.; Wu, F.; et al. A specific LSD1/KDM1A isoform regulates neuronal differentiation through H3K9 demethylation. Mol. Cell 2015, 57, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Toffolo, E.; Rusconi, F.; Paganini, L.; Tortorici, M.; Pilotto, S.; Heise, C.; Verpelli, C.; Tedeschi, G.; Maffioli, E.; Sala, C.; et al. Phosphorylation of neuronal Lysine-Specific Demethylase 1LSD1/KDM1A impairs transcriptional repression by regulating interaction with CoREST and histone deacetylases HDAC1/2. J. Neurochem. 2014, 128, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Wang, Y.; Chen, J.; Li, H.; Kang, L.; Zhang, Y.; Chen, S.; Zhu, B.; Gao, S. RCOR2 Is a Subunit of the LSD1 Complex That Regulates ESC Property and Substitutes for SOX2 in Reprogramming Somatic Cells to Pluripotency. Stem Cells 2011, 29, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Knaupp, A.S.; Mohenska, M.; Larcombe, M.R.; Ford, E.; Lim, S.M.; Wong, K.; Chen, J.; Firas, J.; Huang, C.; Liu, X.; et al. TINC—A Method to Dissect Regulatory Complexes at Single-Locus Resolution—Reveals an Extensive Protein Complex at the Nanog Promoter. Stem Cell Rep. 2020, 15, 1246–1259. [Google Scholar] [CrossRef]
- Jamaladdin, S.; Kelly, R.D.W.; O’Regan, L.; Dovey, O.M.; Hodson, G.E.; Millard, C.; Portolano, N.; Fry, A.M.; Schwabe, J.; Cowley, S.M. Histone deacetylase (HDAC) 1 and 2 are essential for accurate cell division and the pluripotency of embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9840–9845. [Google Scholar] [CrossRef] [Green Version]
- Aguilo, F.; Zhang, F.; Sancho, A.; Fidalgo, M.; Di Cecilia, S.; Vashisht, A.; Lee, D.-F.; Chen, C.-H.; Rengasamy, M.; Andino, B.; et al. Coordination of m 6 A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell 2015, 17, 689–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, Y.; Cui, Y.; Li, Y.; Zhang, M.; Wang, X.; Ji, J.; Zhang, X.; Zhou, M.; Zhang, Z.; Ye, S.-D.; et al. Inhibition of MTA2 and MTA3 induces mesendoderm specification of human embryonic stem cells. Biochem. Biophys. Res. Commun. 2021, 552, 142–149. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.; Wederell, E.; Robertson, G.; Delaney, A.; Morozova, O.; Poon, S.S.; Yap, D.; Fee, J.; Zhao, Y.; McDonald, H.; et al. Retinoblastoma-binding proteins 4 and 9 are important for human pluripotent stem cell maintenance. Exp. Hematol. 2011, 39, 866–879.e1. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Su, T.; Wang, C.; Dong, L.; Liu, S.; Zhu, Y.; Hao, K.; Xia, Y.; Jiang, Q.; Qin, J. Rbbp4 Suppresses Premature Differentiation of Embryonic Stem Cells. Stem Cell Rep. 2021, 16, 566–581. [Google Scholar] [CrossRef]
- Lim, S.; Shparberg, R.A.; Coorssen, J.R.; O’Connor, M.D. Application of the RBBP9 Serine Hydrolase Inhibitor, ML114, Decouples Human Pluripotent Stem Cell Proliferation and Differentiation. Int. J. Mol. Sci. 2020, 21, 8983. [Google Scholar] [CrossRef]
- Lu, Y.; Loh, Y.-H.; Li, H.; Cesana, M.; Ficarro, S.B.; Parikh, J.R.; Salomonis, N.; Toh, C.-X.D.; Andreadis, S.T.; Luckey, C.J.; et al. Alternative Splicing of MBD2 Supports Self-Renewal in Human Pluripotent Stem Cells. Cell Stem Cell 2014, 15, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Hainer, S.; McCannell, K.N.; Yu, J.; Ee, L.-S.; Zhu, L.; Rando, O.J.; Fazzio, T.G. DNA methylation directs genomic localization of Mbd2 and Mbd3 in embryonic stem cells. eLife 2016, 5, e21964. [Google Scholar] [CrossRef] [Green Version]
- Kaji, K.; Caballero, I.M.; MacLeod, R.; Nichols, J.; Wilson, V.A.; Hendrich, B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nature 2006, 8, 285–292. [Google Scholar] [CrossRef]
