
Characterization of RNA Sensing Pathways in Hepatoma Cell Lines and Primary Human Hepatocytes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. PRR Agonist Treatment
2.3. qRT-PCR
2.4. Immunoblot Analysis
2.5. Flow Cytometry Analysis
2.6. Coronavirus Infection
2.7. Arenavirus Infection
2.8. Quantification of Bioactive IFN
2.9. Statistics
3. Results
3.1. Hep3B and HepG2 Cells Express Similar Levels of RNA Sensors as Primary Human Hepatocytes
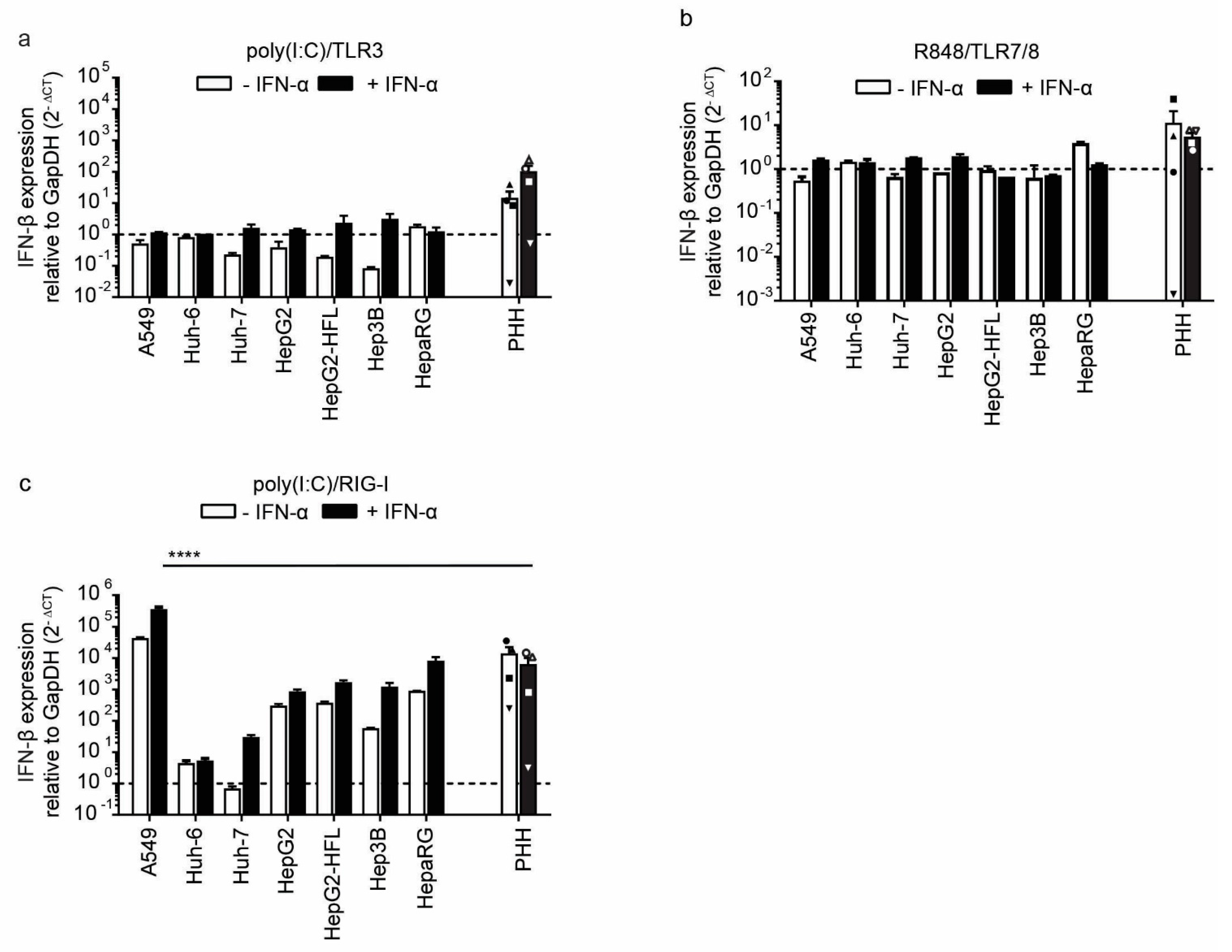
3.2. Liver Cells Primarily Sense RNA by Cytosolic RIG-like Receptors
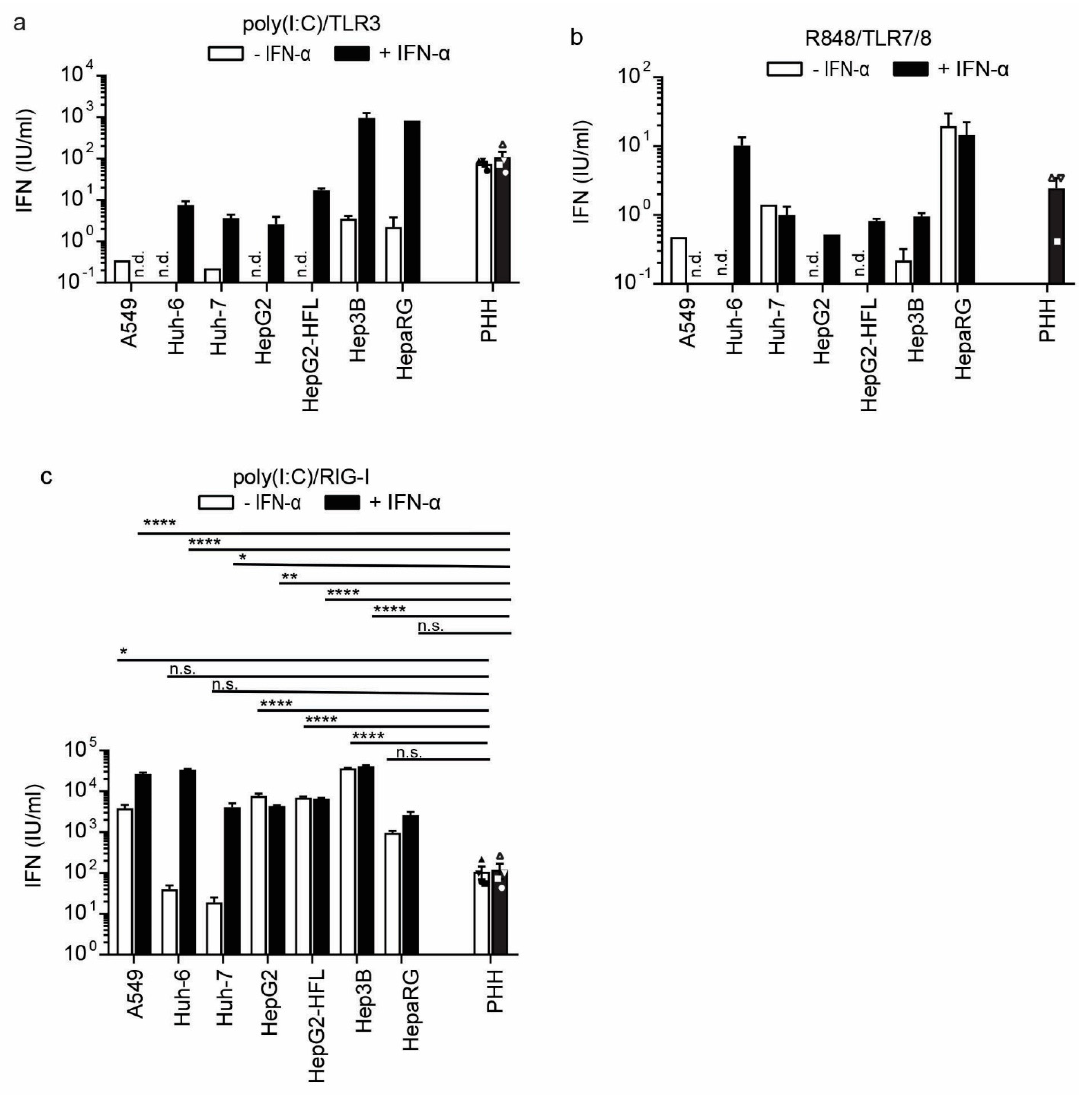
3.3. A Subset of Hepatoma Cells and Primary Hepatocytes Release Bioactive IFN upon Cytosolic RNA Sensing
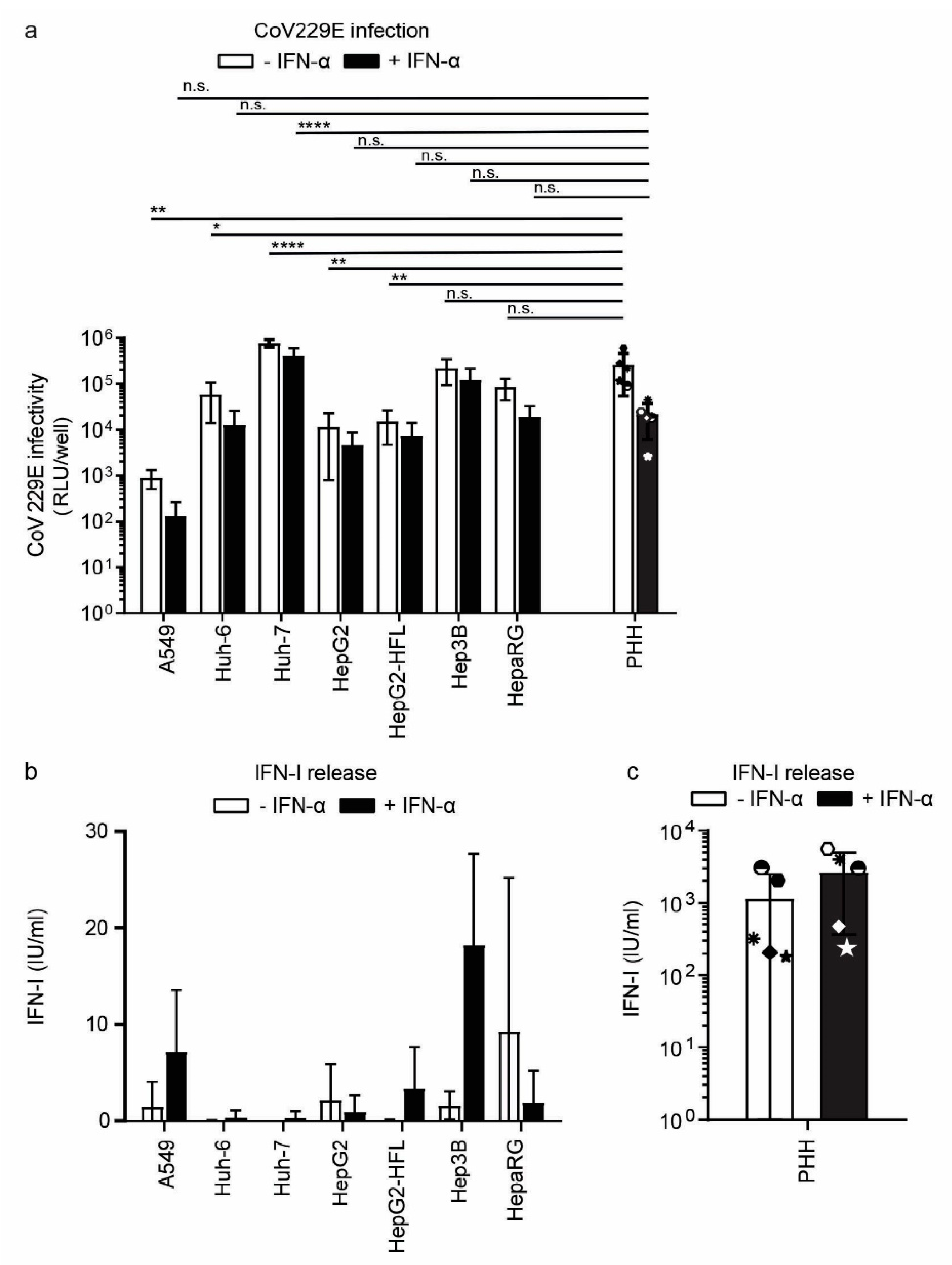
3.4. Sensing of RNA Viruses in Hepatoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef]
- Gray, K.K.; Worthy, M.N.; Juelich, T.L.; Agar, S.L.; Poussard, A.; Ragland, D.; Freiberg, A.N.; Holbrook, M.R. Chemotactic and inflammatory responses in the liver and brain are associated with pathogenesis of Rift Valley fever virus infection in the mouse. PLoS Negl. Trop. Dis. 2012, 6, e1529. [Google Scholar] [CrossRef] [Green Version]
- Lukashevich, I.S.; Tikhonov, I.; Rodas, J.D.; Zapata, J.C.; Yang, Y.; Djavani, M.; Salvato, M.S. Arenavirus-mediated liver pathology: Acute lymphocytic choriomeningitis virus infection of rhesus macaques is characterized by high-level interleukin-6 expression and hepatocyte proliferation. J. Virol. 2003, 77, 1727–1737. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, S.; Liu, H.; Li, W.; Lin, F.; Jiang, L.; Li, X.; Xu, P.; Zhang, L.; Zhao, L.; et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J. Hepatol. 2020, 73, 807–816. [Google Scholar] [CrossRef]
- Sheahan, T.; Imanaka, N.; Marukian, S.; Dorner, M.; Liu, P.; PLoSs, A.; Rice, C.M. Interferon lambda alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe 2014, 15, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandathil, A.J.; Graw, F.; Quinn, J.; Hwang, H.S.; Torbenson, M.; Perelson, A.S.; Ray, S.C.; Thomas, D.L.; Ribeiro, R.M.; Balagopal, A. Use of laser capture microdissection to map hepatitis C virus-positive hepatocytes in human liver. Gastroenterology 2013, 145, 1404–1413.e1. [Google Scholar] [CrossRef] [Green Version]
