
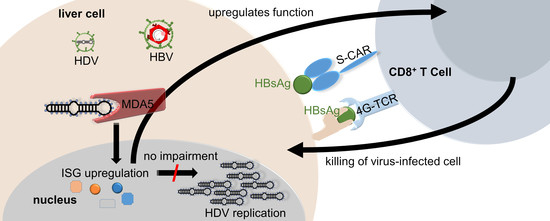
Hepatitis-D Virus Infection Is Not Impaired by Innate Immunity but Increases Cytotoxic T-Cell Activity

 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies, Reagents and Kits
2.2. AAV-HDV Production
2.3. Cells
2.4. CRISPR/Cas9 Mediated KO Generation
2.5. HBV and HDV Infection
2.6. HDV Production
2.7. qPCR for HDV Detection
2.8. qPCR for Gene Expression Analysis
2.9. HDAg Immunofluorescence Staining and Analysis
2.10. Realtime Cell Viability Assay with xCELLigence RTCA
3. Results
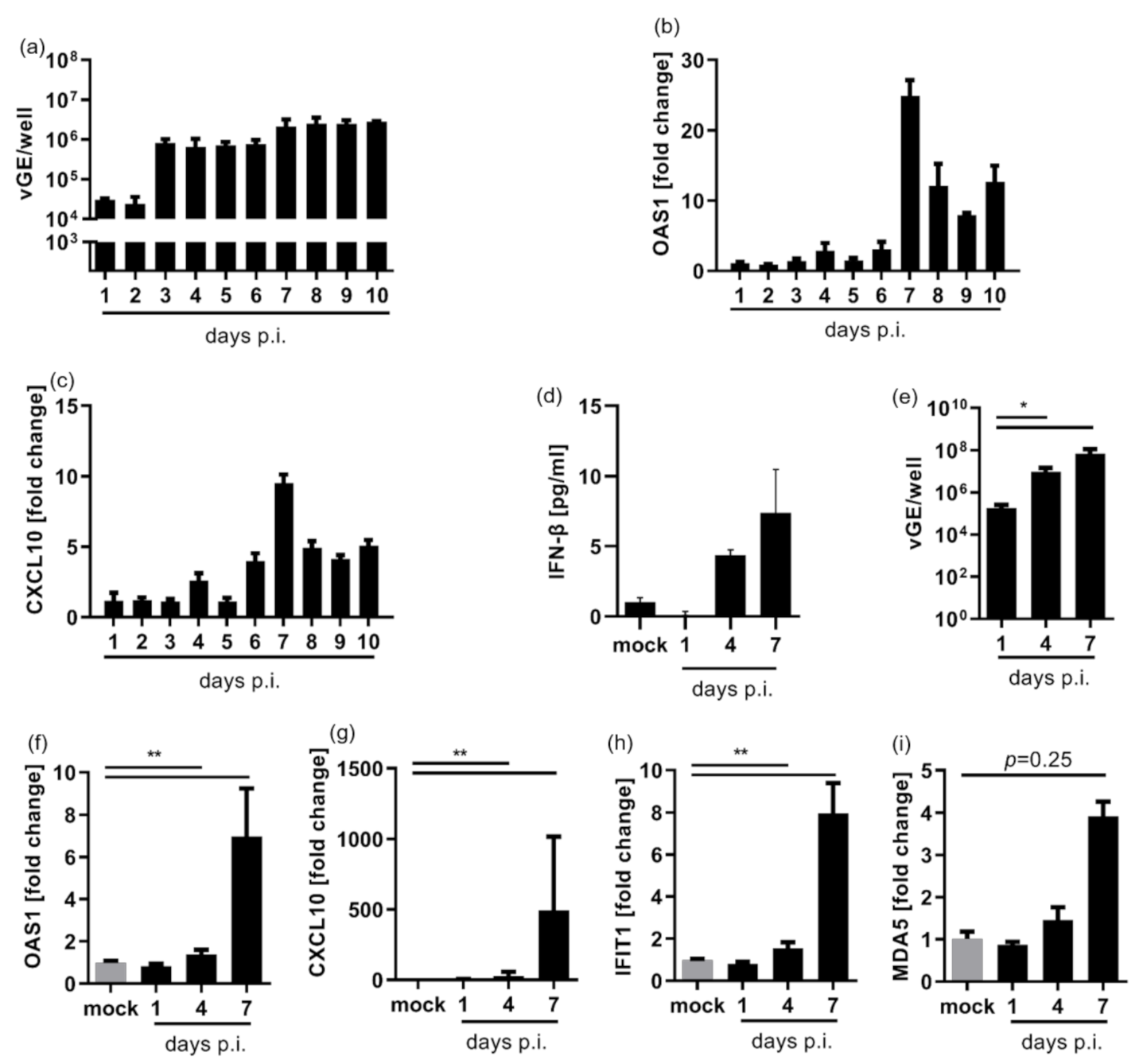
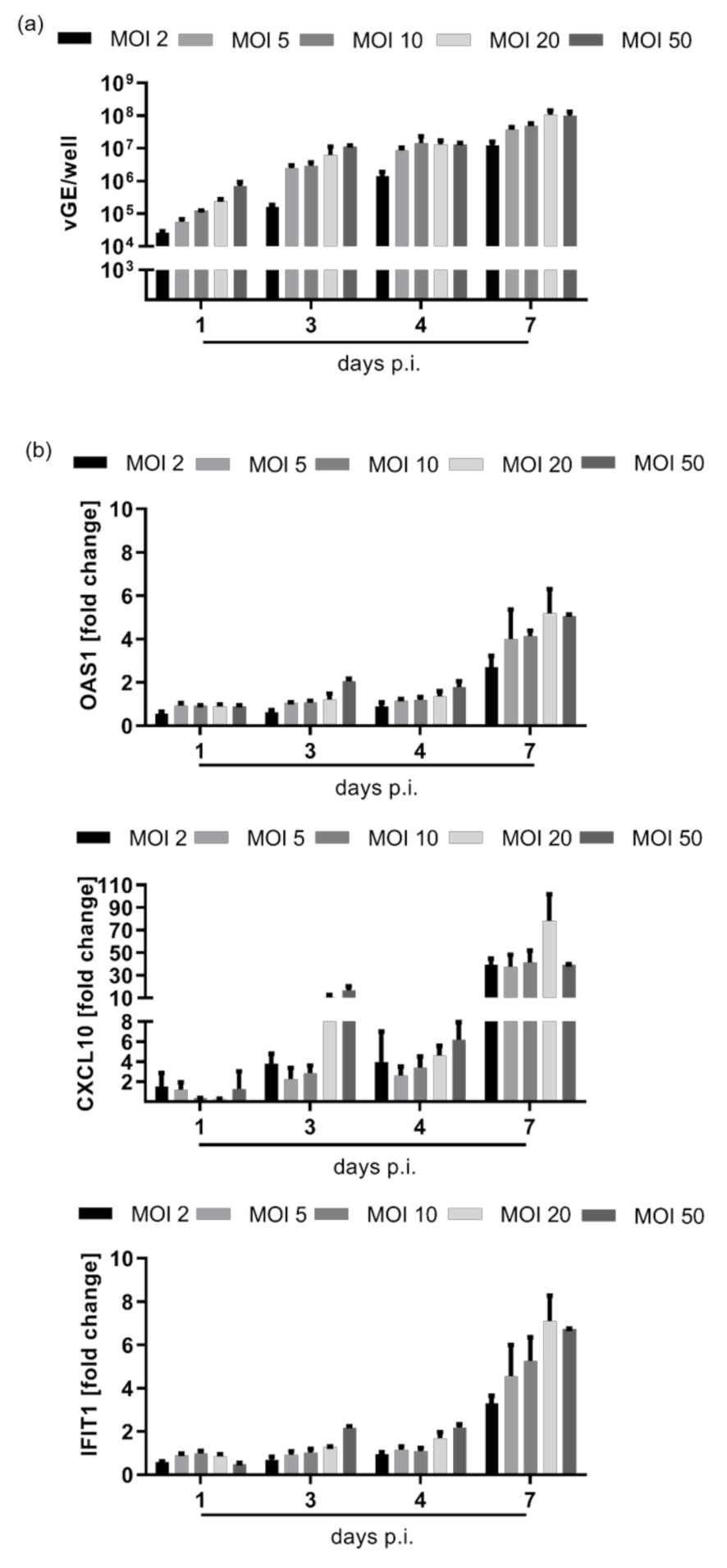
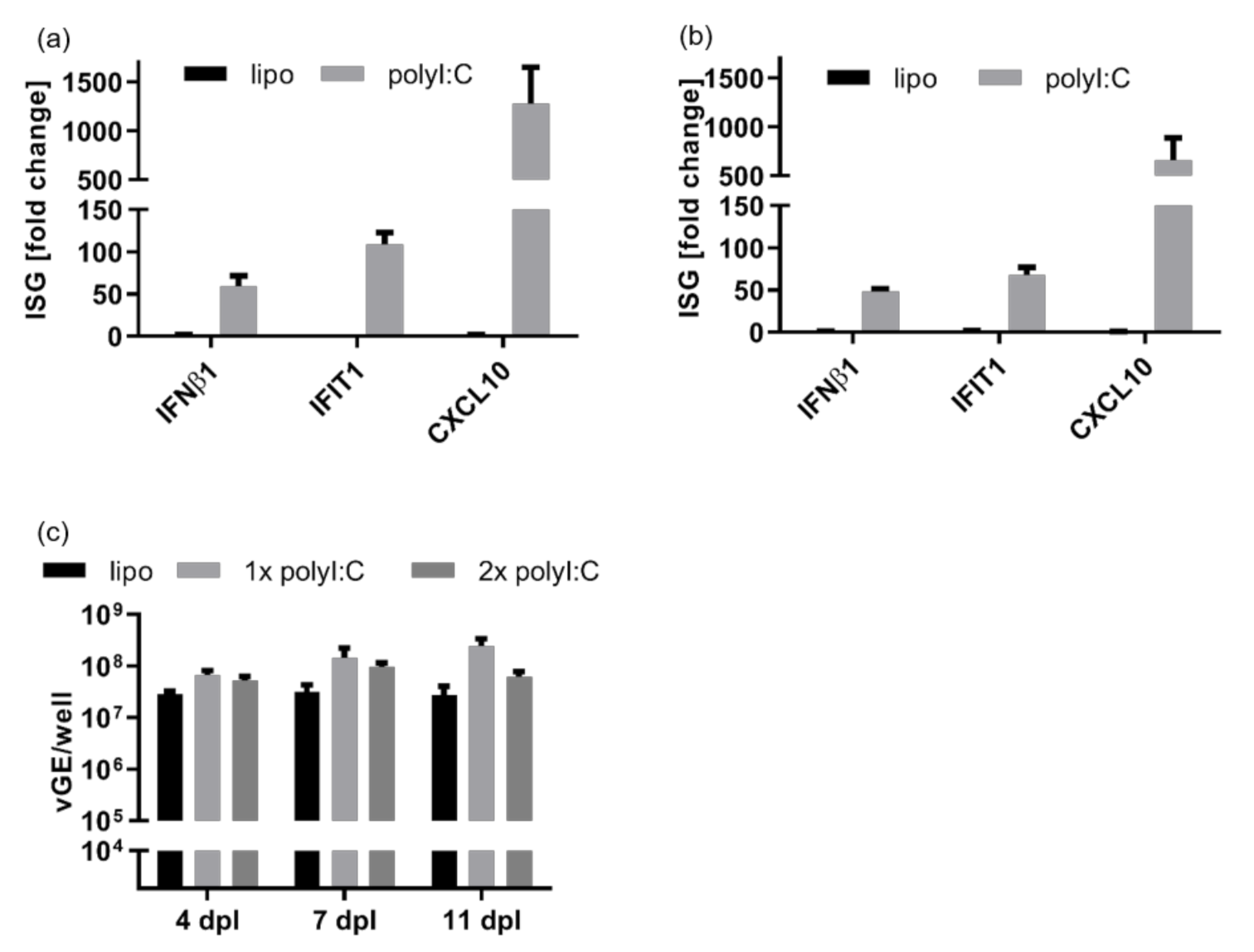
3.1. HDV Infection Induces a Delayed Type I Interferon Response
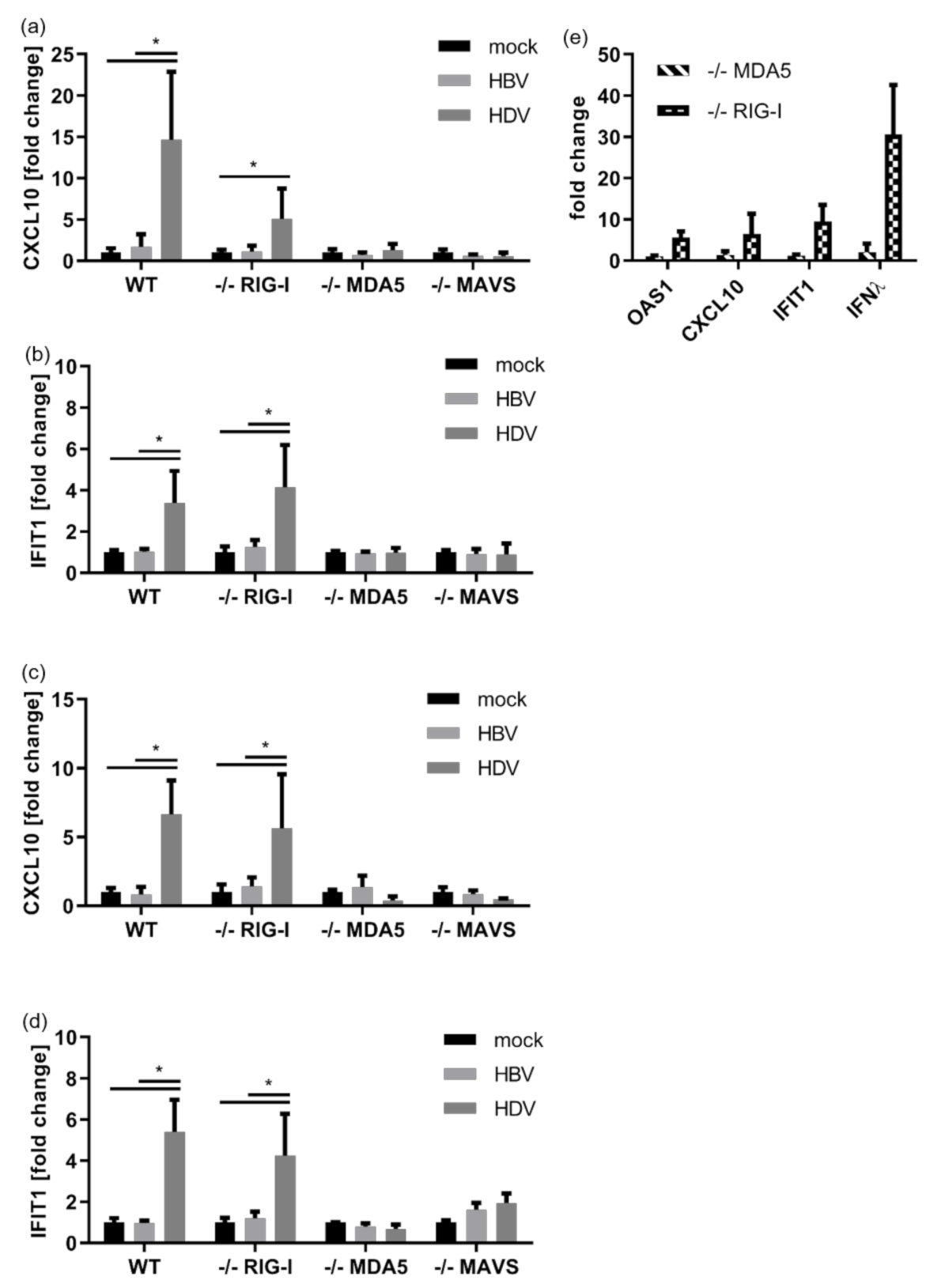
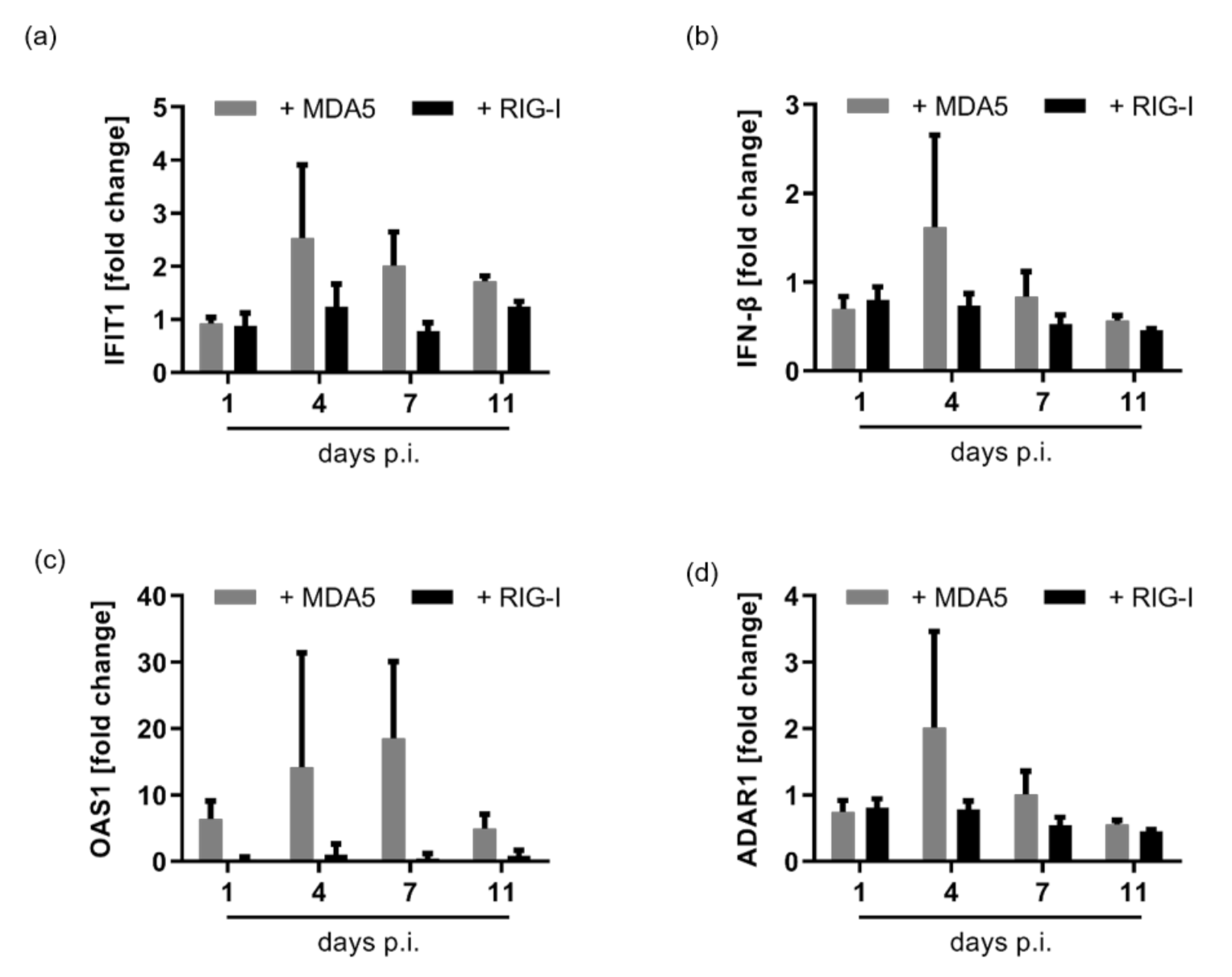
3.2. HDV Is Detected by MDA5
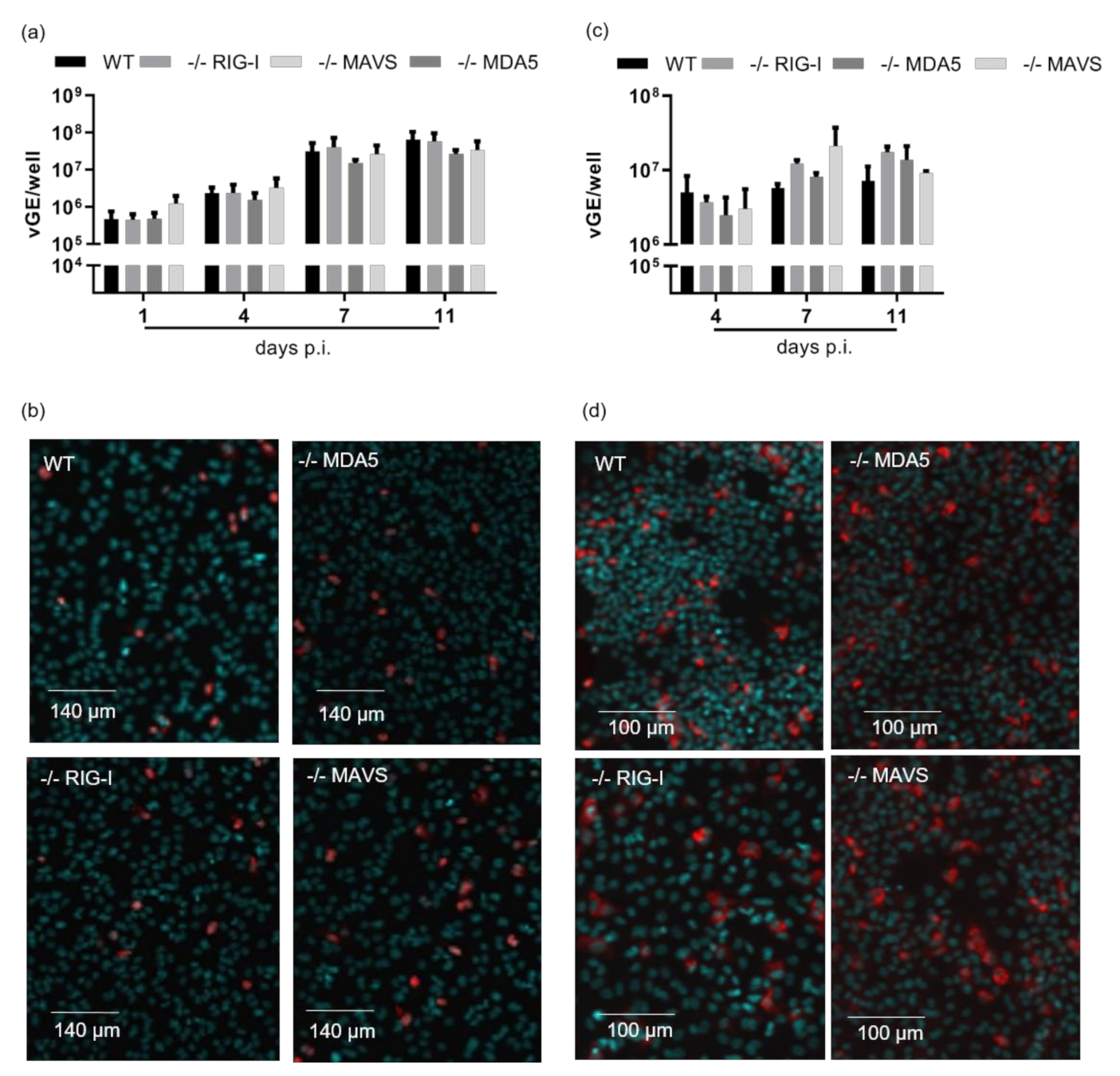
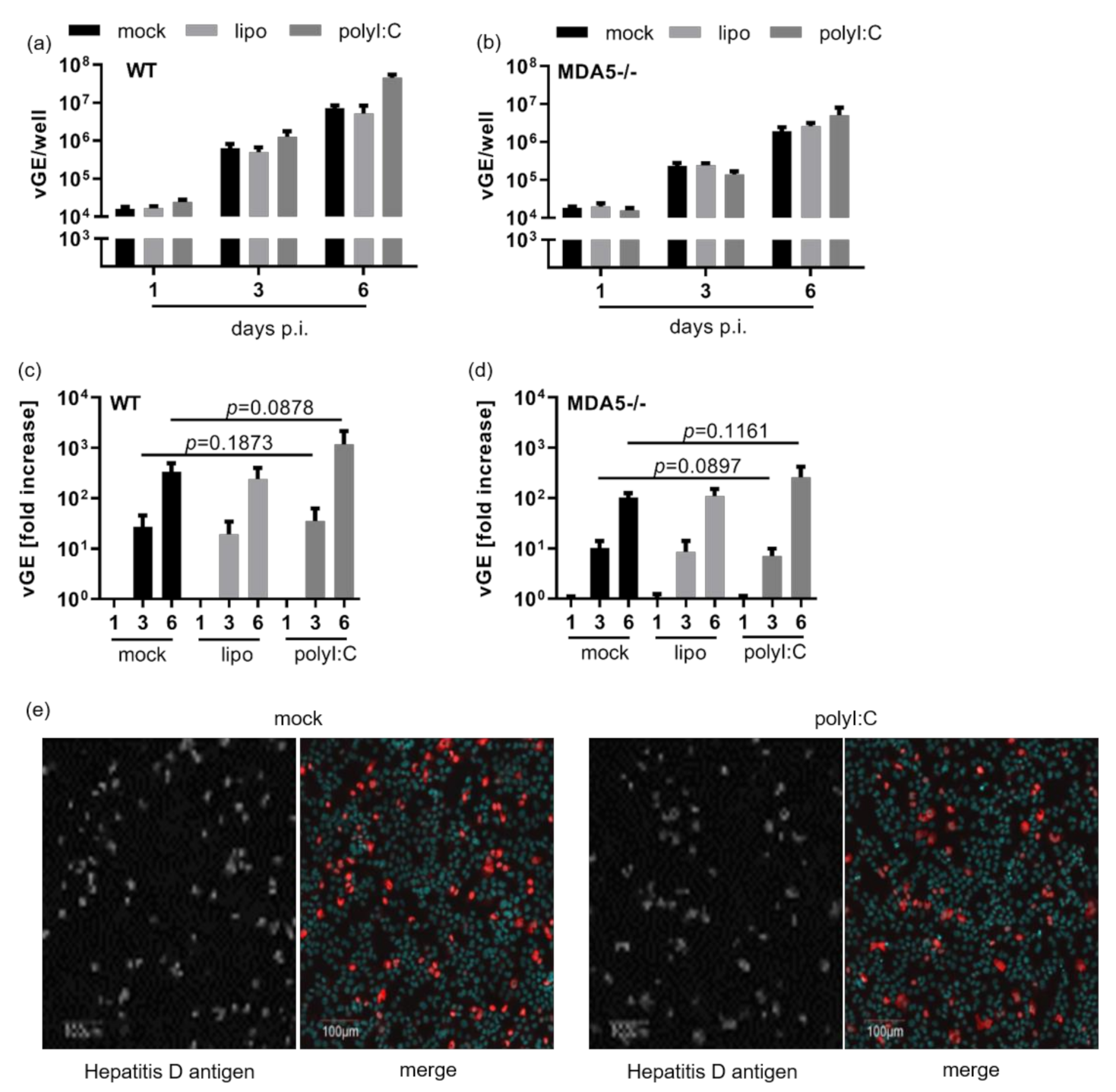
3.3. Intracellular Pattern Recognition of HDV Does Not Impair Virus Replication
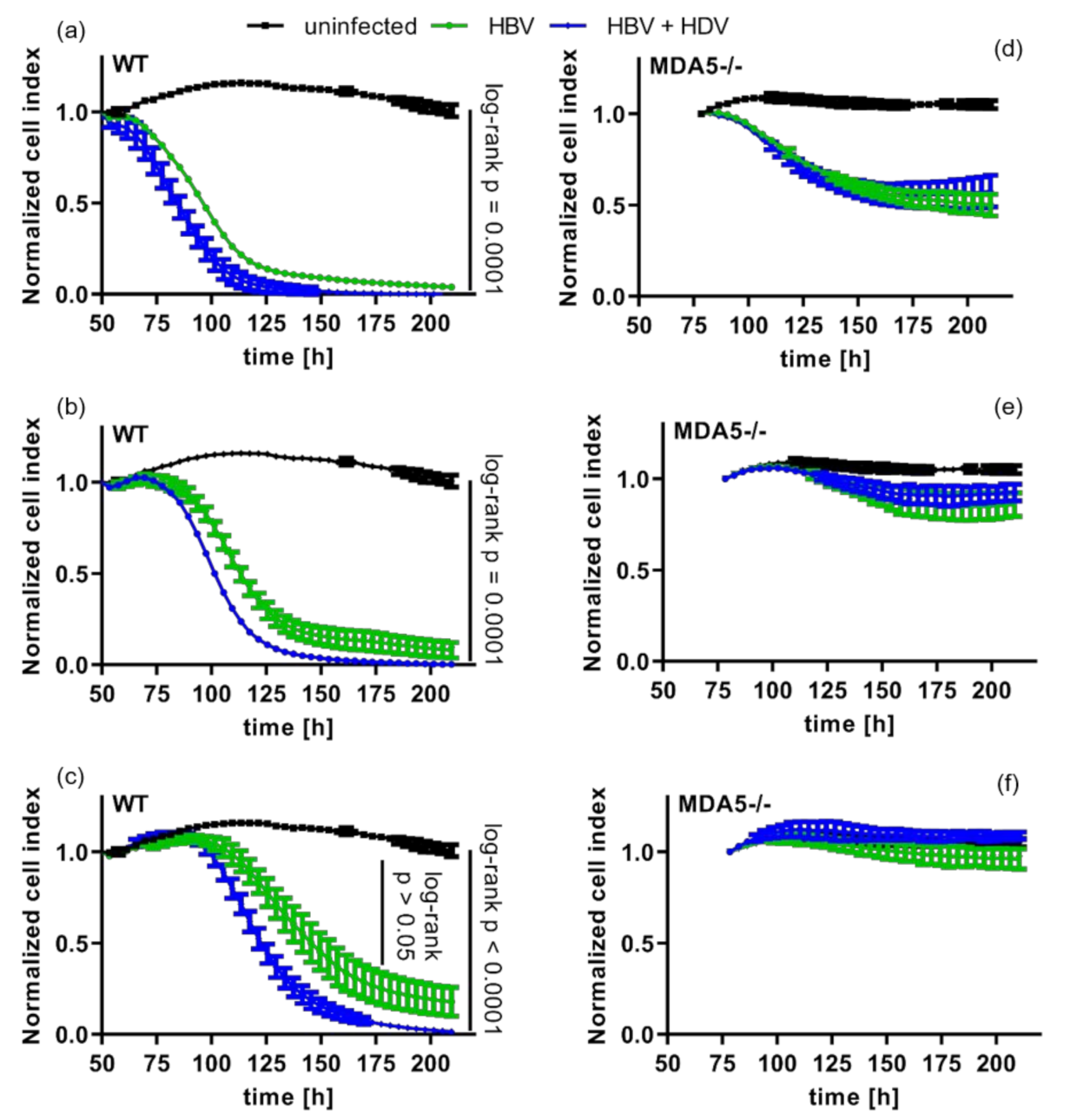
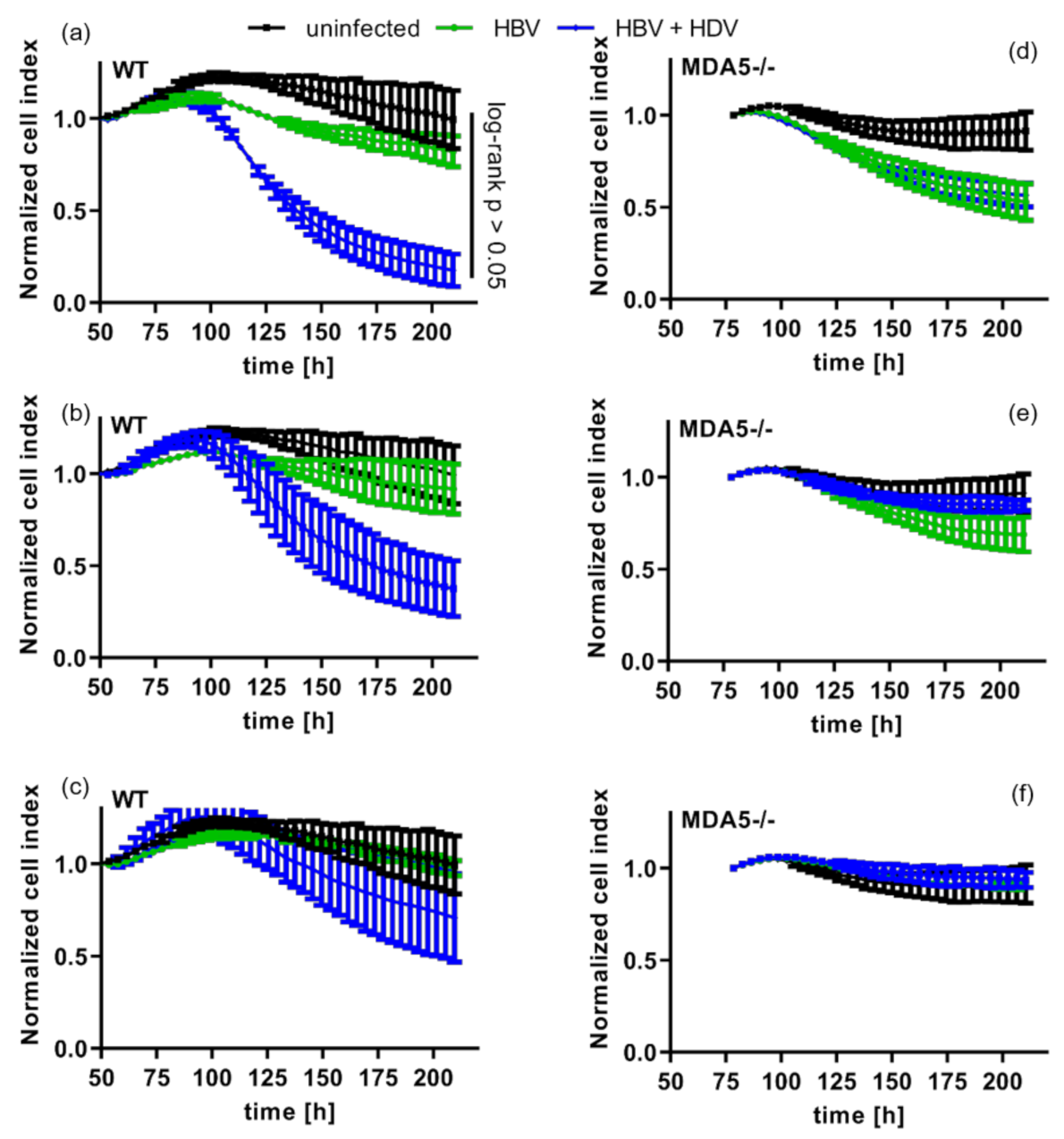
3.4. Pattern Recognition of HDV Increases Cytotoxic T-Cell Killing of Infected Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A




References