- Luo, M.; Ling, T.; Xie, W.; Sun, H.; Zhou, Y.; Zhu, Q.; Shen, M.; Zong, L.; Lyu, G.; Zhao, Y.; et al. NuRD Blocks Reprogramming of Mouse Somatic Cells into Pluripotent Stem Cells. Stem Cells 2013, 31, 1278–1286. [Google Scholar] [CrossRef]
- Rais, Y.; Zviran, A.; Geula, S.; Gafni, O.; Chomsky, E.; Viukov, S.; Mansour, A.A.; Caspi, I.; Krupalnik, V.; Zerbib, M.; et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature 2013, 502, 65–70. [Google Scholar] [CrossRef]
- Dos Santos, R.L.; Tosti, L.; Radzisheuskaya, A.; Caballero, I.M.; Kaji, K.; Hendrich, B.; Silva, J.C. MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 2014, 15, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD Complex Regulates Expression of 5-Hydroxymethylcytosine Marked Genes in Embryonic Stem Cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Han, Z.; Liu, X.; Gu, J.; Tang, F.; Wei, G.; Jin, Y. The chromatin remodeler Chd4 maintains embryonic stem cell identity by controlling pluripotency- and differentiation-associated genes. J. Biol. Chem. 2017, 292, 8507–8519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezmi, E.; Weissbein, U.; Golan-Lev, T.; Nissim-Rafinia, M.; Meshorer, E.; Benvenisty, N. The Chromatin Regulator ZMYM2 Restricts Human Pluripotent Stem Cell Growth and Is Essential for Teratoma Formation. Stem Cell Rep. 2020, 15, 1275–1286. [Google Scholar] [CrossRef]
- Yamamoto, M.; Suwa, Y.; Sugiyama, K.; Okashita, N.; Kawaguchi, M.; Tani, N.; Matsubara, K.; Nakamura, A.; Seki, Y. The PRDM14–CtBP1/2–PRC2 complex regulates transcriptional repression during the transition from primed to naïve pluripotency. J. Cell Sci. 2020, 133, jcs240176. [Google Scholar] [CrossRef]
- Zhang, H.; Gayen, S.; Xiong, J.; Zhou, B.; Shanmugam, A.K.; Sun, Y.; Karatas, H.; Liu, L.; Rao, R.; Wang, S.; et al. MLL1 Inhibition Reprograms Epiblast Stem Cells to Naive Pluripotency. Cell Stem Cell 2016, 18, 481–494. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Li, X.-Y.; Willis, A.L.; Liu, C.; Chen, G.; Weiss, S.J. Snail1-dependent control of embryonic stem cell pluripotency and lineage commitment. Nat. Commun. 2014, 5, 3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unternaehrer, J.J.; Zhao, R.; Kim, K.; Cesana, M.; Powers, J.T.; Ratanasirintrawoot, S.; Onder, T.; Shibue, T.; Weinberg, R.A.; Daley, G.Q. The Epithelial-Mesenchymal Transition Factor SNAIL Paradoxically Enhances Reprogramming. Stem Cell Rep. 2014, 3, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Lin, Y.; Wang, Z.; Wu, X.; Ye, Z.; Wang, Y.; Lan, H. Biological roles of LSD1 beyond its demethylase activity. Cell. Mol. Life Sci. 2020, 77, 3341–3350. [Google Scholar] [CrossRef] [PubMed]
- Dan, S.; Song, Y.; Duan, X.; Pan, X.; Chen, C.; She, S.; Su, T.; Li, J.; Chen, X.; Zhou, Y.; et al. LSD1-mediated demethylation of OCT4 safeguards pluripotent stem cells by maintaining the transcription of PORE-motif-containing genes. Sci. Rep. 2021, 11, 10285. [Google Scholar] [CrossRef] [PubMed]
- Kontaki, H.; Talianidis, I. Lysine Methylation Regulates E2F1-Induced Cell Death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Hallstrom, T.C.; Mori, S.; Nevins, J.R. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 2008, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Karnik, R.; Gu, H.; Ziller, M.J.; Clement, K.; Tsankov, A.M.; Akopian, V.; Gifford, C.A.; Donaghey, J.; Galonska, C.; et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat. Genet. 2015, 47, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nature 2004, 7, 165–171. [Google Scholar] [CrossRef]