- Bellecave, P.; Filipowicz, M.; Donzé, O.; Kennel, A.; Gouttenoire, J.; Meylan, E.; Terracciano, L.; Tschopp, J.; Sarrazin, C.; Berg, T.; et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology 2009, 51, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.-M.; Gale, M.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [Green Version]
- Binder, M.; Eberle, F.; Seitz, S.; Mücke, N.; Hüber, C.M.; Kiani, N.; Kaderali, L.; Lohmann, V.; Dalpke, A.; Bartenschlager, R. Molecular mechanism of signal perception and integration by the innate immune sensor retinoic acid-inducible gene-I (RIG-I). J. Biol. Chem. 2011, 286, 27278–27287. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Research 2018, 7, 309. [Google Scholar] [CrossRef] [Green Version]
- Li, T.-C.; Wakita, T. Small animal models of hepatitis E virus infection. Cold Spring Harb. Perspect. Med. 2019, 9, a032581. [Google Scholar] [CrossRef]
- Billerbeck, E.; de Jong, Y.; Dorner, M.; de la Fuente, C.; PLoSs, A. Animal models for hepatitis C. Curr. Top. Microbiol. Immunol. 2013, 369, 49–86. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-R.; Park, S.-G. Mouse models for hepatitis B virus research. Lab. Anim. Res. 2018, 34, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Hirai-Yuki, A.; Hensley, L.; McGivern, D.R.; González-López, O.; Das, A.; Feng, H.; Sun, L.; Wilson, J.E.; Hu, F.; Feng, Z.; et al. MAVS-dependent host species range and pathogenicity of human hepatitis A virus. Science 2016, 353, 1541–1545. [Google Scholar] [CrossRef] [Green Version]
- Meuleman, P.; Leroux-Roels, G. The human liver-uPA-SCID mouse: A model for the evaluation of antiviral compounds against HBV and HCV. Antivir. Res. 2008, 80, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; de Jong, Y.P.; Meuleman, P. Animal models for the study of HCV. Curr. Opin. Virol. 