- Koh, C.; Heller, T.; Glenn, J.S. Pathogenesis of and New Therapies for Hepatitis D. Gastroenterology 2019, 156, 461–476.e461. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis D virus in 2021: Virology, immunology and new treatment approaches for a difficult-to-treat disease. Gut 2021, 70, 1782–1794. [Google Scholar] [CrossRef] [PubMed]
- Sandmann, L.; Cornberg, M. Experimental Drugs for the Treatment of Hepatitis D. J. Exp. Pharm. 2021, 13, 461–468. [Google Scholar] [CrossRef]
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; Dos Santos, A.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis delta: Virological and clinical aspects. Virol. J. 2017, 14, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L. Fields Virology, 6th Edition. Clin. Infect. Dis. 2014, 59, 613. [Google Scholar] [CrossRef]
- Hosel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef]
- Jung, S.; Altstetter, S.M.; Protzer, U. Innate immune recognition and modulation in hepatitis D virus infection. World J. Gastroenterol. 2020, 26, 2781–2791. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef]
- Zhang, Z.; Filzmayer, C.; Ni, Y.; Sultmann, H.; Mutz, P.; Hiet, M.S.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Hepatitis D virus replication is sensed by MDA5 and induces IFN-beta/lambda responses in hepatocytes. J. Hepatol. 2018, 69, 25–35. [Google Scholar] [CrossRef]
- Suarez-Amaran, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olague, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-beta induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef]