- Choi, J.; Jang, H.; Kim, H.; Lee, J.-H.; Kim, S.-T.; Cho, E.-J.; Youn, H.-D. Modulation of lysine methylation in myocyte enhancer factor 2 during skeletal muscle cell differentiation. Nucleic Acids Res. 2013, 42, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tanaka, K.; Yan, J.; Li, J.; Peng, D.; Jiang, Y.; Yang, Z.; Barton, M.C.; Wen, H.; Shi, X. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc. Natl. Acad. Sci. USA 2013, 110, 17284–17289. [Google Scholar] [CrossRef] [Green Version]
- Bradley, E.; Bieberich, E.; Mivechi, N.F.; Tangpisuthipongsa, D.; Wang, G. Regulation of Embryonic Stem Cell Pluripotency by Heat Shock Protein 90. Stem Cells 2012, 30, 1624–1633. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Lanouette, S.; Elisma, F.; Tremblay, V.; Butson, J.; Figeys, D.; Couture, J.-F. Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J. Mol. Cell Biol. 2011, 3, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.H.; et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018, 174, 549–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Chen, X.; Novak, M.K.; Zhang, S.; Hu, W. Repressing Ago2 mRNA translation by Trim71 maintains pluripotency through inhibiting let 7-microRNAs. Elife 2021, 10, e66288. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Kim, K.I. Regulation of HIF-1α stability by lysine methylation. BMB Rep. 2016, 49, 245–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, P.; Zhang, P.; Zhang, Y.; Sun, L.; Cui, G.; Guo, X.; Wang, H.; Zhang, X.; Shi, Y.; Yu, Z. HIF-1α/Actl6a/H3K9ac axis is critical for pluripotency and lineage differentiation of human induced pluripotent stem cells. FASEB J. 2020, 34, 5740–5753. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Li, D.-Q.; Kumar, R. A Core Chromatin Remodeling Factor Instructs Global Chromatin Signaling through Multivalent Reading of Nucleosome Codes. Mol. Cell 2013, 49, 704–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Wan, M.; Zhang, Y.; Gu, P.; Xin, H.; Jung, S.Y.; Qin, J.; Wong, J.; Cooney, A.J.; Liu, D.; et al. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nature 2008, 10, 731–739. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, P.; McLean, C.; Sheppard, A.; Rivett, D.; Jones, K.; Dalton, S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 2005, 132, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, Y.; Jiang, Z.; Ma, Y.; Lin, C.-J.; Kim, C.; Carter, M.G.; Amano, T.; Park, J.; Kish, S.; et al. Jak/Stat3 Signaling Promotes Somatic Cell Reprogramming by Epigenetic Regulation. Stem Cells 2012, 30, 2645–2656. [Google Scholar] [CrossRef]
- Cho, H.-S.; Suzuki, T.; Dohmae, N.; Hayami, S.; Unoki, M.; Yoshimatsu, M.; Toyokawa, G.; Takawa, M.; Chen, T.; Kurash, J.K.; et al. Demethylation of RB Regulator MYPT1 by Histone Demethylase LSD1 Promotes Cell Cycle Progression in Cancer Cells. Cancer Res. 2010, 71, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Loh, Y.-H.; Wu, Q.; Chew, J.-L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J.; et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006, 38, 431–440. [Google Scholar] [CrossRef]