2015, 13, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PLoSs, A.; Rice, C.M. Towards a small animal model for hepatitis C. EMBO Rep. 2009, 10, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- de Jong, Y.P.; Rice, C.M.; PLoSs, A. New horizons for studying human hepatotropic infections. J. Clin. Investig. 2010, 120, 650–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Schaewen, M.; Dorner, M.; Hueging, K.; Foquet, L.; Gerges, S.; Hrebikova, G.; Heller, B.; Bitzegeio, J.; Doerrbecker, J.; Horwitz, J.A.; et al. Expanding the Host Range of Hepatitis C Virus through Viral Adaptation. mBio 2016, 7, e01915-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitzegeio, J.; Bankwitz, D.; Hueging, K.; Haid, S.; Brohm, C.; Zeisel, M.B.; Herrmann, E.; Iken, M.; Ott, M.; Baumert, T.F.; et al. Adaptation of Hepatitis C Virus to Mouse CD81 Permits Infection of Mouse Cells in the Absence of Human Entry Factors. PLoS Pathog. 2010, 6, e1000978. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Simmonds, P.; Scheel, T.K.H.; Hjelle, B.; Cullen, J.M.; Burbelo, P.D.; Chauhan, L.V.; Duraisamy, R.; Leon, M.S.; Jain, K.; et al. Identification of Rodent Homologs of Hepatitis C Virus and Pegiviruses. mBio 2013, 4, e00216-13. [Google Scholar] [CrossRef] [Green Version]
- Billerbeck, E.; Wolfisberg, R.; Fahnøe, U.; Xiao, J.W.; Quirk, C.; Luna, J.M.; Cullen, J.M.; Hartlage, A.S.; Chiriboga, L.; Ghoshal, K.; et al. Mouse models of acute and chronic hepacivirus infection. Science 2017, 357, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, S.; Murthy, S.; Sharma, H.; Hartlage, A.S.; Kumar, A.; Gadi, S.V.; Simmonds, P.; Chauhan, L.V.; Scheel, T.K.; Billerbeck, E.; et al. Viral persistence, liver disease, and host response in a hepatitis C–like virus rat model. Hepatology 2017, 68, 435–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vondran, F.W.R.; Katenz, E.; Schwartlander, R.; Morgul, M.H.; Raschzok, N.; Gong, X.; Cheng, X.; Kehr, D.; Sauer, I.M. Isolation of primary human hepatocytes after partial hepatectomy: Criteria for identification of the most promising liver specimen. Artif. Organs 2008, 32, 205–213. [Google Scholar] [CrossRef]