- Burwitz, B.J.; Wettengel, J.M.; Muck-Hausl, M.A.; Ringelhan, M.; Ko, C.; Festag, M.M.; Hammond, K.B.; Northrup, M.; Bimber, B.N.; Jacob, T.; et al. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 2017, 8, 2146. [Google Scholar] [CrossRef]
- Schmidt, T.; Schmid-Burgk, J.L.; Hornung, V. Synthesis of an arrayed sgRNA library targeting the human genome. Sci. Rep. 2015, 5, 14987. [Google Scholar] [CrossRef]
- Schmid-Burgk, J.L.; Schmidt, T.; Gaidt, M.M.; Pelka, K.; Latz, E.; Ebert, T.S.; Hornung, V. OutKnocker: A web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014, 24, 1719–1723. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureau, C.; Guerra, B.; Lee, H. The middle hepatitis B virus envelope protein is not necessary for infectivity of hepatitis delta virus. J. Virol. 1994, 68, 4063–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchmann, B.; Döhner, K.; Schirdewahn, T.; Sodeik, B.; Manns, M.P.; Wedemeyer, H.; Ciesek, S.; von Hahn, T. A screening assay for the identification of host cell requirements and antiviral targets for hepatitis D virus infection. Antivir. Res. 2017, 141, 116–123. [Google Scholar] [CrossRef]
- Wisskirchen, K.; Kah, J.; Malo, A.; Asen, T.; Volz, T.; Allweiss, L.; Wettengel, J.M.; Lutgehetmann, M.; Urban, S.; Bauer, T.; et al. T cell receptor grafting allows virological control of Hepatitis B virus infection. J. Clin. Investig. 2019, 129, 2932–2945. [Google Scholar] [CrossRef] [PubMed]