- AlAbdi, L.; Saha, D.; He, M.; Dar, M.S.; Utturkar, S.M.; Sudyanti, P.A.; McCune, S.; Spears, B.H.; Breedlove, J.A.; Lanman, N.A.; et al. Oct4-Mediated Inhibition of Lsd1 Activity Promotes the Active and Primed State of Pluripotency Enhancers. Cell Rep. 2020, 30, 1478–1490.e6. [Google Scholar] [CrossRef]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Shi, G.; Jin, Y. Role of Oct4 in maintaining and regaining stem cell pluripotency. Stem Cell Res. Ther. 2010, 1, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-Y.; Tanaka, Y.; Su, J.; Cakir, B.; Xiang, Y.; Patterson, B.; Ding, J.; Jung, Y.-W.; Kim, J.-H.; Hysolli, E.; et al. Uhrf1 regulates active transcriptional marks at bivalent domains in pluripotent stem cells through Setd1a. Nat. Commun. 2018, 9, 2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Gao, Q.; Tan, S.; You, J.; Lyu, C.; Zhang, Y.; Han, M.; Chen, Z.; Li, J.; Wang, H.; et al. SET8 prevents excessive DNA methylation by methylation-mediated degradation of UHRF1 and DNMT1. Nucleic Acids Res. 2019, 47, 9053–9068. [Google Scholar] [CrossRef] [PubMed]
- Hahm, J.Y.; Kim, J.-Y.; Park, J.W.; Kang, J.-Y.; Kim, K.-B.; Kim, S.-R.; Cho, H.; Seo, S.-B. Methylation of UHRF1 by SET7 is essential for DNA double-strand break repair. Nucleic Acids Res. 2018, 47, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.J.; Boo, K.; Kim, D.; Han, D.-H.; Choe, H.K.; Kim, C.R.; Sun, W.; Kim, H.; Kim, K.; Lee, H.; et al. Phosphorylation of LSD1 by PKCα Is Crucial for Circadian Rhythmicity and Phase Resetting. Mol. Cell 2014, 53, 791–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinyard, M.E.; Su, C.; Siegenfeld, A.P.; Waterbury, A.L.; Freedy, A.M.; Gosavi, P.M.; Park, Y.; Kwan, E.E.; Senzer, B.D.; Doench, J.G.; et al. CRISPR-suppressor scanning reveals a nonenzymatic role of LSD1 in AML. Nat. Chem. Biol. 2019, 15, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Tan, M.; Zhang, Q.; Yang, F.; Wang, S.; Li, H.; Xiong, X.; Sun, Y. LSD1 destabilizes FBXW7 and abrogates FBXW7 functions independent of its demethylase activity. Proc. Natl. Acad. Sci. USA 2019, 116, 12311–12320. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.; Lin, C.-Y.; Chao, A.-N.; Tsai, C.-L.; Chen, M.-Y.; Lee, L.-Y.; Chang, T.-C.; Wang, T.-H.; Lai, C.-H.; Wang, H.-S. Lysine-specific demethylase 1 (LSD1) destabilizes p62 and inhibits autophagy in gynecologic malignancies. Oncotarget 2017, 8, 74434–74450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnesecchi, J.; Cerutti, C.; Vanacker, J.-M.; Forcet, C. ERRα protein is stabilized by LSD1 in a demethylation-independent manner. PLoS ONE 2017, 12, e0188871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaoki, J.; Takeuchi, M.; Abe, R.; Kaneko, H.; Wada, T.; Hino, S.; Nakao, M.; Furukawa, Y.; Kobayashi, M. Splicing- and demethylase-independent functions of LSD1 in zebrafish primitive hematopoiesis. Sci. Rep. 2020, 10, 8521. [Google Scholar] [CrossRef] [PubMed]
- Macfarlan, T.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B.; et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, C.T.; Dovey, O.M.; Lezina, L.; Luo, J.L.; Gant, T.; Barlev, N.; Bradley, A.; Cowley, S.M. Lysine-Specific Demethylase 1 Regulates the Embryonic Transcriptome and CoREST Stability. Mol. Cell. Biol. 2010, 30, 4851–4863. [Google Scholar] [CrossRef] [Green Version]
- Adamo, A.; Sesé, B.; Boue, S.; Castaño, J.; Paramonov, I.; Barrero, M.; Belmonte, J.C.I. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nature 2011, 13, 652–659. [Google Scholar] [CrossRef]