- Carpentier, A.; Sheldon, J.; Vondran, F.W.R.; Brown, R.J.; Pietschmann, T. Efficient acute and chronic infection of stem cell-derived hepatocytes by hepatitis C virus. Gut 2020, 69, 1659–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumpter, R.; Loo, Y.-M.; Foy, E.; Li, K.; Yoneyama, M.; Fujita, T.; Lemon, S.M.; Gale, M. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 2005, 79, 2689–2699. [Google Scholar] [CrossRef] [Green Version]
- Grünvogel, O.; Colasanti, O.; Lee, J.-Y.; Klöss, V.; Belouzard, S.; Reustle, A.; Esser-Nobis, K.; Hesebeck-Brinckmann, J.; Mutz, P.; Hoffmann, K.; et al. Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll-Like Receptor 3 in Hepatocytes. Gastroenterology 2018, 154, 2237–2251.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israelow, B.; Narbus, C.M.; Sourisseau, M.; Evans, M.J. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014, 60, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Y.; Urban, S. Hepatitis B Virus Infection of HepaRG Cells, HepaRG–hNTCP Cells, and Primary Human Hepatocytes. Methods Mol. Biol. 2017, 1540, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Ait–Goughoulte, M.; Michelet, M.; Floriot, O.; Bonnin, M.; Gruffaz, M.; Rivoire, M.; Fletcher, S.; Javanbakht, H.; Lucifora, J.; et al. Expression and functionality of Toll– and RIG–like receptors in HepaRG cells. J. Hepatol. 2015, 63, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Preiss, S.; Thompson, A.; Chen, X.; Rodgers, S.; Markovska, V.; Desmond, P.; Visvanathan, K.; Li, K.; Locarnini, S.; Revill, P. Characterization of the innate immune signalling pathways in hepatocyte cell lines. J. Viral Hepat. 2008, 15, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, Z.; Kato, N.; Gale, M.; Lemon, S.M. Distinct poly(I–C) and virus–activated signaling pathways leading to interferon–beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef] [Green Version]
- Windisch, M.P.; Frese, M.; Kaul, A.; Trippler, M.; Lohmann, V.; Bartenschlager, R. Dissecting the interferon–induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 2005, 79, 13778–13793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar] [PubMed]