- Wisskirchen, K.; Metzger, K.; Schreiber, S.; Asen, T.; Weigand, L.; Dargel, C.; Witter, K.; Kieback, E.; Sprinzl, M.F.; Uckert, W.; et al. Isolation and functional characterization of hepatitis B virus-specific T-cell receptors as new tools for experimental and clinical use. PLoS ONE 2017, 12, e0182936. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.; Eberle, F.; Seitz, S.; Mucke, N.; Huber, C.M.; Kiani, N.; Kaderali, L.; Lohmann, V.; Dalpke, A.; Bartenschlager, R. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 2011, 286, 27278–27287. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R.; Ackrill, A.M.; Goldin, R.D.; Kerr, I.M.; Thomas, H.C.; Stark, G.R. Expression of the terminal protein region of hepatitis B virus inhibits cellular responses to interferons alpha and gamma and double-stranded RNA. Proc. Natl. Acad. Sci. USA 1991, 88, 2888–2892. [Google Scholar] [CrossRef] [Green Version]
- Christen, V.; Duong, F.; Bernsmeier, C.; Sun, D.; Nassal, M.; Heim, M.H. Inhibition of alpha interferon signaling by hepatitis B virus. J. Virol. 2007, 81, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, Y.; Lin, S.; Zhang, X.; Xiang, L.; Yuan, Z. Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 2007, 88, 3260–3269. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Durantel, D.; Testoni, B.; Hantz, O.; Levrero, M.; Zoulim, F. Control of hepatitis B virus replication by innate response of HepaRG cells. Hepatology 2010, 51, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Lutgehetmann, M.; Bornscheuer, T.; Volz, T.; Allweiss, L.; Bockmann, J.H.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Dandri, M. Hepatitis B virus limits response of human hepatocytes to interferon-alpha in chimeric mice. Gastroenterology 2011, 140, 2074–2083.e2. [Google Scholar] [CrossRef] [PubMed]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef] [Green Version]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [Green Version]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Altstetter, S.; Wilsch, F.; Shein, M.; Schütz, A.; Protzer, U. Extracellular vesicles derived from Hepatitis-D Virus infected cells induce a proinflammatory cytokine response in human peripheral blood mononuclear cells and macrophages. Matters 2020, 1–10. [Google Scholar] [CrossRef]
- Preiss, S.; Thompson, A.; Chen, X.; Rodgers, S.; Markovska, V.; Desmond, P.; Visvanathan, K.; Li, K.; Locarnini, S.; Revill, P. Characterization of the innate immune signalling pathways in hepatocyte cell lines. J. Viral Hepat. 2008, 15, 888–900. [Google Scholar] [CrossRef]