- Plusa, B.; Hadjantonakis, A.-K. Embryonic stem cell identity grounded in the embryo. Nature 2014, 16, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Daley, G.Q. From fibroblasts to iPS cells: Induced pluripotency by defined factors. J. Cell. Biochem. 2008, 105, 949–955. [Google Scholar] [CrossRef]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Xu, X.; Li, J.; Liu, J.; Gu, H.; Zhang, R.; Chen, J.; Kuang, Y.; Fei, J.; Jiang, C.; et al. Lithium, an anti-psychotic drug, greatly enhances the generation of induced pluripotent stem cells. Cell Res. 2011, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zhang, Q.; Yin, X.; Yang, W.; Du, Y.; Hou, P.; Ge, J.; Liu, C.; Zhang, W.; Zhang, X.; et al. Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 2010, 21, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent Stem Cells Induced from Mouse Somatic Cells by Small-Molecule Compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Cacchiarelli, D.; Trapnell, C.; Ziller, M.J.; Soumillon, M.; Cesana, M.; Karnik, R.; Donaghey, J.; Smith, Z.D.; Ratanasirintrawoot, S.; Zhang, X.; et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell 2015, 162, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Neelamegam, R.; Ricq, E.L.; Malvaez, M.; Patnaik, D.; Norton, S.; Carlin, S.M.; Hill, I.T.; Wood, M.A.; Haggarty, S.J.; Hooker, J.M. Brain-Penetrant LSD1 Inhibitors Can Block Memory Consolidation. ACS Chem. Neurosci. 2011, 3, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xia, P.; Ye, B.; Huang, G.; Liu, J.; Fan, Z. Transient Activation of Autophagy via Sox2-Mediated Suppression of mTOR Is an Important Early Step in Reprogramming to Pluripotency. Cell Stem Cell 2013, 13, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Liang, L.; Li, Y.; Feng, C.; Li, L.; Zhang, Y.; He, S.; Pei, D.; Guo, Y.; Zheng, H. Lysine-specific histone demethylase 1 inhibition promotes reprogramming by facilitating the expression of exogenous transcriptional factors and metabolic switch. Sci. Rep. 2016, 6, 30903. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic Oxidative Bioenergetics Transitions into Pluripotency-Dependent Glycolysis to Facilitate Nuclear Reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Leitch, H.G.; McEwen, K.R.; Turp, A.; Encheva, V.; Carroll, T.; Grabole, N.; Mansfield, W.; Nashun, B.; Knezovich, J.G.; Smith, A.; et al. Naive pluripotency is associated with global DNA hypomethylation. Nat. Struct. Mol. Biol. 2013, 20, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Li, E. Structure and Function of Eukaryotic DNA Methyltransferases. Curr. Top. Dev. Biol. 2004, 60, 55–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ueda, Y.; Dodge, J.E.; Wang, Z.; Li, E. Establishment and Maintenance of Genomic Methylation Patterns in Mouse Embryonic Stem Cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 2003, 23, 5594–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsumura, A.; Hayakawa, T.; Kumaki, Y.; Takebayashi, S.-I.; Sakaue, M.; Matsuoka, C.; Shimotohno, K.; Ishikawa, F.; Li, E.; Ueda, H.R.; et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells 2006, 11, 805–814. [Google Scholar] [CrossRef]
- Bostick, M.; Kim, J.K.; Estève, P.O.; Clark, A.; Pradhan, S.; Jacobsen, S.E. UHRF1 Plays a Role in Maintaining DNA Methylation in Mammalian Cells. Science 2007, 317, 1760–1764. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.-I.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Karagianni, P.; Amazit, L.; Qin, J.; Wong, J. ICBP90, a Novel Methyl K9 H3 Binding Protein Linking Protein Ubiquitination with Heterochromatin Formation. Mol. Cell. Biol. 2008, 28, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Tsai, H.-C.; Yen, R.