- Narbus, C.M.; Israelow, B.; Sourisseau, M.; Michta, M.L.; Hopcraft, S.E.; Zeiner, G.M.; Evans, M.J. HepG2 cells expressing microRNA miR–122 support the entire hepatitis C virus life cycle. J. Virol. 2011, 85, 12087–12092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen–Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzé, G.; Di Marco, S.; Mouchel–Vielh, E.; Monneron, D.; Bandu, M.T.; Horisberger, M.A.; Dorques, A.; Lutfalla, G.; Mogensen, K.E. Domains of interaction between alpha interferon and its receptor components. J. Mol. Biol. 1994, 243, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Hueging, K.; Doepke, M.; Vieyres, G.; Bankwitz, D.; Frentzen, A.; Doerrbecker, J.; Gumz, F.; Haid, S.; Wölk, B.; Kaderali, L.; et al. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell–to–cell transmission by contributing to a postenvelopment step of assembly. J. Virol. 2014, 88, 1433–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine, M.; Riemer, M.; Krech, T.; DeTemple, D.; Jäger, M.D.; Lehner, F.; Manns, M.P.; Klempnauer, J.; Borlak, J.; Bektas, H.; et al. Explanted diseased livers–A possible source of metabolic competent primary human hepatocytes. PLoS ONE 2014, 9, e101386. [Google Scholar] [CrossRef]
- Pitkäranta, A.; Hovi, T. Induction of interferon in human leukocyte cultures by natural pathogenic respiratory viruses. J. Interferon. Res. 1993, 13, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Möller, R.; Fedeli, C.; Gerold, G.; Kunz, S. Comparison of the innate immune responses to pathogenic and nonpathogenic clade B new world arenaviruses. J. Virol. 2019, 93, e00148-19. [Google Scholar] [CrossRef]
- Moreno, H.; Kunz, S. The Protein Kinase Receptor Modulates the Innate Immune Response against Tacaribe Virus. Viruses 2021, 13, 1313. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Jokinen, J.D.; Holz, G.E.; Whang, P.S.; Martin, A.M.; Warner, N.L.; Arteel, G.E.; Lukashevich, I.S. Novel mechanism of arenavirus-induced liver pathology. PLoS ONE 2015, 10, e0122839. [Google Scholar] [CrossRef]