- Faure-Dupuy, S.; Vegna, S.; Aillot, L.; Dimier, L.; Esser, K.; Broxtermann, M.; Bonnin, M.; Bendriss-Vermare, N.; Rivoire, M.; Passot, G.; et al. Characterization of Pattern Recognition Receptor Expression and Functionality in Liver Primary Cells and Derived Cell Lines. J. Innate Immun. 2018, 10, 339–348. [Google Scholar] [CrossRef]
- Ebert, G.; Poeck, H.; Lucifora, J.; Baschuk, N.; Esser, K.; Esposito, I.; Hartmann, G.; Protzer, U. 5′ Triphosphorylated small interfering RNAs control replication of hepatitis B virus and induce an interferon response in human liver cells and mice. Gastroenterology 2011, 141, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Perez-Vargas, J.; Amirache, F.; Boson, B.; Mialon, C.; Freitas, N.; Sureau, C.; Fusil, F.; Cosset, F.L. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 2019, 10, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 2011, 85, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, Y.; Mao, A.; Li, C.; Li, Y.; Tien, P. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell Mol. Immunol. 2010, 7, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar] [CrossRef]
- Hou, Z.; Zhang, J.; Han, Q.; Su, C.; Qu, J.; Xu, D.; Zhang, C.; Tian, Z. Hepatitis B virus inhibits intrinsic RIG-I and RIG-G immune signaling via inducing miR146a. Sci. Rep. 2016, 6, 26150. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis δ virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.H.; Chen, Y.S.; Chen, P.J. Nucleolar targeting of hepatitis delta antigen abolishes its ability to initiate viral antigenomic RNA replication. J. Virol. 2008, 82, 692–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Tham, C.Y.L.; Tan, A.T.; Kah, J.; Chia, A.; Volz, T.; Giersch, K.; Ladiges, Y.; Loglio, A.; Borghi, M.; Sureau, C.; et al. Hepatitis Delta Virus Infection Increases the Efficacy of HBV-Specific T Cell-Based Therapies. Cell Rep. Med. 2020. [Google Scholar] [CrossRef]
- Kaser, A.; Nagata, S.; Tilg, H. Interferon alpha augments activation-induced T cell death by upregulation of Fas (CD95/APO-1) and Fas ligand expression. Cytokine 1999, 11, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.T., Jr.; Gale, M., Jr.; Loo, Y.-M. RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Malathi, K.; Dong, B.; Gale, M., Jr.; Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 2007, 448, 816–819. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.-P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Jung, S.; von Thülen, T.; Yang, I.; Laukemper, V.; Rupf, B.; Janga, H.; Panagiotidis, G.-D.; Schoen, A.; Nicolai, M.; Schulte, L.N.; et al. A ribosomal RNA fragment with 2′,3′-cyclic phosphate and GTP-binding activity acts as RIG-I ligand. Nucleic Acids Res. 2020, 48, 10397–10412. [Google Scholar] [CrossRef]