-W.C.; Zhang, Y.W.; Kong, X.; Wang, W.; Xia, L.; Baylin, S.B. Critical threshold levels of DNA methyltransferase 1 are required to maintain DNA methylation across the genome in human cancer cells. Genome Res. 2017, 27, 533–544. [Google Scholar] [CrossRef]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S.-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Petell, C.J.; Alabdi, L.; He, M.; San Miguel, P.; Rose, R.; Gowher, H. An epigenetic switch regulates de novo DNA methylation at a subset of pluripotency gene enhancers during embryonic stem cell differentiation. Nucleic Acids Res. 2016, 44, 7605–7617. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interactor | Function in ESCs | References |
|---|---|---|
| RCOR2 | RCOR2 regulates pluripotency via suppressing lineage-specific genes and the reprogramming of somatic cells to iPSCs. | [41,42] |
| HDAC1 | HDAC1/2 induce the transcriptional program of self-renewal-associated genes such as Oct4, Nanog, Esrrb, and Rex1, thereby regulates the pluripotency of ESCs. | [43] |
| ZNF217 | ZNF217 has a critical role in ESC self-renewal by restricting the METTL3 methyltransferase activity. | [44] |
| MTA | MTA2 and MTA3, but not MTA1, preserve human ESCs from differentiating into the mesodermal lineage. | [45] |
| RBBP4 and 9 | RBBP4 and 9 regulate ESC self-renewal by sustaining the transcription of core pluripotency factors and inhibiting the genes involved in organogenesis. | [46,47,48] |
| MBD2 | Two isoforms of MBD2, MBD2a and MBD2c, with contrasting roles: MBD2a enhances ESC differentiation through recruitment of the NuRD complex while MBD2c facilitates reprogramming. Mbd2/NuRD is also essential to maintain normal chromatin structure and gene regulation in ESCs. | [49,50] |
| MBD3 | MBD3 is a scaffolding protein essential for NuRD complex assembly. MBD3/NuRD hinders the expression of pluripotency and preimplantation transcripts allowing cells to exit self-renewal for proper lineage-commitment. It is important to maintain normal chromatin structure and gene regulation in ESCs. Conflictive data in enhancing and suppressing reprogramming. | [35,50,51,52,53,54,55] |
| CHD4 | CHD4 suppresses the aberrant expression of Tbx3, which mainly impairs endoderm differentiation. | [56] |
| ZMYM2 | ZMYM2 plays a central role in transcriptional regulation of ESCs. It represses the expression of NANOG and OCT4 during early differentiation allowing ESCs to exit from the pluripotency state. | [57] |
| CTBP1 | CTBP1/2 is a core regulator of PRDM14-mediated transcriptional repression which is a prerequisite for transition from primed to the naïve state. | [58] |
| MLL1 | MLL1-mediated H3K4me1 deposition at enhancers regulates cell-fate determination and its blockage reinforces naïve reprogramming. | [59] |
| Snail1 | Snail1 is dispensable for ESC self-renewal, however, it steers EpiSC exit and modulates neuroectodermal, endodermal and mesodermal specification. It also enhances reprogramming. | [60,61] |
| Substrate | K Position | Effect | Role in ESCs | References |
|---|---|---|---|---|
| E2F1 | 185 | Stabilization of E2F1 and activation of proapoptotic genes. | N/A | [64,65] |
| DNMT1 | 1096 and 142 | Removal of the methyl group from K1096 (mouse), K1094 (human), and K142 of DNMT1 increases stability. K142 demethylation in the S-phase promotes stability by restricting L3MBTL3-CRL4DCAF5-mediated proteolysis. | DNMT1 is essential for ESCs cell viability and surveillance by controlling DNA methylation. | [10,66] |