- Ikeda, M.; Sugiyama, K.; Mizutani, T.; Tanaka, T.; Tanaka, K.; Sekihara, H.; Shimotohno, K.; Kato, N. Human hepatocyte clonal cell lines that support persistent replication of hepatitis C virus. Virus Res. 1998, 56, 157–167. [Google Scholar] [CrossRef]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Roelandt, P.; Obeid, S.; Paeshuyse, J.; Vanhove, J.; Van Lommel, A.; Nahmias, Y.; Nevens, F.; Neyts, J.; Verfaillie, C.M. Human pluripotent stem cell-derived hepatocytes support complete replication of hepatitis C virus. J. Hepatol. 2012, 57, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Robotham, J.M.; Lee, E.; Dalton, S.; Kneteman, N.M.; Gilbert, D.M.; Tang, H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012, 8, e1002617. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; PLoSs, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrion, R.; Brasky, K.; Mansfield, K.; Johnson, C.; Gonzales, M.; Ticer, A.; Lukashevich, I.; Tardif, S.; Patterson, J. Lassa virus infection in experimentally infected marmosets: Liver pathology and immunophenotypic alterations in target tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolay, W.; Moeller, R.; Kahl, S.; Vondran, F.W.R.; Pietschmann, T.; Kunz, S.; Gerold, G. Characterization of RNA Sensing Pathways in Hepatoma Cell Lines and Primary Human Hepatocytes. Cells 2021, 10, 3019. https://doi.org/10.3390/cells10113019
Nicolay W, Moeller R, Kahl S, Vondran FWR, Pietschmann T, Kunz S, Gerold G. Characterization of RNA Sensing Pathways in Hepatoma Cell Lines and Primary Human Hepatocytes. Cells. 2021; 10(11):3019. https://doi.org/10.3390/cells10113019
Chicago/Turabian StyleNicolay, Wiebke, Rebecca Moeller, Sina Kahl, Florian W. R. Vondran, Thomas Pietschmann, Stefan Kunz, and Gisa Gerold. 2021. "Characterization of RNA Sensing Pathways in Hepatoma Cell Lines and Primary Human Hepatocytes" Cells 10, no. 11: 3019. https://doi.org/10.3390/cells10113019
APA StyleNicolay, W., Moeller, R., Kahl, S., Vondran, F. W. R., Pietschmann, T., Kunz, S., & Gerold, G. (2021). Characterization of RNA Sensing Pathways in Hepatoma Cell Lines and Primary Human Hepatocytes. Cells, 10(11), 3019. https://doi.org/10.3390/cells10113019