- Steinberg, J.; Wadenpohl, T.; Jung, S. The Endogenous RIG-I Ligand Is Generated in Influenza A-Virus Infected Cells. Viruses 2021, 13, 1564. [Google Scholar] [CrossRef]
- Mentha, N.; Clément, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altstetter, S.M.; Quitt, O.; Pinci, F.; Hornung, V.; Lucko, A.M.; Wisskirchen, K.; Jung, S.; Protzer, U. Hepatitis-D Virus Infection Is Not Impaired by Innate Immunity but Increases Cytotoxic T-Cell Activity. Cells 2021, 10, 3253. https://doi.org/10.3390/cells10113253
Altstetter SM, Quitt O, Pinci F, Hornung V, Lucko AM, Wisskirchen K, Jung S, Protzer U. Hepatitis-D Virus Infection Is Not Impaired by Innate Immunity but Increases Cytotoxic T-Cell Activity. Cells. 2021; 10(11):3253. https://doi.org/10.3390/cells10113253
Chicago/Turabian StyleAltstetter, Sebastian Maximilian, Oliver Quitt, Francesca Pinci, Veit Hornung, Aaron Michael Lucko, Karin Wisskirchen, Stephanie Jung, and Ulrike Protzer. 2021. "Hepatitis-D Virus Infection Is Not Impaired by Innate Immunity but Increases Cytotoxic T-Cell Activity" Cells 10, no. 11: 3253. https://doi.org/10.3390/cells10113253
APA StyleAltstetter, S. M., Quitt, O., Pinci, F., Hornung, V., Lucko, A. M., Wisskirchen, K., Jung, S., & Protzer, U. (2021). Hepatitis-D Virus Infection Is Not Impaired by Innate Immunity but Increases Cytotoxic T-Cell Activity. Cells, 10(11), 3253. https://doi.org/10.3390/cells10113253