| p53 | 370 | Inhibition of the transcriptional activity of p53. | Upon DNA damage, activated p53 represses the core ESC transcriptome and induces the expression of lineage-specific markers. p53 is a transcriptional regulator which suppresses Nanog expression during ESCs differentiation. | [9,67] |
| MEF2D | 267 | Enhances its transcriptional activity. | Promotes myogenic differentiation. | [68] |
| ERa | 266 | Demethylation of K266 allows subsequent acetylation leading to activating of ERα target genes. | N/A | [69] |
| HSP90 | 615 | It promotes HSP90 degradation. | It regulates pluripotency by: (i) regulating OCT4, NANOG and pSTAT3 expression and prevention of proteasomal-mediated degradation of OCT4 and NANOG; (ii) modulating Oct4 mRNA, particularly restraining ESC from mesoderm differentiation. | [70,71] |
| AGO2 | 726 | Stabilization | Its expression promotes an accelerated differentiation by increasing let-7 microRNAs which inhibits Trim71 translation. | [72,73] |
| HIF-1a | 391 | Demethylation of HIF1α at K391 prevents proteasomal-mediated degradation and PHD2-induced hydroxylation, thereby enhancing transcriptional activity of HIF1α to facilitate VEGF expression. | Activated HIF1α enhances the glycolytic program leading to efficient reprogramming. It also sustains self-renewal of iPSCs through regulating Actl6a and acetylation. Inhibition of HIF1α promotes endoderm and mesoderm differentiation. | [74,75] |
| MTA1 | 532 | K532 demethylation disorganizes the formation of the NuRD repressor complex. Unmethylated MTA1 promotes acetylation of demethylated histone H3K9 shifting gene repression to activation. | MTA1 forms a complex with NANOG and POU5F1 known as a NODE. MTA1 deficiency upregulates the expression of endoderm-associated markers. | [76,77] |
| STAT3 | 140 | K140 demethylation enhances transcriptional activity in response to IL-6. | STAT 3 controls Myc expression, promoting self-renewal and pluripotency in ESCs. Its activation is essential for the reprogramming of terminally differentiated cells. | [78,79,80] |
| MYPT1 | 442 | K442 demethylation destabilizes MYPT1 and increases RB1 phosphorylation leading to cell cycle progression. | N/A | [81] |
| OCT4 | 222 | Prevents proteasome independent degradation and refrains the ‘locked-in’ mode binding of OCT4 homodimers which enhances the expression of target genes. | OCT4 is a core pluripotency factor. | [82,83,84,85] |
| UHRF1 | 385 | K385 demethylation stabilizes UHRF1. | It associates with Setd1a/COMPASS complex to maintain mesoderm and neuroectoderm histone marks, ensuring a proper differentiation in stem cells. In association with the Setd1a/COMPASS complex, UHRF1 aids in the regulation of H3K4me3 and H3K27me3 methylation. The maintenance of bivalent histone marks ensures efficient mesoderm and ectoderm differentiation. | [86,87,88] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Gamero, C.; Malla, S.; Aguilo, F. LSD1: Expanding Functions in Stem Cells and Differentiation. Cells 2021, 10, 3252. https://doi.org/10.3390/cells10113252
Martinez-Gamero C, Malla S, Aguilo F. LSD1: Expanding Functions in Stem Cells and Differentiation. Cells. 2021; 10(11):3252. https://doi.org/10.3390/cells10113252
Chicago/Turabian StyleMartinez-Gamero, Carlos, Sandhya Malla, and Francesca Aguilo. 2021. "LSD1: Expanding Functions in Stem Cells and Differentiation" Cells 10, no. 11: 3252. https://doi.org/10.3390/cells10113252
APA StyleMartinez-Gamero, C., Malla, S., & Aguilo, F. (2021). LSD1: Expanding Functions in Stem Cells and Differentiation. Cells, 10(11), 3252. https://doi.org/10.3390/cells10113252